

Abstract
Chronic Inflammatory Demyelinating Polyneuropathy is a debilitating autoimmune disease characterized by peripheral nerve demyelination and dysfunction. How the autoimmune response is initiated, identity of provoking antigens, and pathogenic effector mechanisms are not well-defined. The Autoimmune Regulator (Aire) plays a critical role in central tolerance by promoting thymic expression of self-antigens and deletion of self-reactive T cells. Here, we utilized mice with hypomorphicAire function and two patients with Aire mutations to define how Aire deficiency results in spontaneous autoimmune peripheral neuropathy. Autoimmunity against peripheral nerves in both mice and humans targets Myelin Protein Zero (P0), an antigen whose expression is Aire-regulated in the thymus. Consistent with a defect in thymic tolerance, CD4+ T cells are sufficient to transfer disease in mice and produce IFN-gamma in infiltrated peripheral nerves. Our findings suggest that defective Aire-mediated central tolerance to P0 initiates an autoimmune Th1 effector response toward peripheral nerves.
Introduction
Chronic Inflammatory Demyelinating Polyneuropathy (CIDP) is the most common acquired chronic autoimmune neuropathy and affects 1 in 10,000 individuals (1). The pathogenic steps resulting in immune destruction of the peripheral nervous system (PNS) are not well-understood, in part because of the scarcity of robust animal models. We recently reported that spontaneous autoimmune peripheral neuropathy develops in NOD mice harboring a G228W point mutation in the Autoimmune Regulator (Aire) gene (NOD.AireGW/+ mice) (2). Aire plays a critical role in central tolerance by upregulating the ectopic expression of a wide array of tissue-specific self-antigens in medullary thymic epithelial cells (mTECs) (3) and promoting the negative selection of developing thymocytes that recognize these antigens with high affinity (4). NOD.AireGW/+ mice have hypomorphicAire function in that mTECs express tissue-specific self-antigens at approximately 10% of normal levels (2). NOD.AireGW/+ mice are protected from early-lethal autoimmune diseases (e.g. exocrine pancreatitis, pneumonitis) but remain susceptible to a distinct set of autoimmune diseases that include autoimmune peripheral neuropathy.
Patients with Autoimmune Polyendocrinopathy Syndrome Type 1 (APS1) have genetic mutations in Aire and develop autoimmunity. Recently, CIDP was recognized as a potential novel component of APS1 in two unrelated children with progressive sensory loss, motor weakness, and confirmed mutations in Aire(5). We show here that NOD.AireGW/+ mice develop autoimmune peripheral neuropathy that strongly resembles CIDP. We utilize NOD.AireGW/+ mice to identify Myelin Protein Zero (P0) as a major Aire-regulated PNS antigen and demonstrate defective tolerance to P0 in both Aire-deficient mice and humans.
Materials and Methods
Mice
NOD.AireGW/+ mice were generated as previously described (2). NOD.scid mice were purchased from the Jackson Laboratory. Mice were housed in a pathogen free barrier facility at University of California, San Francisco (UCSF) and at the University of North Carolina, Chapel Hill (UNC Chapel Hill). Clinical neuropathy and diabetes were assessed as described in (6). For the group of mice used in neuropathy incidence studies, mice that developed diabetes were maintained on insulin (I.P. injection daily) until 22 weeks of age. NOD.AireGW/+ mice with clinical neuropathy were used as serum and splenocyte donors.Experiments complied with the Animal Welfare Act and the National Institute of Health (NIH) guidelines for the ethical care and use of animals in biomedical research.
Histology/Electron Microscopy
H&E staining was performed as described in (2). Immune infiltration was scored in a blinded fashion with 0, 1, 2, 3, 4 scores indicating no, <25%, 25-50%, 50-75%, and >75% infiltration respectively. Luxol fast blue staining was performed as described in (7). Toluidine blue staining and transmission electron microscopy was performed on sciatic nerve sections from mice perfused with 2% paraformaldehyde, 2.5% gluteraldehyde in 0.1M cacodylate buffer. Toluidine blue staining was performed on semi-thin sections of epon embedded sciatic nerves. Transmission electron microscopy was performed as described in (8).
Indirect immunofluorescence
Immunofluorescence staining was performed using diluted (1:600) mouse and human sera on OCT-embedded NOD.scid sciatic nerve sections as described in (9).
Immunohistochemistry
Stains for immune cells in OCT-embedded sciatic nerves were performed with anti-CD4 (BioXcell, GK1.5), anti-CD8 (BioXcell, YTS169), and anti-F4/80 (eBioscience BM8) antibodies as described in (9).
Adoptive Transfer
Adoptive transfer of whole spleen and lymph node cells from neuropathic NOD.AireGW/+ mice were performed as described in (6). Diabetic NOD.AireGW/+ mice were excluded as donors. For each NOD.scid recipient mouse, 10×106 whole spleen and lymph node cells were transferred. CD4+ and CD8+ T cells were isolated by staining with CD4-FITC (Southern Biotech) and CD8a-APCCy7 (BD). Populations were purified using a MoFlo (Dako) cell sorter and plated in the presence of anti-CD3 and anti-CD28 (1 microgram per mL in PBS) for 96 hours. For some experiments, anti-CD4 and anti-CD8 microbeads (MiltenyiBiotec) were used for positive selection on a MS or LS column. For the last 24 hours, 1 microgram/mL of IL-2 (20 U/mL) was added to culture. 7×106 CD4 T cells, 7×106 CD8, or 3×106 CD8 T cells were transferred. Co-transfers of 7×106 CD4 and 7×106 CD8 T cells were also performed.
Intracellular Cytokine Staining
Infiltrating immune cells were isolated from sciatic nerves by mincing and digestion in 2 mg/ml collagenase. Cells were stimulated with PMA/Ionomycin and stained as described in (9).
Immunoblot/Immunoaffinity purification
Sciatic nerve extracts were prepared in Chaps buffer. Immunoblots and immunoaffinity purification were performed as described in (10). Bound antigen was eluted using 100 mM TEA, pH 11.5 and neutralized with 1M phosphate buffer, pH 6.8.
Mass Spectrometry
Antigen samples were excised and submitted to the Stanford University Protein and Nucleic Acid facility. Samples were subjected to tryptic digestion and resulting peptides subjected to mass analysis using a 4700 Proteomics Analyzer (Applied Biosystems).
Fusion Protein Purification/Competition Studies
P0-maltose binding protein (MBP) fusion protein was produced as described in (10). Human P0 cDNA clone (IMAGE) (>90% homology to mouse P0) was subcloned into pMAL-c2X (New England Biolabs) using EcoRI (5′) and HindIII (3′) restriction sites. These sites were introduced onto human P0 amplicons generated with primers 5′ GAATTCATGGCTCCCGGGGCTC CCT 3′ and 5′ AAGCTTCTATTTCTTATCCTTGCGAGACTCC 3′. Competition studies were performed by pre-incubating sera with serial dilution of recombinant P0-MBP or MBP prior to immunoblotting. ELISAs were performed using 1:600 serum dilutions and 50 ng/well of recombinant protein.
ELISA
Recombinant P0-MBP or MBP alone (50 ng per well) were immobilized in wells and diluted (1:600) serum specimens were used for assays. Rat anti-mouse alkaline phosphatase was used to detect mouse immunoglobulin.
Real Time RT-PCR
Thymic stromal preparations were performed as described in (10). mTECs were sorted using the markers Propidium Iodide (PI)-, CD45- G8.8+, Ly51int. P0 primer/probe was purchased from Applied Biosystems (Mm00485139_m1). 6-8 week old mice were used for mTEC purification.
Proliferation Assay
Proliferation assay using 3H thymidine incorporation was performed as described in (11). P0 180-199 SSKRGRQTPVLYAMLDHSRS and HEL 11-25 AMKRHGLDNYRGYSL peptides were purchased from Genemed Synthesis. 5 × 105splenocytes/well were cultured in HL-1 medium with nonessential amino acids (BioWhittaker), 2mM L-glutamine, 1mM sodium pyruvate, 55 mM b-mercaptoethanol with peptide at 3 different concentrations and without peptide. Cultures were pulsed with 1 microCurie of 3H thymidine and incubated for an additional 18 hours. Cells were harvested on a glass fiber filter and incorporation of 3H thymidine measured using a liquid scinitillation counter. Stimulation index was calculated by dividing counts per minute (cpm) with antigen by cpm without antigen.
Radioligand Binding Assay
Human P0 cDNA clone (IMAGE) was in vitro transcribed and translated with 35S labeling, and assays performed as described in (9). Autoantibody index was calculated as [cpm sample-cpm negative control] divided by [cpm positive standard-cpm negative standard] x100. Samples were positive if >3 SD above mean for healthy control group.
Human subjects
Patients and controls were included in the study after written informed consent was obtained. Study protocol was approved by the institutional review board at UCSF.
Statistics
Data was analyzed with Prism software (GraphPad) using unpaired t-tests. Log rank tests were used for comparison of survival curves. p≤0.05 was considered significant.
Results
HypomorphicAire mice spontaneously develop autoimmune peripheral neuropathy
By 22 weeks of age, approximately 80% of female NOD.AireGW/+ mice develop spontaneous neuropathy (Fig. 1A, (2)) that is not seen in wildtype (NOD.WT) littermates. Affected mice display bilateral weakness of the hind limbs that progresses to severe paralysis affecting all limbs (Fig. 1B and Supplemental Movie 1) This neuropathy is associated with immune cell infiltration in NOD.AireGW/+ sciatic nerves (Fig. 1C and D), but not in brain or spinal cord (Supplemental Fig. 1A).
Figure 1. HypomorphicAire mice spontaneously develop autoimmune peripheral neuropathy.

A) Neuropathy curve of female NOD.AireGW/+ (AireGW/+) mice and female NOD.WT (WT) littermates. B) Clasping of hind limbs in neuropathic NOD.AireGW/+ mouse indicating bilateral weakness that is not seen in NOD.WT mice. C) Representative H&E stained, formalin fixed, longitudinal sections of sciatic nerves from an NOD.WT and NOD.AireGW/+ mouse at 22 weeks of age (20x magnification). D) Cumulative infiltration scores of sciatic nerves from NOD.WT and NOD.AireGW/+ mice. Each symbol represents an individual mouse. Asterix (*) indicates significant difference between the two groups. E) Luxol fast blue stained longitudinal sections of sciatic nerves from NOD.WT and NOD.AireGW/+ mice for the presence of myelin. Arrows point to areas of demyelination (20x magnification). Images are representative of 4 independent experiments. F) Toluidine blue stained cross sections of NOD.WT and NOD.AireGW/+ sciatic nerves (40x magnification) for the presence of myelin. Images are representative of at least 3 independent experiments. G) Electron micrograph of sciatic nerves shown in (F). Arrows point to myelinated axon in NOD.WT, and demyelinated axon in NOD.AireGW/+ nerve (1100x magnification). Images are representative of at least 3 independent experiments.
Progression of neuropathy is associated with upregulation in sciatic nerves of CXCL10 (IP-10), CCL2 (MCP-1), and CCL5 (RANTES) (Supplemental Fig. 1B), three chemokines which have been linked with inflammatory neuropathies (11, 12). Multifocal areas of demyelination are seen on Luxol fast blue stained sections of NOD.AireGW/+ sciatic nerve (Fig. 1E). Additionally, decreased density of myelinated axons is seen on toluidine blue stained cross sections of sciatic nerve in neuropathic NOD.AireGW/+ mice (Fig. 1F). Electron microscopy of these same sciatic nerves demonstrates demyelination of individual axons in NOD.AireGW/+ mice (Fig. 1G). The symmetric nerve dysfunction, mononuclear cells infiltrating peripheral nerves, and multifocal demyelination recapitulate many of the features of CIDP.
In addition to developing autoimmune peripheral neuropathy, NOD.AireGW/+ mice also develop spontaneous autoimmune diabetes. The incidence of diabetes in NOD.AireGW/+ mice is the same as in NOD.WT mice (Supplemental Fig. 2A and (2)). Histology of sciatic nerves from 5 diabetic (non-neuropathic) NOD.AireGW/+ mice did not demonstrate signs of immune infiltration in the sciatic nerve (Supplemental Fig. 2B and C). Sera from 2 diabetic NOD.AireGW/+ mice were negative for P0 autoantibodies by ELISA (data not shown).Rarely, diabetic NOD.AireGW/+ mice maintained on insulin also develop neuropathy. Histology of sciatic nerves from 2 mice with both diabetes and neuropathy demonstrated moderate immune infiltration (Supplemental Fig. 2C).
CD4+ T cells transfer autoimmune peripheral neuropathy and produce IFN-gamma in peripheral nerves
To determine the cellular composition of immune cells infiltrating the PNS of NOD.AireGW/+ mice, we stained sciatic nerves from NOD.WT and NOD.AireGW/+ mice with antibodies against CD4, CD8, F4/80, B220, and CD11c cell surface markers. Within NOD.AireGW/+ nerve infiltrates, CD4+ helper T cells and F4/80+ macrophages were more frequent than CD8+ cytotoxic T cells, B220+ B cells, or CD11c+ dendritic cells (Fig. 2A and data not shown). Few cells expressing these markers were seen in NOD.WT nerves.
Figure 2. CD4+ T cells are sufficient to transfer autoimmune peripheral neuropathy.

A) Immunohistochemical stains of sciatic nerve from either neuropathic NOD.AireGW/+ (AireGW/+) mouse or NOD.WT (WT) littermate with antibodies against CD4, CD8, and F4/80. Images shown are representative of 2 separate experiments. B) Representative H&E stained, formalin fixed, longitudinal sections of sciatic nerve from neuropathic mouse (T936) used as donor in adoptive transfer experiments (20x magnification). Arrow points to areas of dense immune cell infiltration.C) Donor NOD.AireGW/+ mice used in adoptive transfer experiments, sex, age, clinical finding, and histological score of sciatic nerves at time of spleen and lymph node harvest. D) Neuropathy curve of NOD.scid mice receiving activated whole spleen and lymph node cells (10×106 cells per recipient; n=8), sorted CD4+ T cells (7×106 cells per recipient; n=12), sorted CD8+ T cells (3×106; n=4 or 7×106; n=7), or combined CD4+ and CD8+ T cells (7×106 of each, n=8) from neuropathic NOD.AireGW/+ donor mice. Cumulative data from 4 independent experiments are shown. E) Histological scores of sciatic nerves from recipient mice at the time of sacrifice. Each shape represents an individual mouse. Cumulative data from 4 independent experiments are shown. Line represents average score. Asterix (*) represents significant difference from whole spleen and lymph node transfer (p ≤0.05). n.s.= not significant.
To demonstrate a cellular basis for the pathogenesis of autoimmune peripheral neuropathy, we transferred spleen and lymph node cells from neuropathic NOD.AireGW/+ mice (between 19 and 34 weeks of age) into immunodeficientPrkdcscid/scid NOD mice (NOD.scid mice). Histology of donor sciatic nerves demonstrated immune infiltration in all nerves tested (Fig. 2B and 2C). 8 out of 8 recipients of whole spleen and lymph node cells (10×106 cells per recipient) from neuropathic NOD.AireGW/+ mice developed neuropathy by 10 weeks post transfer (Fig. 2D), suggesting that immune cells are a pathogenic entity.
To determine the relative importance of CD4+ helper and CD8+ cytotoxic T cell subsets, we transferred purified CD4+ and CD8+ populations into NOD.scid recipients. Similar to recipients of whole spleen and lymph node cells, 12 out of 12 recipients of CD4+ cells (7×106 cells per recipient) developed neuropathy by 10 weeks post transfer (Fig. 2D). This neuropathy incidence in recipients of 7×106 CD4+ cells was not significantly different from recipients of 10×106 whole spleen and lymph node cells. Thus, CD4+ cells are sufficient to transfer neuropathy.
None of the 4 recipients of 3×106 CD8+ cells were neuropathic up to 12 weeks post transfer (Fig. 2D). However, 3 out of 7 recipients of 7×106 CD8+ cells developed neuropathy within 12 weeks (Fig. 2D). The incidence of neuropathy in recipients of 7×106 CD8+ cells was significantly less than recipients of 10×106 whole spleen and lymph node cells (p= 0.008) and significantly less than recipients of 7×106 CD4+ cells (p= 0.0193). This finding suggests that CD8+ cells can also transfer neuropathy, but less effectively than CD4+ cells. Finally, 8 out of 8 recipients of both 7×106 CD4+ and 7×106 CD8+ cells combined developed neuropathy by 10 weeks post transfer. The incidence of neuropathy in recipients of both CD4+ and CD8+ cells was not significantly different from either recipients of 10×106whole spleen and lymph node cells or from recipients of 7×106 CD4+ cells (Fig. 2D). Histological examination of sciatic nerves from recipients demonstrated immune infiltration within sciatic nerves in all mice with clinical neuropathy at the time of sacrifice (Fig. 2E).
We stained CD4+ T cells infiltrating NOD.AireGW/+ sciatic nerves for intracellular IFN-gamma, IL-4, and IL-17 cytokines to determine the frequency of subsets producing these cytokines. IFN-gamma production was seen in approximately 40% of CD4+ T cells (Fig. 3A and 3B), and IL-4 and IL-17 production was rarely seen (Fig. 3B). No infiltrating CD4+ T cells were detected in NOD.WT sciatic nerves. A comparable percentage of CD4+ T cell subsets were seen in the spleen of NOD.AireGW/+ and NOD.WT mice (Fig. 3C).
Figure 3. CD4+ T cells infiltrating the sciatic nerve predominantly produce IFN-gamma.

A) Representative flow cytometry plot of CD4+ T cells infiltrating the sciatic nerves of 18 week old neuropathic NOD.AireGW/+ mouse stained for intracellular IFN-gamma after activation with PMA/ionomycin. Negative control is CD4+ T cells from same sciatic nerve preparation that have not been activated. Plot is representative of 4 mice from 3 independent experiments. B) Average percent of CD4+ T cells producing IFN-gamma, IL4, or IL17A in sciatic nerves of NOD.WT or neuropathic NOD.AireGW/+ mice (n=4 for each group). Each organ was processed separately (not pooled). Data are included from 3 independent experiments. C) Average percent of CD4+ T cells producing IFN-gamma, IL4 or IL17A in spleen of NOD.WT or neuropathic NOD.AireGW/+ mice (n=4 for each group). Each organ was processed separately (not pooled). Data are average of 3 independent experiments. D) Absolute numbers of CD4+ T cells producing IFN-gamma, IL4, or IL17A in sciatic nerves of NOD.WT or neuropathic NOD.AireGW/+ mice. E) Absolute numbers of CD4+ T cells producing IFN-gamma, IL4, or IL17A in spleens of NOD.WT or neuropathic NOD.AireGW/+ mice.
Within the sciatic nerves, an average of 1,700 IFN-gamma producing CD4+ T cells were isolated from each neuropathic NOD.AireGW/+ mouse (Figure 3D). On the other hand, fewer than 100 IL-4 and IL-17 producing CD4+ T cells were present on average in the sciatic nerves. Within the spleen, an average of 5.9×106 IFN-gamma producing CD4+ T cells was seen in NOD.AireGW/+ mice, compared to 2×106 IFN-gamma producing CD4+ T cells in NOD.WT mice (Figure 2G). Fewer than 1×106 IL-4 and IL-17 producing CD4+ T cells were seen in NOD.AireGW/+ and NOD.WT spleens. Taken together, this data is consistent with a strong Th1 effector response in the sciatic nerves in the model.
Myelin Protein Zero (P0) is a major Aire-regulated PNS autoantigen
Serum autoantibodies from Aire-deficient mice have been successfully utilized to identify tissue-specific self-antigens that are targets of autoimmunity (9, 10). On indirect immunofluorescence, serum autoantibodies in neuropathic NOD.AireGW/+ mice react against sciatic nerves in a streak-like pattern (Fig. 4A). Immunoblot with sera from neuropathic NOD.AireGW/+ mice against whole sciatic nerve extract demonstrated an oligoclonal pattern of reactivity in NOD.AireGW/+ sera which was predominantly against a 25-30 kD antigen (Fig. 4B). This 25-30 kD antigen was immunopurified using autoantibodies from sera of NOD.AireGW/+ mice to bind antigens from whole sciatic nerve extract and subjected to peptide mass fingerprinting. Microsequenced peptides derived from the 28kD PNS-specific protein Myelin Protein Zero (P0) with a confidence score approaching 100% (Fig. 4C).
Figure 4. P0 is a dominant Aire-regulated PNS antigen in mice.

A) Indirect immunofluorescence of sciatic nerve sections using sera from either NOD.WT (WT) or NOD.AireGW/+ (AireGW/+) mice. Representative staining pattern shown of 3 independent experiments. B) Multiscreen immunoblot of whole sciatic nerve extract using sera from 4 NOD.WT (left) and 4 neuropathic NOD.AireGW/+ (right) mice at 15-25 weeks of age. Each lane corresponds to serum from an individual mouse. Arrow points to 25-30 kD bands predominantly recognized by 4 individual NOD.AireGW/+ mice. Blot shown is representative of 5 experiments. C) Amino acid sequence of P0 protein, with sequences identified by mass spectrometry underlined and bolded. D) Competition blot in which serum from a NOD.AireGW/+ mouse was preincubated with recombinant P0 fused with an MBP tag (left panel) or with the MBP tag alone (right panel) at increasing concentrations from 0 (left lane) to 750 (right lane) nanograms of recombinant protein. Data is representative of 2 independent experiments. E) P0 ELISA using sera from either NOD.WT or NOD.AireGW/+ mice. Each circle represents an individual mouse. Asterix (*) indicates significant difference between the two groups. Data shown is cumulative of 3 independent experiments, with each well done at least in duplicate. F) Real-Time RT-PCR assay for relative transcriptional expression of Ins2, GAD67, and P0 in sorted mTECs from NOD.WT and NOD.AireGW/+ mice. Data is normalized to cyclophilin A and relative to NOD.WT. Representative data from at least 2 independent experiments assayed in duplicate. G) Proliferation assay using 3H thymidine incorporation of splenocytes from NOD.WT and NOD.AireGW/+ mice pulsed with irrelevant HEL peptide (left) or P0 180-199 peptide (right) at three different peptide concentrations. Averages with standard deviations of three independent experiments are shown. Within each experiment, three replicates for each genotype and peptide concentration were performed. Splenocytes from individual mice (not pooled) were used in each experiment. Asterix (*) indicates significant difference between the two groups. n.s.= not significant.
To confirm the specificity of autoantibodies in NOD.AireGW/+ sera for P0, we performed a competition assay in which recombinant P0-MBP tag fusion protein was incubated with serum samples prior to immunoblotting. Titration of increasing concentrations of recombinant P0-MBP fusion protein into serum samples resulted in decreasing intensity of the 25-30 kD band on immunoblot, while the equivalent concentrations of negative control protein (MBP tag alone) did not (Fig. 4D). Furthermore, autoantibodies in sera from NOD.AireGW/+ mice had significantly increased reactivity to P0-MBP fusion protein by ELISA than autoantibodies in sera from NOD.WT littermates (Fig. 4E).
NOD.AireGW/+ mice have a quantitative decrease in the expression of Aire-regulated tissue-specific self-antigens in mTECs(2). Since P0 is a major PNS autoantigen in NOD.AireGW/+ mice, we sought to determine whether P0 is quantitatively decreased in NOD.AireGW/+ mTECs. cDNA generated from sorted mTECs was used to interrogate the relative expression levels of tissue-specific self-antigens in NOD.AireGW/+ compared to NOD.WT mTECs. To validate our cDNA samples, we demonstrated that insulin 2 (Ins2), but not glutamic acid decarboxylase (GAD) 67, is quantitatively decreased in NOD.AireGW/+mTECs compared to NOD.WT (Fig. 4F), as has previously been reported (4). Similar to Ins2, P0 expression in NOD.AireGW/+mTECs was approximately 10% of NOD.WT expression (Fig. 4F).
The decreased expression of P0 in NOD.AireGW/+mTECs led us to hypothesize that autoimmune peripheral neuropathy results from defective negative selection of P0 specific T cells in the thymus. To test this hypothesis, splenocytes from neuropathic NOD.AireGW/+ mice (between 20 and 22 weeks of age) were incubated with irrelevant hen egg lysozyme (HEL) peptide or P0 peptide 180-199. Splenocytes from NOD.AireGW/+ and wildtype mice proliferated to the same extent in response to HEL peptide (Fig. 4G, left). At all three concentrations of P0 180-199, on the other hand, NOD.AireGW/+ immune cells proliferated to a significantly greater degree than wildtype immune cells (Fig. 4G, right).
Autoreactivity to P0 in APS1 patients
CIDP was recently reported in two unrelated APS1 patients with confirmed mutations in Aire(5). One patient harbored compound heterozygous Aire mutations (R257X/R203X) and the other patient was homozygous for an Aire R139X mutation. The diagnosis of CIDP was based on 1) weakness, sensory loss, and absent reflexes; 2) evidence of demyelination; and 3) the presence of infiltrating CD4+ T cells and macrophages on nerve biopsy. Surprisingly, autoantibodies against a number of neuropathy-associated antigens were negative (5). Autoantibodies against P0, however, were not previously tested in these patients.
Indirect immunofluorescence using sera from these two patients demonstrated autoantibodies that bound sciatic nerve in a streak-like pattern (Fig. 5A) reminiscent of that seen in NOD.AireGW/+ mice. We then employed a radioligand binding assay to test for P0 specific autoantibodies in the sera from these two patients. While none of the sera from the healthy controls reacted against P0, sera from the two APS1 patients with CIDP were positive for autoantibodies against P0 (Fig. 5B). Additionally, 2 out of 12 patients with APS1 but without a clinical history of neuropathy also demonstrated serum reactivity to P0, raising the possibility that these patients may be at risk for developing CIDP.
Figure 5. Autoantibodies to P0 in APS1 patients.
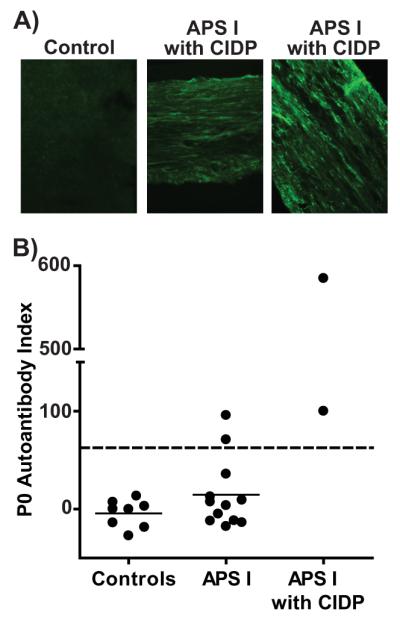
A) Indirect immunofluorescence using sera from 2 APS1 patients with CIDP (middle and right) and a healthy control subject (left) against sciatic nerve from an immunodeficient mouse. Images shown are representative of 3 independent experiments. B) Relative titers of P0 reactive autoantibodies in sera from healthy controls (left), APS1 patients without a history of CIDP (middle), and APS1 with a history of CIDP (right) detected by radioligand binding assay. Each symbol represents an individual subject. Dashed line=upper limit of normal, defined as 3 SDs from mean of healthy controls. Solid lines represent means of each group. Representative experiment is shown. Wells were performed in at least duplicate, and at least 6 independent experiments were performed.
Discussion
In this study, we show that mice and humans with defective Aire function share loss of tolerance to P0, which correlates with the development of spontaneous autoimmune peripheral neuropathy. We show for the first time that P0 is an Aire-regulated antigen in mTECs and that decreased P0 expression in mTECs is linked to increased immune cell reactivity toward P0 in the periphery. Finally, we show that CD4+ T cells are sufficient to transfer neuropathy and that the autoimmune response in the peripheral nerves is dominated by a Th1 effector response.
The spontaneous autoimmune peripheral neuropathy in NOD.AireGW/+ mice shares a number of features with human CIDP. First, CD4+ T cells and F4/80 macrophages are abundant immune cell types in CIDP patient nerve biopsies (13, 14). Similarly, CD4+ T cells and F4/80 expressing macrophages are also prevalent in the immune infiltrates of NOD.AireGW/+ sciatic nerves (Fig. 2A). Second, CD4+ helper T cells in CIDP patients are skewed toward the production of IFN-gamma (15). Similarly, approximately 40% of CD4+ helper T cells in the sciatic nerve infiltrates of neuropathic NOD.AireGW/+ mice produce IFN-gamma after in vitro stimulation (Fig. 3A and 3B) and less than 1% produce IL-17 or IL-4. Third, increased expression of the chemokine CXCL-10 in sural nerve biopsies is associated with CIDP (12). In neuropathic NOD.AireGW/+ mice, CXCL-10 expression is also increased in the sciatic nerves (Supplemental Fig. 1B). Finally, demyelination is a hallmark of CIDP, and extensive demyelination is characteristic of sciatic nerves from neuropathic NOD.AireGW/+ mice (Fig. 1E-G). Thus, autoimmune peripheral neuropathy in NOD.AireGW/+ mice resembles human CIDP in immune response and pathological changes in affected nerves.
The identification of P0 as a peripheral nerve autoantigen in Aire deficiency is significant because dominant protein antigen(s) in human autoimmune peripheral neuropathy are not well-defined. Alpha and beta tubulin, P0, P2, PMP22, and connexin 32 have all been reported to be candidate antigens targeted by the autoimmune response in CIDP (reviewed in (16). P0 is a peripheral nerve-specific protein that makes up the majority of myelin in the PNS (17). Loss of P0 due to genetic mutations (rather than autoimmune destruction) can result in Charcot Marie Tooth Type IB, a heritable demyelinating peripheral neuropathy (OMIM #118200). In addition, P0 can induce EAN upon immunization with adjuvant in mice (18) and is an autoantigen in other models of autoimmune peripheral neuropathy (11, 19).
We demonstrate in this study that P0 is an Aire-regulated tissue-specific self-antigen in the thymus and that immune cells in the spleen of Aire-deficient mice have increased reactivity towards P0. These data delineate a model a model in which tolerance toward peripheral nerves is maintained by Aire-mediated upregulation of P0 antigen in the thymus. Negative selection of P0-specific T cells in the thymus prevents escape of P0-reactive T cells into the periphery which can cause autoimmune peripheral neuropathy. In this study, we did not test whether T cells infiltrating the sciatic nerves of NOD.AireGW/+ mice are also reactive to P0 because of the low number of immune cells that we are able to isolate from sciatic nerves. Further study, including the generation of antigen-specific clones from infiltrating T cells,will be needed to define the myelin-specific T cell response in the model.
We found that CD4+ T cells were sufficient to transfer neuropathy to immunodeficient mice in a 6 to10 week time frame. Previously, P0 specific T cell lineshave been shown to transfer neuropathy into Lewis rats by 6 days (20). The slower kinetics of neuropathy development in our study can potentially be due to multiple factors, including the polyclonal repertoire of the T cells, species-specific differences between mice and rats, and lack of B cells in NOD.scid recipients.
A caveat of this study is the small number of patients with APS1 and CIDP. APS1 is a rare condition with a frequency of much less than 1/100,000 in the North American population. In addition, it appears that CIDP is an infrequent manifestation within APS1 subjects. We have attempted to identify additional subjects with APS1 and biopsy-proven CIDP within larger patient collections available in Scandinavia, but were unable to identify additional patients. Importantly, however, the two patients in this study are unrelated and have different Aire mutations, which strengthen the possibility that our findings are associated with Aire.
Finally, findings in this study may inform the development of more specific and effective immune therapies for autoimmune peripheral neuropathy. Current treatments consist primarily of glucocorticoids, intravenous immunoglobulin (IVIG), and plasmapheresis which result in non-specific immunosuppression (21). Furthermore, these treatments are ineffective in one third of patients (22). The demonstration in this study that CD4+ T cells are sufficient to transfer disease and that infiltrating CD4+ T cells produce IFN gamma suggests that therapies targeting this cell lineage or Th1 subsets may be efficacious in treating CIDP. Also, the identification of P0 as an important autoantigen in Aire-mediated autoimmune peripheral neuropathy may help guide the development of antigen-based therapies, such as antigen-specific DNA vaccination or infusion of antigen-coupled cells (23). Such therapeutic approaches that address underlying pathogenesis may be more efficacious in treating inflammatory neuropathies.
Supplementary Material
Acknowledgments
Source of Support: This work is supported by the NIH, the Neuropathy Association (MSA), and the Burroughs Wellcome Fund (MSA).
Glossary
Abbreviations
- CIDP
Chronic Inflammatory Demyelinating Polyneuropathy
- PNS
peripheral nervous system
- Aire
Autoimmune Regulator
- P0
Myelin Protein Zero
- mTECs
medullary thymic epithelial cells
- APS
Autoimmune Polyendocrinopathy Syndrome
- MBP
maltose binding protein
References
- 1.Laughlin RS, Dyck PJ, Melton LJ, 3rd, Leibson C, Ransom J, Dyck PJ. Incidence and prevalence of CIDP and the association of diabetes mellitus. Neurology. 2009;73:39–45. doi: 10.1212/WNL.0b013e3181aaea47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Su MA, Giang K, Zumer K, Jiang H, Oven I, Rinn JL, Devoss JJ, Johannes KP, Lu W, Gardner J, Chang A, Bubulya P, Chang HY, Peterlin BM, Anderson MS. Mechanisms of an autoimmunity syndrome in mice caused by a dominant mutation in Aire. J Clin Invest. 2008;118:1712–1726. doi: 10.1172/JCI34523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anderson MS, Venanzi ES, Klein L, Chen Z, Berzins SP, Turley SJ, von Boehmer H, Bronson R, Dierich A, Benoist C, Mathis D. Projection of an immunological self shadow within the thymus by the aire protein. Science. 2002;298:1395–1401. doi: 10.1126/science.1075958. [DOI] [PubMed] [Google Scholar]
- 4.Anderson MS, Venanzi ES, Chen Z, Berzins SP, Benoist C, Mathis D. The cellular mechanism of Aire control of T cell tolerance. Immunity. 2005;23:227–239. doi: 10.1016/j.immuni.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 5.Valenzise M, Meloni A, Betterle C, Giometto B, Autunno M, Mazzeo A, Cao A, De Luca F. Chronic inflammatory demyelinating polyneuropathy as a possible novel component of autoimmune poly-endocrine-candidiasis-ectodermal dystrophy. European journal of pediatrics. 2009;168:237–240. doi: 10.1007/s00431-008-0736-8. [DOI] [PubMed] [Google Scholar]
- 6.Salomon B, Rhee L, Bour-Jordan H, Hsin H, Montag A, Soliven B, Arcella J, Girvin AM, Padilla J, Miller SD, Bluestone JA. Development of spontaneous autoimmune peripheral polyneuropathy in B7-2-deficient NOD mice. J Exp Med. 2001;194:677–684. doi: 10.1084/jem.194.5.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Taylor LC, Gilmore W, Ting JP, Matsushima GK. Cuprizone induces similar demyelination in male and female C57BL/6 mice and results in disruption of the estrous cycle. Journal of neuroscience research. 88:391–402. doi: 10.1002/jnr.22215. [DOI] [PubMed] [Google Scholar]
- 8.Newbern JM, Li X, Shoemaker SE, Zhou J, Zhong J, Wu Y, Bonder D, Hollenback S, Coppola G, Geschwind DH, Landreth GE, Snider WD. Specific functions for ERK/MAPK signaling during PNS development. Neuron. 2011;69:91–105. doi: 10.1016/j.neuron.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shum AK, DeVoss J, Tan CL, Hou Y, Johannes K, O’Gorman CS, Jones KD, Sochett EB, Fong L, Anderson MS. Identification of an autoantigen demonstrates a link between interstitial lung disease and a defect in central tolerance. Sci Transl Med. 2009;1:9ra20. doi: 10.1126/scitranslmed.3000284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Devoss J, Hou Y, Johannes K, Lu W, Liou GI, Rinn J, Chang H, Caspi R, Fong L, Anderson MS. Spontaneous autoimmunity prevented by thymic expression of a single self-antigen. J Exp Med. 2006;203:2727–2735. doi: 10.1084/jem.20061864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim HJ, Jung CG, Jensen MA, Dukala D, Soliven B. Targeting of myelin protein zero in a spontaneous autoimmune polyneuropathy. J Immunol. 2008;181:8753–8760. doi: 10.4049/jimmunol.181.12.8753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kieseier BC, Tani M, Mahad D, Oka N, Ho T, Woodroofe N, Griffin JW, Toyka KV, Ransohoff RM, Hartung HP. Chemokines and chemokine receptors in inflammatory demyelinating neuropathies: a central role for IP-10. Brain. 2002;125:823–834. doi: 10.1093/brain/awf070. [DOI] [PubMed] [Google Scholar]
- 13.Cornblath DR, Griffin DE, Welch D, Griffin JW, McArthur JC. Quantitative analysis of endoneurial T-cells in human sural nerve biopsies. Journal of neuroimmunology. 1990;26:113–118. doi: 10.1016/0165-5728(90)90082-x. [DOI] [PubMed] [Google Scholar]
- 14.Bouchard C, Lacroix C, Plante V, Adams D, Chedru F, Guglielmi JM, Said G. Clinicopathologic findings and prognosis of chronic inflammatory demyelinating polyneuropathy. Neurology. 1999;52:498–503. doi: 10.1212/wnl.52.3.498. [DOI] [PubMed] [Google Scholar]
- 15.Csurhes PA, Sullivan AA, Green K, Pender MP, McCombe PA. T cell reactivity to P0, P2, PMP-22, and myelin basic protein in patients with Guillain-Barre syndrome and chronic inflammatory demyelinating polyradiculoneuropathy. J Neurol Neurosurg Psychiatry. 2005;76:1431–1439. doi: 10.1136/jnnp.2004.052282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Allen D, Giannopoulos K, Gray I, Gregson N, Makowska A, Pritchard J, Hughes RA. Antibodies to peripheral nerve myelin proteins in chronic inflammatory demyelinating polyradiculoneuropathy. J Peripher Nerv Syst. 2005;10:174–180. doi: 10.1111/j.1085-9489.2005.0010207.x. [DOI] [PubMed] [Google Scholar]
- 17.Martini R. Animal models for inherited peripheral neuropathies. Journal of anatomy. 1997;191(Pt 3):321–336. doi: 10.1046/j.1469-7580.1997.19130321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zou LP, Ljunggren HG, Levi M, Nennesmo I, Wahren B, Mix E, Winblad B, Schalling M, Zhu J. P0 protein peptide 180-199 together with pertussis toxin induces experimental autoimmune neuritis in resistant C57BL/6 mice. Journal of neuroscience research. 2000;62:717–721. doi: 10.1002/1097-4547(20001201)62:5<717::AID-JNR11>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 19.Louvet C, Kabre BG, Davini DW, Martinier N, Su MA, DeVoss JJ, Rosenthal WL, Anderson MS, Bour-Jordan H, Bluestone JA. A novel myelin P0-specific T cell receptor transgenic mouse develops a fulminant autoimmune peripheral neuropathy. J Exp Med. 2009;206:507–514. doi: 10.1084/jem.20082113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Linington C, Lassmann H, Ozawa K, Kosin S, Mongan L. Cell adhesion molecules of the immunoglobulin supergene family as tissue-specific autoantigens: induction of experimental allergic neuritis (EAN) by P0 protein-specific T cell lines. European journal of immunology. 1992;22:1813–1817. doi: 10.1002/eji.1830220721. [DOI] [PubMed] [Google Scholar]
- 21.Magy L, Vallat JM. Evidence-based treatment of chronic immune-mediated neuropathies. Expert Opin Pharmacother. 2009;10:1741–1754. doi: 10.1517/14656560903036095. [DOI] [PubMed] [Google Scholar]
- 22.Hughes RA, Allen D, Makowska A, Gregson NA. Pathogenesis of chronic inflammatory demyelinating polyradiculoneuropathy. J Peripher Nerv Syst. 2006;11:30–46. doi: 10.1111/j.1085-9489.2006.00061.x. [DOI] [PubMed] [Google Scholar]
- 23.Miller SD, Turley DM, Podojil JR. Antigen-specific tolerance strategies for the prevention and treatment of autoimmune disease. Nat Rev Immunol. 2007;7:665–677. doi: 10.1038/nri2153. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.