

Abstract
Specificity within the pathways of ubiquitin conjugation are defined by protein-binding affinities among the components. Enzyme kinetics provides a facile high-resolution experimental approach for quantitating such protein-binding affinities and yields additional mechanistic insights into the transition state of the enzyme-catalyzed reaction. Most ubiquitin ligases form free polyubiquitin chains at a slow rate in the absence of their cognate target protein as a normal step in their overall catalytic cycle. Rates of polyubiquitin chain formation can, therefore, be used as a reporter function kinetically to characterize binding interactions within the ligation pathway. We describe experimental approaches for: (1) precisely quantitating functional E1 and E2 concentrations by their stoichiometric formation of 125I-ubiquitin thiolester; (2) semiquantitative screens to define the cognate E2(s) for ubiquitin ligases based on their ability to support polyubiquitin chain formation; (3) initial rate studies to quantify Km and kcat as a measure of the ability of specific E2-ubiquitin thiolester substrates to support ligase-catalyzed polyubiquitin chain formation; and (4) an isopeptidase T-based technique for distinguishing between free and conjugated polyubiquitin chains formed in the functional assays. These kinetic methods provide mechanistic insights that are otherwise inaccessible by other experimental approaches and yield a precision in characterizing protein interactions that exceeds that of other techniques.
Keywords: Ubiquitin, Ligase, Ubiquitin carrier protein, Ubiquitin-activating enzyme, Kinetics, Polyubiquitin, Chain, Michaelis–Menten
1. Introduction
Enzyme reactions catalyzing new bond formation follow a thermodynamically defined two-step mechanism that generates a high-energy intermediate in the first half reaction (activation) at the expense of ATP or a related nucleotide triphosphate, the cleavage of which is exploited to drive creation of the new bond in the second half reaction (ligation). Mechanisms for the conjugation of ubiquitin and other ubiquitin-like peptides to protein targets follow this obligate mechanism, which is summarized in Fig. 1 for ubiquitin as the archetype for this family of posttranslational modifications (1). Ubiquitin-activating enzyme (E1/Uba1) utilizes ATP (formally as the ATP·Mg2+ chelate) to catalyze formation of a ternary complex comprising two forms of activated ubiquitin: a covalently bound high-energy ubiquitin thiolester to the active site cysteine of the enzyme that serves as the immediate donor of activated ubiquitin for subsequent isopeptide bond formation and a tightly bound ubiquitin adenylate intermediate that serves as precursor for ubiquitin thiolester formation (2–4). Ubiquitin-protein isopeptide ligase (E3) couples the aminolysis of the high-energy ubiquitin thiolester to formation of the isopeptide bond on the target protein or within an elongating polyubiquitin chain (5).
Fig. 1.

Schematic mechanism of ubiquitin conjugation. Ubiquitin-activating enzyme (E1) couples hydrolysis of ATP to the formation of a ternary complex composed of a covalently bound ubiquitin thiolester and a tightly bound ubiquitin adenylate intermediate. The activating enzyme subsequently binds a ubiquitin-specifi c carrier protein (E2) and catalyzes the transfer of the former intermediate to the E2 to form a corresponding E2-ubiquitin thiolester. The ubiquitin-protein isopeptide ligase (E3) binds its cognate E2-ubiquitin thiolester among the total pool of such cellular intermediates and catalyzes a reaction that couples aminolytic cleavage of the E2-ubiquitin thiolester to formation of the new isopeptide bond on the target protein (P).
The half reactions in most ligation reactions of metabolism are catalyzed by the same enzyme, as in the case of the aminoacyl tRNA synthetases which follow a catalytic cycle of carboxyl group activation and subsequent transfer to a ribose hydroxyl moiety on the tRNA that parallels the chemistry of ubiquitin isopeptide bond formation. In contrast, the half reactions of ubiquitin conjugation are segregated into separate enzymes that provide broad specificity through the divergent evolution of hundreds of E3 ligases that target selected proteins. The half reactions of activation and ligation are functionally linked through a large superfamily of ubiquitin carrier proteins (E2/Ubc) that shuttle activated ubiquitin as a thiolester to a conserved cysteine within the E2 catalytic core domain (6). Clearly, a single E2 species would have sufficed to fill this mechanistic role; in contrast, eukaryotic cells contain dozens of different E2 species that segregate into distinct families (1, 7). Speciation of the E2 superfamily suggests that new E2 families have arisen in response to the emergence of novel roles for ubiquitination and that E3 ligases are supported by a narrow range of specific E2 families.
The hierarchical architecture of the ubiquitin ligation mechanism allows us to consider ubiquitin-dependent cell regulation as a set of parallel pathways defined by cognate E2–E3 pairs. Within this hierarchy, the E1 defines specificity for the correct ubiquitin-like protein in order to activate only its cognate polypeptide, which occurs with great fidelity (8). The activating enzyme also functions as a licensing factor to bind and charge only the correct E2(s) associated with the ubiquitin-like protein among the large superfamily of paralogs present within cells (4). The E3 in turn must identify and bind its cognate E2-ubiquitin thiolester (the actual cosubstrate for the ligase) and the correct target protein prior to conjugation of the latter. A fundamental question, thus, relates to identifying the cognate E2(s) for a given E3, recognizing that functional spec-ificity of an E3 for its cognate E2 may arise from the inherent binding affinity for the E2-ubiquitin thiolester, reflecting differences in the intrinsic Km and/or differences in catalytic competence to support isopeptide bond formation, reflected in kcat (Vmax). The simplest approach to answering this question is through an “E2 screen” in which representative E2 family members are tested in parallel for their relative abilities to support the E3 ligation reaction.
Although such screens are straightforward in principle, they prove to be technically demanding in practice. Ideally, one wishes the various candidate E2 species to be present at the same concentration of active protein in the assays; however, recombinant E2 family proteins and isoforms within families vary significantly in their relative stability during expression, purification, storage, and subsequent freeze–thaw cycles. Because of such differences in protein stability and the resulting fraction of active protein present in E2 preparations, comparing E2 activity at constant total protein invariably yields erroneous results. Another potential problem arises in choosing the fixed E2 concentration to be used in the screen. The E2 superfamily shares a common 150 amino acid core catalytic domain and similar binding surfaces for E3 Ring and Hect domain interactions that are the direct consequence of the evolutionary divergence of E2 and E3 families over time (9, 10). Such similarities in E2 structure and E2–E3 interactions allow functionally irrelevant binding interactions to be favored at sufficiently high concentrations of the E2-ubiquitin thiolester, leading to ambiguous conclusions regarding the identity of the cognate E2 for the ligase. A particularly good example of this problem is the ability of UbcH8, an ISG15-specific E2 (11, 12), to support selected ubiquitin conjugation reactions at micromolar concentrations that exceeds its intracellular concentration following interferon induction (13–17). A related problem confounding the determination of the cog-nate E2 relates to the assay conditions with respect to incubation time and E3 concentration since prolonged incubation times and high concentrations of ligase can accentuate trace activities arising from low E2-ubiquitin thiolester binding affinity and/or kcat for noncognate E2 species. Finally, conjugation of the target protein by the ligase may be difficult to reproduce in vitro due to the absence of requisite posttranslational modifications, auxiliary protein subunits, and/or target protein-docking adapters.
1.1. Fundamentals of Enzyme Kinetic Studies
All physical methods used to measure binding affinity rely on a “readout” that is proportional to the concentration of the bound species. In enzyme kinetics, the rate of the reaction is proportional to the concentration of the bound substrate and represents a sensitive “readout” of complex formation, provided that the rate of product accumulation is measured under valid conditions (discussed below). The familiar Michaelis–Menten scheme (Scheme 1) summarizes this relationship between free (S) and bound (ES) substrate for a single-substrate reaction. For most enzymes, rates of substrate binding to form ES (defined by k1) and rates of ES dissociation (defined by k–1) are much faster than the rate of product formation defined by kcat (k1, k–1 ≫ kcat). Under these conditions, ES formation is in equilibrium and the concentration of the bound substrate increases with a hyperbolic dependence with respect to the concentration of free S. In this scheme, the Michaelis–Menten constant (Km) is defined as k–1/k1 and is equivalent to a binding dissociation constant (Kd), which at equilibrium is a direct measure of the standard Gibbs energy change for substrate association (or dissociation, depending on sign since Kassociation = 1/ Kdissociation) according to the equation ΔGo = –RT·ln Kd, where R is the gas constant (8.31 J/K mol or 1.99 cal/K mol) and temperature (T) is in K. Therefore, enzyme kinetics provide a convenient functional assay that allows one to quantify substrate binding affinity as Km, catalytic competence as kcat (Vmax), and the resulting changes in both due to orthologous components or targeted mutations. In addition, because kinetic studies are functional assays that reflect the physiological role of the enzyme within the cell, the contribution of different components and auxiliary effectors can be readily tested.
Scheme 1.
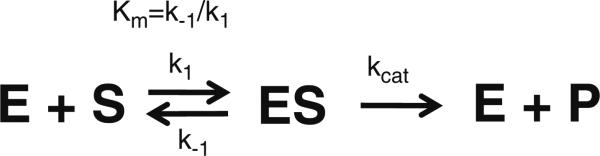
Michaelis–Menten scheme
Of equal importance, kcat provides a window on the transition state of the enzyme-catalyzed reaction since its magnitude is inversely related to the energy of activation (ΔG‡) for the enzyme-bound transition state by ΔG‡ = –RT·ln kcat, where the constants are the same as for the Gibbs free energy equation at equilibrium. The kcat reflects the overall geometry of the enzyme-bound transition state and the contributions of functional groups involved in catalysis or binding of the transition state, either in the active site or present on the substrate, that contribute to its stabilization. Mutations or other changes that perturb the optimal geometry of the wild-type enzyme-bound transition state or alter the contribution of catalytic groups result in destabilization of the transition state (larger ΔG‡) and correspondingly lower values for kcat. Kinetic evidence that Asp576 of human ubiquitin-activating enzyme is both a binding group for ATP·Mg2+ and, more critically, a catalytic group stabilizing the incipient pentacoordinate transition state provides an excellent example of the differences in the level of information available from quantitative versus qualitative approaches to enzyme function (18).
If one measures the time-dependent accumulation of product (the progress curve), the instantaneous rate defined as the tangent to the progress curve decreases progressively with time because the instantaneous rate is proportional to the remaining substrate, which is also decreasing. By convention, this problem can be circumvented by measuring the initial rate or velocity (vo) of the reaction, defined as the time interval in the early part of the progress curve for which the instantaneous rate appears to be constant. In reality, the initial velocity changes with time even in the early segments of the progress curve because substrate is constantly being consumed; however, the minute continuous changes in velocity over the observation interval are sufficiently small that they are undetectable under the conditions of the assay. Under such conditions, substrate remaining approximately equals the original concentration of substrate, (S)t = (S)o. The latter condition directly follows from the initial velocity assumption and has the important consequence that the accumulation of product is also negligible and can be assumed not to influence the initial rate through potential formation of nonproductive enzyme–product complexes.
In practical terms, kinetic studies intended to quantitate substrate binding are conducted by measuring vo at different initial concentrations of substrate ((S)o). As a rule of thumb, the measurement interval is over the initial 5–10% of total product formation (defined as being equal to (S)o), although the validity of this assumption must be empirically tested by demonstrating that product accumulation is linear over the measurement interval (usually, by measuring product accumulation at two time points). A plot of vo versus (S)o should show a hyperbolic dependence, which is confirmed by the linearity of a double-reciprocal (Lineweaver–Burk) plot of 1/vo versus 1/(S)o. The values of Km and Vmax, from which kcat can be approximated by Vmax = kcat (E)o where (E)o is the total enzyme concentration, can in principle be determined from the double-reciprocal plot. However, because the double-reciprocal transformation distorts measurement errors (particularly at low substrate concentration), determination of both Km and Vmax is skewed. Values of Km and Vmax are more accurately determined by nonlinear regression analysis, which avoids distortion of measurement error through fitting the data directly to the hyperbolic Michaelis–Menten equation. Although the Lineweaver–Burk plot is not used to determine values of Km and Vmax, it is important tool for graphical analysis in confirming that an enzyme conforms to hyperbolic kinetics, identifying deviations from the mathematical model due to substrate inhibition at high concentrations and detecting allosteric cooperativity.
1.2. Application of Initial Velocity Kinetics to Polyubiquitin Chain Formation Assays
The target protein and the cognate E2-ubiquitin thiolester are both cosubstrates of E3 in the ubiquitin ligase-catalyzed conjugation reaction. One expects each to show a hyperbolic concentration dependence on the rate of target protein conjugation, measured as the addition of ubiquitin moieties, versus either target protein and E2-ubiquitin thiolester concentrations. Each cosubstrate has a corresponding Km associated with its respective binding to the ligase. Depending on the kinetic mechanism of the reaction, each cosubstrate can bind independently of the other (random addition) or the relative binding affinity of each substrate can be influenced by binding of the other cosubstrate (ordered or pseudo-ordered addition). Properly designed initial rate studies can determine the binding affinities of the target protein and the E2-ubiquitin thiolester, as well as the kcat of the corresponding ternary complex of target protein and E2-ubiquitin thiolester bound to the ligase.
There remain several technical considerations in designing initial rate studies to measure E3-catalyzed reactions. Of particular importance is the manner in which the reaction products are monitored. During the conjugation reaction, ligases typically assemble polyubiquitin chains on the substrate either by sequential addition or transfer of intact chains to the target protein. For this reason, it is most practical to follow the conjugation reaction using labeled ubiquitin so that the initial velocity is expressed as ubiquitins conjugated per unit time. Covalent modification of ubiquitin with fluorescent tags or similar moieties is not practical since these groups can sterically hinder the E1-catalyzed activation and/or E2-dependent transthiolation reactions, which in turn can alter the rate-limiting step. Another approach is genetically to append a peptide harboring a protein kinase motif to the N terminus of recombinant ubiquitin which can then be 32P labeled with (γ-32P)ATP (19). A second approach uses chloramine T-mediated radioiodination to modify the single Tyr59 residue of wild-type ubiquitin (2, 3). Both methods produce a radiolabeled peptide that is easily quantitated and for which an accurate specific radioactivity can be determined for precise quantitation of product accumulation; however, only the radioiodinated protein has been kinetically validated as being functionally indistinguishable from wild-type ubiquitin (3).
Because ubiquitin conjugation is a multistep process, there has been some confusion regarding conditions under which E3 function can be reliably measured. In some instances, investigators have first formed the E2-ubiquitin thiolester, isolated the intermediate, and then added it back at known concentrations to assays containing the ligase and protein substrate (20, 21). Such single-turnover experiments are kinetically valid and obviate potential contributions from the E1-catalyzed transthiolation step. Single-turnover experiments also have the advantage that E3 assays can be conducted under conditions that might inhibit E1-catalyzed transthiolation of E2. However, such approaches are laborious and complicated by the fact that the E2-ubiquitin thiolester is labile and that spontaneous inactivation of the E2 moiety may occur during isolation of the charged intermediate or during storage. An alternative experimental approach recognizes that the mechanism of ubiquitin conjugation summarized in Fig. 1 can be viewed as a coupled reaction in which the E1 step is used to form a cosubstrate of E3. Since rate studies only monitor the slowest step of a multistep process, the concentration of E1 in the assay can be empirically set to maintain E3 as the rate-limiting step under all conditions. The latter approach has the advantage that the labile E2-ubiquitin thiolester cosubstrate is produced in situ, the concentration of which is equal to the initial concentration of E2 ((E2)o) under E3-limiting conditions. This negates technical concerns of E2 inactivation or thiolester hydrolysis during formation and storage of the activated intermediate that are inherent in the single turnover approach. One need only confirm that the central criterion of coupled reactions be satisfied that the rate remains independent of the coupling step. Coupled E3 assays have been used previously to examine the kinetics of E3α/Ubr1-dependent conjugation of N-end rule substrates and to determine the relative binding affinities of E2-ubiquitin thiolester (both directly and indirectly through competition kinetics using E2-ubiquitin oxyester) and the uncharged E2 product, as the E2 active site Cys → Ala mutant (22–25).
The final technical issue in designing an E3 conjugation assay is the choice of target protein. When a suitable natural target protein or model substrate is available, initial rate studies of ligase reactions are relatively straightforward (22, 24); however, frequently the substrate is unknown or difficult to obtain in sufficiently pure form for in vitro assays, requires posttranslational modification(s) to be recognized by the ligase, or binds the ligase only through additional docking proteins. Many Ring and Hect domain ligases assembly polyubiquitin chains as thiolesters attached to the cognate E2 or active site cysteine, respectively, that are then transferred onto the target protein (26–28). The ability of ligases to form polyubiquitin chains can, thus, be used as a functional readout for kinetic studies in the absence of target protein. The latter readout obviates problems associated with posttranslational modifications and docking proteins; however, the resulting kcat reflects chain formation and not transfer to the target protein, which is likely to be rate limiting in the overall reaction. Nonetheless, rate studies monitoring polyubiquitin chain formation can be of significant utility in the characterization of enzyme activity and substrate affinity. For this reason, we focus on this readout in subsequent discussions.
2. Materials
125I-ubiquitin, obtained by labeling FPLC-purified commercial bovine ubiquitin (Sigma) with Na 125I as described previously (29) (see Note 1). Typically, the specific radioactivity should be at least 5,000 cpm/pmol, determined by gamma counting, and ca. 20 μM concentration or greater, determined spectrophotometrically using a 280 nM extinct coefficient of 0.16 (mg/ml)–1 (30).
Uba1 (E1), purified from human erythrocytes as described previously (29) or obtained from a commercial source. Active E1 should be quantitated by its stoichiometric formation of an 125I-ubiquitin thiolester (29) (see Note 2).
10× assay buffer, 0.5 M Tris–HCl (pH 7.5), 100 mM MgCl2, 20 mM ATP, 10 mM dithiothreitol (DTT), 100 mM creatine phosphate, and 40 IU/ml creatine phosphokinase (CPK) (see Note 3). The CPK is added to the assay buffer stock immediately before setting up the incubations.
CPK stock, 103 IU/ml enzyme in 50 mM Tris–HCl (pH 7.5) and 1 mM DTT (see Note 4).
Protein diluent solution, 50 mM Tris–HCl (pH 7.5), 1 mM DTT, and 1 mg/ml bovine serum albumin.
Recombinant ubiquitin carrier protein stock solutions at 20–100 μM active E2 protein in 50 mM Tris–HCl (pH 7.5) and 1 mM DTT stored at –80°C in small aliquots (see Note 5).
E3 ubiquitin ligase in 50 mM Tris–HCl (pH 7.5) and 1 mM DTT.
Recombinant isopeptidase T (IsoT), 10 mg/ml in 50 mM Tris–HCl (pH 7.5) and 1 mM DTT.
Apyrase stock at 103 IU/ml in 50 mM Tris–HCl (pH 7.5) and 1 mM DTT.
1 M DTT stock solution.
2× SDS sample buffer, 50 mM Tris–HCl (pH 6.8), 4% (w/v) SDS, 20% (v/v) glycerol, 0.5% (w/v) bromophenol blue. Prior to use, the sample buffer is adjusted to 0.2% (v/v) 2-mercaptoethanol (reducing gels for 125I-ubiquitin conjugate resolution) or used without addition (nonreducing gels for 125I-ubiquitin thiolester assay of E1 and E2).
Standard 12% (w/v) polyacrylamide gels (16 × 18-cm format).
Whatman filter paper, Saran Wrap, Glow-In-the-Dark paint (Duncan).
Vacuum gel dryer.
Kodak BioMax XAR X-ray film.
X-ray film cassette equipped with an L Plus intensifying screen.
X-ray film developer.
Gamma counter.
3. Methods
3.1. Stoichiometric Determination of E1 and E2 Concentrations
The ability of Uba1 (E1) to form a stoichiometric ternary complex containing ubiquitin adenylate and ubiquitin thiolester followed by the quantitative transfer of the thiolester to E2 serves as the basis for an important quantitative functional assay of active E1 and E2 enzymes based on measuring the corresponding 125I-ubiquitin thiolester (2, 3, 31). Analogous assays can be employed to quantitate other ubiquitin-like protein-activating enzymes, their cognate E2 proteins, and HECT domain ubiquitin ligases. However, in the latter case, one should be cautious since such Hect domain quantitation may underestimate the actual amount of active enzyme due to rapid hydrolysis of the Hect domain-125I-ubiquitin thiolester relative to the rate of transthiolation from the E2 125I-ubiquitin thiolester. The quantitative end-point stoichiometry of Hect domain ligases has never been rigorously confirmed.
Prepare a standard 12% (w/v) SDS-PAGE gel and running buffer, equilibrated to 4°C (see Note 6).
An incubation of 25-μl final volume should contain 2.5 μl of 10× assay buffer (see Subheading 2, item 3), ca. 50 nM E1, and ca. 50 nM E2 made to volume (less the subsequent addition of 125I-ubiquitin) with protein diluent solution (see Subheading 2, item 5). Because the formation of the E1 ternary complex as well as its subsequent transfer of the activated ubiquitin to the E2 is rapid, the incubations should be equilibrated for 2–3 min at 37°C to reach thermal equilibrium before initiating the assay by the addition of 125I-ubiquitin to a final concentration of 5 μM. The amount of E1 and E2 in the reaction should be approximately equimolar to allow the rapid stoichiometric formation of the labeled intermediates. Consequently, two sets of reactions should be prepared: one containing a serial dilution of E1 for accurate quantitation of the activating enzyme, since E1 exhibits half-sites reactivity at higher concentrations which can result in underestimation of active enzyme concentration, and another with a single E1 concentration of approximately 50 nM and a serial dilution of the E2. The experimental conditions should allow the reaction to reach the end point within 1–2 min for the accurate quantitation of active E1 and E2 protein. Longer incubation times should be avoided because the concomitant autoubiquitination of both enzymes leads to overestimation of their actual active concentrations.
After 1 min at 37°C, the reaction is quenched by adding 25 μl of 2× SDS sample buffer without 2-mercaptoethanol. The sample is allowed to stand on ice for 5 min to allow the proteins to unfold. Do not boil the sample for an extended period as for reducing gels because this destroys the thiolester linkage (see Note 7). As a control, a parallel set of incubations can be run that are quenched with SDS sample buffer containing 2-mercaptoethanol, for which no thiolester bands should be observed after autoradiography.
Immediately load 40 μl of the quenched incubation per lane on the SDS-PAGE gel and resolve under standard conditions at 4°C. To prevent heating of the gel and hydrolysis of the thiolester linkage during electrophoresis, immerse the gel completely in ice-cold running buffer for good heat transfer and stir the buffer in the lower chamber during the run.
After the SDS-PAGE is completed, float the gel onto a piece of Whatman filter paper, overlay with Saran Wrap, and dry the gel using a standard vacuum gel drier. Mark the filter paper with glow-in-the-dark paint in two or more places so that the resulting autoradiogram can be accurately superimposed onto the gel for later quantitation. Expose the autoradiogram overnight at –80°C using Kodak X film and an appropriate intensifying screen. The next day, overlay the developed autoradiogram over the dried gel and excise the corresponding E1- and E2-125I-ubiquitin thiolester bands for quantitation of associated radioactivity by gamma counting. Determine the absolute amount of E1 and E2 thiolester by using the specific radioactivity of the 125I-ubiquitin determined previously, correcting for decay of the radionuclide as needed (29).
Figure 2 illustrates an autoradiogram of data typically obtained from the 125I-ubiquitin thiolester assay described in the text. By positioning the autoradiogram over the dried gel and using the orientation spots precisely to align the gel, one can then excise the bands corresponding to the respective 125I-ubiquitin thiolester bands. Quantitation of associated radioactivity by gamma counting and calculation of the absolute amount of associated 125I-ubiquitin, using the specific radioactivity of the latter, allow a facile quantitation of active enzyme for each component. That the bands are thiolesters can be confirmed by resolving parallel samples under reducing conditions, resulting in cleavage of the thiolester bonds. Mobility artifacts can arise in nonreducing SDS-PAGE that are not otherwise encountered, but that one should be aware (see Note 8).
Fig. 2.

Autoradiogram of stoichiometric quantitation of ubiquitin conjugation components. Incubations were performed as described in Subheading 3.1 for 125I-ubiquitin in the absence (lane 1) or presence of the indicated components. Human Uba1 and recombinant UbcH7 E2 carrier protein were present at 50 nM. Recombinant GST–E6AP Hect domain fusion protein was present at 10 nM. Shown are the autoradiographic densities for the corresponding 125I-ubiquitin thiolesters. Quantitation of these thiolester intermediates is achieved by excising the corresponding bands and quantitating associated 125I by gamma counting.
3.2. Semiquantitative Screen for Cognate E2–E3 Specificity
The E3 ubiquitin ligases are functionally grouped by the domains through which they interact with their cognate E2 isoforms; however, there is insufficient information currently available in order to predict a priori the specificity of a given E3 for its cognate E2. Consequently, functional E2 screens are most frequently performed in order to identify the cognate E2(s) for a ligase. The screens can be made semiquantitative by following a few simple guidelines.
The E2 concentration should be identical for each E2 species and determined empirically by 125I-ubiquitin thiolester assays (see Subheading 3.1).
The E2 concentration chosen should be at or below the Km for binding of the cognate E2-ubiquitin thiolester to the E3. An E2 (actually, E2-125I-ubiquitin thiolester) concentration at or below Km provides the greatest sensitivity in detecting potential differences in Km and kcat (Vmax) among the paralogs and obviates favoring otherwise low-affinity-binding interactions found at higher concentrations. Obviously, satisfying this criterion requires prior knowledge of the Km for the cognate E2; however, it is convenient arbitrarily to set the E2 concentration at 50–100 nM since such Km values typically fall within this region for most ligases.
The incubation time chosen should fall within the initial velocity region of the progress curve and be E3 limiting. Satisfying this criterion guarantees that the autoradiographic intensities of the 125I-ubiquitin conjugates are proportional to the E3-catalyzed rate, allowing one to identify subtle differences in activity among the E2 paralogs.
In lieu of a physiological substrate, E3-catalyzed polyubiquitin chain formation can be measured as the functional readout. The latter criterion makes the E2 screen amenable to a wide variety of E3 ligases since almost all can be shown to catalyze free chain formation in the absence of substrate. However, some care must be exercised since some E2 paralogs show modest autoubiquitination and chain formation in the absence of an E3 ligase, requiring that suitable controls be performed in the absence of ligase.
A typical E2 screen can be performed using the following protocol and the appropriate enzyme components. A separate experiment with IsoT distinguishes free chains from polyubiquitin chains conjugated to proteins of the assay, typically the ligase (see Subheading 3.4).
Prepare a standard 12% (w/v) SDS-PAGE gel and running buffer, equilibrated to 4°C.
A series of incubations of 25-μl final volume are prepared containing 2.5 μl of 10× assay buffer (see Subheading 2, item 3 and Note 4), 100 nM Uba1, 100 nM E2, and sufficient E3 to observe polyubiquitin chain formation under the assay conditions (see Note 9). The incubations are made to final volume with protein diluent solution (see Subheading 2, item 5).
The incubations are allowed thermally to equilibrate for 2–3 min at 37°C before initiating the assay by addition of 125I-ubiquitin to a final concentration of 5 μM.
After 20 min at 37°C, the reaction is quenched by addition of 25 μl of 2× SDS sample buffer containing 2-mercaptoethanol and briefly mixed by vortex.
Samples are boiled for 5 min, placed on ice briefly, centrifuged for 1 min to remove the condensate from the top of the Eppendorf tube, and then vortexed briefly to mix the contents of the tube.
Immediately load 40 μl of each assay per lane on the SDS-PAGE gel and resolve under standard reducing conditions at 4°C. After the SDS-PAGE is completed, float the gel onto a piece of Whatman filter paper, overlay with Saran Wrap, and dry the gel using a standard vacuum gel drier. Mark the filter paper with glow-in-the-dark paint in two or more places so that the orientation of the gel relative to the lanes can be determined later. Autoradiograph overnight at –80°C using Kodak BioMax XAR film and an L Plus intensifying screen.
Figure 3 illustrates typical data from a semiquantitative E2 screen conducted with the Trim25/Efp Ring finger ligase. When the screen is conducted with 100 nM of each E2, quantitated by its stoichiometric formation of 125I-ubiquitin thiolester as outlined in Subheading 3.1 to guarantee that each is present at the same concentration of active protein, it is obvious that the ligase is specific for the Ubc5 family of E2 carrier proteins. Because the incubations are conducted under initial velocity conditions, the differences in autoradiographic intensity for the polyubiquitin chains formed with each of the E2 isozymes indicate that the ligase shows greatest activity with Ubc5B (lane 5) and the least with Ubc5A (lane 4). Frequently, a ligase can be observed to function with several related families of E2 paralogs in these assays, but with differing activities revealed by the extent of chain formation. Under these conditions, identifying the cognate E2(s) requires knowledge of the actual affinity between the ligase and the E2-ubiquitin thiolester as well as the resulting kcat (Vmax).
Fig. 3.

Autoradiogram of an E2 screen conducted in the presence of Trim25 ligase. Incubations were performed as described in Subheading 3.2 in 10-min incubations containing 100 nM Uba1 and 1 μM recombinant human GST-Trim25 (determined as total protein) in the absence (lane 2) or presence of 100 nM of the indicated recombinant E2 proteins. The concentrations of active E1 and E2 proteins were determined by the stoichiometric formation of 125I-ubiquitin thiolester, as described in Subheading 3.1.
3.3. Initial Rate Studies of E3-Catalyzed Chain Formation
Since the E3 ubiquitin ligases can transfer ubiquitin to a growing polyubiquitin chain in the absence of the corresponding cognate substrate, the initial rate of polyubiquitin chain formation under E3-limiting conditions is a valid readout of ligase activity for kinetic studies intended to analyze the mechanism of E2–E3 interaction and to determine the corresponding cognate E2 (1, 22, 23). After the initial E2 screen (see Subheading 3.2), the functional E2 paralogs can be further examined to define their Km and kcat (Vmax) for polyubiquitin chain formation. Such studies determine the E2 concentration dependence on the initial rate for polyubiquitin chain formation by measuring vo at different (E2)o. The incubations are a variation on those used for the E2 screen in Subheading 3.2. Preliminary experiments should be conducted to confirm empirically that the initial rate is E3 dependent, demonstrated by independence of vo on (E1)o. Other preliminary experiments are needed to confirm that the incubation time chosen remains within the initial velocity region of the progress curve. Finally, a parallel thiolester gel at the time of the rate study provides precise values for (E1)o, (E2)o, and (E3)o, if the latter is a Hect domain ligase.
Prepare a standard 12% (w/v) SDS-PAGE gel and running buffer, equilibrated to 4°C.
A series of incubations of 25-μl final volume are prepared containing 2.5 μl of 10× assay buffer (see Subheading 2, item 3 and Note 4), 50–100 nM Uba1, sufficient E3 to observe polyubiquitin chain formation under the assay conditions chosen, and a series of E2 concentrations within the optimal range for rate studies (see Note 10). Incubations are made to final volume with protein diluent solution (see Subheading 2, item 5). Control incubations should contain E1 alone and E1 in combination with E3. At the highest E2 concentration, include an incubation in which (E1)o is doubled as a control to confirm E3-limiting conditions (see Note 11).
The incubations are thermally equilibrated for 2–3 min at 37°C and then the reactions are initiated by addition of 125I-ubiquitin to a final concentration of 5 μM.
Reactions are continued for 10 min at 37°C (see Note 12) and then quenched with 25 μl of SDS sample buffer containing 2-mercaptoethanol, as in Subheading 3.2, step 4. Samples are boiled for 5 min, placed on ice briefly, centrifuged for 1 min to remove the condensate from the top of the Eppendorf tube, and then vortexed briefly to mix the contents of the tube.
Immediately load 40 μl of each incubation onto the SDS-PAGE gel and resolve under standard conditions at 4°C.
After the SDS-PAGE is completed, float the gel onto a piece of Whatman filter paper, overlay with Saran Wrap, and dry the gel using a standard vacuum gel drier. Mark the filter paper with glow-in-the dark paint in two or more places so that the resulting autoradiogram can be superimposed accurately onto the gel for later quantitation. Autoradiograph overnight at –80°C using Kodak BioMax XAR film and an L Plus intensifying screen. The next day, overlay the developed autoradiogram over the dried gel and excise regions of the lanes corresponding to 125I-ubiquitin conjugates.
Quantitate associated radioactivity by gamma counting. Determine the absolute amount of radioiodinated ubiquitin conjugated using the specific radioactivity of the 125I-ubiquitin determined previously, correcting for decay of the radionuclide as needed (29).
Figure 4a illustrates typical data from an initial rate study conducted as described above using a GST–E6AP fusion protein containing a point mutant in which Phe849 has been changed to tyrosine. The mutant shows robust conjugation with increasing UbcH7. Polyubiquitin chains are formed of sufficient molecular weight that they fail to migrate into the stacker gel (indicated). Lane 2, containing Uba1 and ligase but no E2, shows a prominent band at 108 kDa that represents Uba1 auto-monoubiquitination. The gel is analyzed by excising the bands above 25 kDa (including the stacker gel) and quantitating 125I-ubiquitin by gamma counting. The data is corrected for radioactivity present in an identical region of lane 2. After correcting the counts for having loaded only 80% of the total incubation, then dividing by the corrected specific radioactivity for the 125I-ubiquitin, the resulting absolute amount of conjugated radioiodinated ubiquitin is divided by the incubation time to yield the initial rate. Figure 4b illustrates the dependence of vo on (UbcH7)o. That the data follows Michaelis–Menten hyperbolic kinetics is demonstrated by the linearity of the double-reciprocal plot shown in the inset to Fig. 4b; however, since the double-reciprocal plot has an intrinsic bias in the estimation of Km and Vmax, these kinetic constants are determined by nonlinear regression analysis using the GraFit© suite of software (see Note 13). There is excellent agreement between the nonlinear regression fit and the data. Frequently, we have observed that some ligases exhibit substrate inhibition with respect to E2-125I-ubiquitin thiolester at high concentrations in the micromolar range. Substrate inhibition is indicative of E2 thiolester binding to a lower affinity site and is detected by an upward deflection of the data points (lower vo) near the Y axis (highest (S)o). Such points should be omitted from the nonlinear regression fit since they do not conform to the mathematical model for hyperbolic binding. In contrast, allosteric cooperativity with respect to the E2 thiolester is revealed by curvature over the entire concentration range.
Fig. 4.

Initial rate kinetic study of a GST-E6APF849Y point mutant. Incubations containing 100 nM Uba1, 30 nM GST-E6APF849Y, and the indicated concentrations of recombinant UbcH7 were incubated for 10 min at 37°C as outlined in Subheading 3.3. Conjugated 125I-ubiquitin was quantitated by excising each lane above the 25-kDa relative molecular weight marker and used to calculate the initial velocity as described in the text. (a) Autoradiogram of the resulting SDS-PAGE resolution of the incubations. (b) Dependence of initial rate (vo) on (UbcH7)o (solid line represents the nonlinear regression fit for Km = 52 ± 8 nM and kcat = 1.4 ± 0.1 × 10–3 s–1). Inset – Double-reciprocal plot of the rate data.
3.4. Identification of Free Versus Conjugated Polyubiquitin Chains
One final technique involves distinguishing free polyubiquitin chains from those attached to a target protein. The method relies on the ability of IsoT (gene name USP5) specifically to disassemble free but not conjugated polyubiquitin chains (32). This technique is useful in characterizing the product of the conjugation reactions and is best done in conjunction with the E2 screen experiment of Subheading 3.2.
A reaction of 200-μl final volume is prepared containing 50 mM Tris–HCl (pH 7.5), 2 mM ATP, 10 mM MgCl2, 1 mM DTT, 1 mg/ml of carrier protein BSA, 10 mM creatine phosphate, 1 IU CPK (see Note 4), 50 nM E1, 100 nM E2, and sufficient E3 to observe polyubiquitin chain formation under the assay conditions chosen.
The incubation is allowed thermally to equilibrate for 2–3 min and then the reaction is initiated by addition of 125I-ubiquitin to a final concentration of 5 μM.
After 20 min at 37°C, DTT is added to a final concentration of 10 mM to cleave polyubiquitin chains present as thiolesters on E2 or the Hect domain. Apyrase is added to a final concentration of 25 IU/ml to deplete the reaction of ATP.
The reaction is incubated for an additional 10 min at 37°C to insure that ATP is depleted. A sample of 25 μl is then removed and quenched with an equal volume of 2× sample buffer containing 2-mercaptoethanol (zero-time sample).
Recombinant IsoT is added to a final concentration of 20 nM and the incubation is continued at 37°C. Aliquots of 25 μl are taken at 10, 20, and 30 min after addition of IsoT and quenched with an equal volume of 2× sample buffer containing 2-mercaptoethanol. After the 30-min sample is collected, a second aliquot of IsoT is added to the remaining incubation and the reaction allowed to continue for an additional 10 min at 37°C.
Samples are boiled for 5 min, placed on ice briefly, centrifuged for 1 min to remove the condensate from the top of the Eppendorf tube, and then vortexed briefly to mix the contents of the tube.
Immediately load 40 μl of each incubation onto the SDS-PAGE gel and resolve under standard conditions at 4°C.
After the SDS-PAGE is completed, float the gel onto a piece of Whatman filter paper, overlay with Saran Wrap, and dry the gel using a standard vacuum gel drier. Mark the filter paper with glow-in-the-dark paint in two or more places so that the resulting autoradiogram can be superimposed accurately onto the gel for later quantitation. Autoradiograph overnight at –80°C using Kodak BioMax XAR film and an L Plus intensifying screen.
Upon addition of IsoT to the reaction, the ubiquitin-specific protease disassembles free polyubiquitin chains from the free carboxyl (distal) end. Therefore, free chains disappear with time, leaving only the conjugated chains. In practice, the free chains are generally those present in the stacker gel while conjugated chains are those present at the top and within the resolving gel. Addition of the extra aliquot of IsoT and additional incubation is a control to guarantee that all free chains have been disassembled.
Acknowledgments
The authors thank Patrick Connick for providing the data presented in Fig. 3. This work was supported by National Institutes of Health grant GM034009 (to A.L.H.).
Footnotes
From experience, we prefer to conduct these and other experiments using 125I-ubiquitin since it can be reproducibly prepared, yields a high-specific activity, and has a longer half-life than the 32P-labeled recombinant protein. In addition, radioiodinated ubiquitin has been kinetically validated as indistinguishable from wild-type peptide in rate studies (3, 22, 24).
Throughout this chapter, concentrations of E1 and E2 are expressed as active protein determined empirically by the stoichiometric formation of their corresponding 125I-ubiquitin thiolester (2, 3, 31). Expressing these components as active protein is preferable to calculation from total protein since the latter is less reproducible and the enzymes are subject to progressive loss of activity on storage and with accumulated freeze–thaw cycles. After the thiolester assay to determine the active protein concentration is completed, the enzymes are ali-quoted into small fractions suitable for a single use, flash frozen in liquid nitrogen, and stored at –80°C. The enzymes show no significant loss of activity for over a year under such conditions. Repeated freeze–thaw cycles progressively decrease the amount of active protein; consequently, a thiolester assay should be performed with each new kinetic experiment.
In these assays, a fivefold excess of Mg2+ over ATP should be maintained in order to ensure that the nucleotide is quantitatively present as ATP·Mg2+ since free ATP is a competitive inhibitor of E1 (33).
CPK is extremely labile to freeze–thaw cycles, so the enzyme should be divided into 50-μl aliquots, then flash frozen in liquid nitrogen, and stored at –20°C. Aliquots should be thawed by hand and immediately placed on ice until used. Aliquots should be used only once and any excess should be discarded.
Recombinant human ubiquitin carrier proteins (E2) are expressed from pGEX plasmids as recombinant GST-E2 in an Escherichia coli BL21 DE3 strain. The culture is grown at 37°C with shaking until an OD600 of 0.6 is reached and then protein expression is induced by addition of isopropyl-β-d-thiogalactopyranoside to a final concentration of 0.4 mM. After 3 h, the cells are harvested at 103× g for 15 min and then resuspend in 50 mM Tris–HCl (pH 7.5), 150 mM NaCl, and 5 mM DTT. Cells are lysed by Emulsiflex and then centrifuged at 105 × g for 30 min at 4°C. Most E2s are soluble and can be purified directly from the resulting high-speed supernatant using a glutathione-sepharose affinity column. The column is equilibrated in 50 mM Tris–HCl (pH 7.5), 150 mM NaCl, and 5 mM DTT. The GST-E2 is eluted from the column with 50 mM Tris–HCl (pH 7.5), 20 mM GSH, and 1 M NaCl. The eluted fraction is dialyzed overnight at 4°C against 4 L of buffer containing 50 mM Tris–HCl (pH 7.5) and 1 mM DTT. It is essential that the GST moiety is removed before use since the domain sterically hinders binding to E1, significantly altering the binding affinity to the activating enzyme (Km effect) and lowering the kcat for transthiolation, the net effect of which is to shift the rate-limiting step to transthiolation. The GST is removed by thrombin cleavage (50 IU/ml final concentration) and the E2 is purified using a glutathione-sepharose 4B column equilibrated with 50 mM Tris–HCl (pH 7.5) and 1 mM DTT to removed unprocessed fusion protein and free GST. The protein is concentrated using Centricon spin concentrator (Amicon), divided into small aliquots, flash frozen in liquid nitrogen, and stored at –80°C. The E2 protein concentration is calculated based on 280-nm absorbance using the theoretical extinction coefficient for the protein. Active E2 protein is assayed by 125I-ubiquitin thiolester assay (2, 3, 31). Typical percent active protein consistently ranges from >90% for human Ubc2b/E214Kb/Rad6 (gene name UBE2B) to <20% for human UbcH7 (gene name UBE2L3). Aliquots of the purified E2 should be flash frozen in liquid nitrogen and stored at –80°C, where the enzymes are stable for at least 1 year, although all are labile to repeated freeze–thaw cycles.
The temperature during nonreducing SDS-PAGE should be maintained at 4°C to quantitatively preserve the thiolester bond (2, 3, 31). Under no circumstances should 2-mercaptoethanol be added to the SDS sample buffer.
Some E2s exhibit a partial unfolding artifact resulting in two bands of 125I-ubiquitin thiolester. Unfolding can be promoted by briefly heating the quenched sample at 100°C for no more than 30 s prior to loading the gel.
Under nonreducing conditions, some streaking of label is observed that corresponds to thiolester that hydrolyzes during electrophoresis, but this should be negligible. In other cases, as shown for the UbcH7-125I-ubiquitin thiolester (Fig. 2), a minor running artifact can result from partial unfolding of the E2 (indicated by the smaller band beneath the main UbcH7-125I-ubiquitin thiolester band). Occasionally, the presence of E2 appears to decrease the amount of E1 thiolester observed compared to the lane containing E1 alone. This is an indication that the transthiolation reaction has not reached an end point and that the E1 thiolester is in steady state since it is being depleted by charging of the remaining E2. Under these conditions, one can extend the incubation time or decrease the amount of E2 in the assay.
Typically, 100 nM each of Uba1 and E2 yield an assay that is E3 limiting over a 10-min assay period. The E3 concentration in the assay is chosen empirically by first conducting a series of incubation at different E3 concentrations to pick a concentration at which product formation is linear with (E3)o.
When the Km is not known, one typically performs an initial range study to obtain an approximate value for Km. This can conveniently be done with a “decade study” in which (E2)o is varied by tenfold over several concentrations. Once an approximate value of Km has been determined, the final experiment is conducted with concentrations of E2 that represent a geometric progression about the estimated Km (i.e., ¼ Km, ½ Km, Km, 2Km, 4Km, 8Km). Such a geometric progression provides optimal spacing of data points for greatest accuracy in the subsequent double-reciprocal plot and nonlinear regression analysis.
This is a critical control since the results are not valid unless conducted under E3-limiting conditions, indicated by the absence of an effect on vo when doubling (E1)o.
An incubation time of 10–20 min is optimal for rate studies. Longer incubation times run the risk of deviating from initial velocity conditions by depletion of ATP due to contaminating ATPases or thermal denaturation of one or more components in the assay.
There are a number of different software packages for performing nonlinear regression analysis. All use similar algorithms and differ principally in their ease of use and the utility of the graphics interface.
References
- 1.Haas AL, Siepmann TJ. Pathways of ubiquitin conjugation. Faseb J. 1997;11:1257–1268. doi: 10.1096/fasebj.11.14.9409544. [DOI] [PubMed] [Google Scholar]
- 2.Haas AL, Warms JV, Hershko A, Rose IA. Ubiquitin-activating enzyme. Mechanism and role in protein-ubiquitin conjugation. J Biol Chem. 1982;257:2543–2548. [PubMed] [Google Scholar]
- 3.Haas AL, Rose IA. The mechanism of ubiquitin activating enzyme. A kinetic and equilibrium analysis. J Biol Chem. 1982;257:10329–10337. [PubMed] [Google Scholar]
- 4.Streich FC, Haas AL. Activation of ubiquitin and ubiquitin-like proteins. Subcell. Biochem. 2010;54:1–16. doi: 10.1007/978-1-4419-6676-6_1. [DOI] [PubMed] [Google Scholar]
- 5.Pickart CM. Mechanism underlying ubiquitination. Annu Rev Biochem. 2001;70:503–533. doi: 10.1146/annurev.biochem.70.1.503. [DOI] [PubMed] [Google Scholar]
- 6.Ye Y, Rape M. Building ubiquitin chains: E2 enzymes at work. Nat Rev Mol Cell Biol. 2009;10:755–764. doi: 10.1038/nrm2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ying M, Zhan Z, Wang W, Chen D. Origin and evolution of ubiquitin-conjugating enzymes from Guillardia theta nucleomorph to hominoid. Gene. 2009;447:72–85. doi: 10.1016/j.gene.2009.07.021. [DOI] [PubMed] [Google Scholar]
- 8.Bohnsack RN, Haas AL. Conservation in the mechanism of Nedd8 activation by the human AppBp1-Uba3 heterodimer. J Biol Chem. 2003;278:26823–26830. doi: 10.1074/jbc.M303177200. [DOI] [PubMed] [Google Scholar]
- 9.Huang L, Kinnucan E, Wang G, et al. Structure of an E6AP-UbcH7 complex: Insights into ubiquitination by the E2-E3 enzyme cascade. Science. 1999;286:1321–1326. doi: 10.1126/science.286.5443.1321. [DOI] [PubMed] [Google Scholar]
- 10.Zheng N, Wang P, Jeffrey PD, Pavletich NP. Structure of a c-Cbl-UbcH7 complex: RING domain function in ubiquitin-protein ligases. Cell. 2001;102:533–539. doi: 10.1016/s0092-8674(00)00057-x. [DOI] [PubMed] [Google Scholar]
- 11.Haas AL. ISG15-dependent regulation. In: Mayer RJ, Ciechanover A, Rechsteiner M, editors. Protein Degradation. Chapt. 5. Wiley-VCH Verlag; Weinheim, Germany: 2006. pp. 103–131. [Google Scholar]
- 12.Durfee LA, Kelley ML, Huibregtse JM. The basis for selective E1-E2 interactions in the ISG15 conjugation system. J Biol Chem. 2008;283:23895–23902. doi: 10.1074/jbc.M804069200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kumar S, Kao WH, Howley PM. Physical interaction between specific E2 and Hect E3 enzymes determines functional cooperativity. J Biol Chem. 1997;272:13548–13554. doi: 10.1074/jbc.272.21.13548. [DOI] [PubMed] [Google Scholar]
- 14.Moynihan TP, Ardley HC, Nuber U, et al. The ubiquitin-conjugating enzymes UbcH7 and UbcH8 interact with RING finger/IBR motif-containing domains of HHARI and H7-AP1. J Biol Chem. 1999;274:30963–30968. doi: 10.1074/jbc.274.43.30963. [DOI] [PubMed] [Google Scholar]
- 15.Zhao C, Beaudenon SL, Kelley ML, et al. The UbcH8 ubiquitin E2 enzyme is also the E2 enzyme for ISG15, an IFN-α/β-induced ubiquitin-like protein. Proc Natl Acad Sci USA. 2004;101:7578–7582. doi: 10.1073/pnas.0402528101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim KI, Giannakopoulos NV, Virgin HW, Zhang DE. Interferon-inducible ubiquitin E2, Ubc8, is a conjugating enzyme for protein ISGylation. Mol Cell Biol. 2004;24:9592–9600. doi: 10.1128/MCB.24.21.9592-9600.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang Y, Gao J, Chung KK, et al. Parkin functions as an E2-dependent ubiquitin-protein ligase and promotes the degradation of the synaptic vesicle-associated protein, CDCrel-1. Proc Natl Acad Sci USA. 2005;97:13354–13359. doi: 10.1073/pnas.240347797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tokgöz Z, Bohnsack RN, Haas AL. Pleiotropic effects of ATP•Mg2+ binding in the catalytic cycle of ubiquitin activating enzyme. J Biol Chem. 2006;281:14729–14737. doi: 10.1074/jbc.M513562200. [DOI] [PubMed] [Google Scholar]
- 19.Huang DT, Paydar A, Zhuang M, et al. Structural basis for recruitment of Ubc12 by an E2 binding domain in NEDD8's E1. Mol Cell. 2005;17:341–350. doi: 10.1016/j.molcel.2004.12.020. [DOI] [PubMed] [Google Scholar]
- 20.Petroski MD, Deshaies RJ. Mechanism of lysine 48-linked ubiquitin-chain synthesis by the cullin-RING ubiquitin-ligase complex SCF-Cdc34. Cell. 2005;123:1107–1120. doi: 10.1016/j.cell.2005.09.033. [DOI] [PubMed] [Google Scholar]
- 21.Yunus AA, Lima CD. Purification of SUMO conjugating enzymes and kinetic analysis of substrate conjugation. Methods Mol Biol. 2009;497:167–186. doi: 10.1007/978-1-59745-566-4_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baboshina OV, Crinelli R, Siepmann TJ, Haas AL. N-end rule specificity within the ubiquitin/proteasome pathway is not an affinity effect. J Biol Chem. 2001;276:39428–39437. doi: 10.1074/jbc.M106967200. [DOI] [PubMed] [Google Scholar]
- 23.Lawson TG, Sweep ME, Schlax PE, et al. Kinetic analysis of the conjugation of ubiquitin to picornavirus 3c proteases catalyzed by the mammalian ubiquitin-protein ligase E3 α. J Biol Chem. 2001;276:39629–39637. doi: 10.1074/jbc.M102659200. [DOI] [PubMed] [Google Scholar]
- 24.Siepmann TJ, Bohnsack RN, Tokgöz Z, et al. Protein interactions within the N-end rule ubiquitin ligation pathway. J Biol Chem. 2003;278:9448–9457. doi: 10.1074/jbc.M211240200. [DOI] [PubMed] [Google Scholar]
- 25.Kumar B, Lecompte KG, Klein JM, Haas AL. Ser120 of Ubc2/Rad6 regulates ubiquitin-dependent N-end rule targeting by E3α/Ubr1. J Biol Chem. 2010;285:41300–41309. doi: 10.1074/jbc.M110.169136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang M, Pickart CM. Different HECT domain ubiquitin ligases employ distinct mechanisms of polyubiquitin chain synthesis. Embo J. 2005;24:4324–4333. doi: 10.1038/sj.emboj.7600895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang M, Cheng D, Peng J, Pickart CM. Molecular determinants of polyubiquitin link-age selection by an HECT ubiquitin ligase. Embo J. 2006;25:1710–1719. doi: 10.1038/sj.emboj.7601061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li W, Tu D, Brunger AT, Ye Y. A ubiquitin ligase transfers preformed polyubiquitin chains from a conjugating enzyme to a substrate. Nature. 2007;446:333–337. doi: 10.1038/nature05542. [DOI] [PubMed] [Google Scholar]
- 29.Haas AL. Purification of E1 and E1-like enzymes. Methods Mol Biol. 2005;301:23–35. doi: 10.1385/1-59259-895-1:023. [DOI] [PubMed] [Google Scholar]
- 30.Haas AL, Wilkinson KD. The large scale purification of ubiquitin from human erythrocytes. Prep Biochem. 1985;15:49–60. doi: 10.1080/00327488508062433. [DOI] [PubMed] [Google Scholar]
- 31.Haas AL, Bright PM. The resolution and characterization of putative ubiquitin carrier protein isozymes from rabbit reticulocytes. J Biol Chem. 1988;263:13258–13267. [PubMed] [Google Scholar]
- 32.Wilkinson KD, Tashayev VL, O'Connor LB, et al. Metabolism of the polyubiquitin degradation signal- Structure, mechanism, and role of isopeptidase T. Biochemistry. 1995;34:14535–14546. doi: 10.1021/bi00044a032. [DOI] [PubMed] [Google Scholar]
- 33.Haas AL, Warms JV, Rose IA. Ubiquitin adenylate: Structure and role in ubiquitin activation. Biochemistry. 1983;22:4388–4394. doi: 10.1021/bi00288a007. [DOI] [PubMed] [Google Scholar]