

Abstract
Substantial evidence indicates that the disease-associated conformer of the prion protein (PrPTSE) constitutes the etiological agent in prion diseases. These diseases affect multiple mammalian species. PrPTSE has the ability to convert the conformation of the normal prion protein (PrPC) into a β-sheet rich form resistant to proteinase K digestion. Common immunological techniques lack the sensitivity to detect PrPTSE at sub-femtomole levels while animal bioassays, cell culture, and in vitro conversion assays offer ultrasensitivity but lack the high-throughput the immunological assays offer. Mass spectrometry is an attractive alternative to the above assays as it offers high-throughput, direct measurement of a protein’s signature peptide, often with sub-femtomole sensitivities. Although a liquid chromatography-multiple reaction monitoring (LC-MRM) method has been reported for PrPTSE, the chemical composition and lack of amino acid sequence conservation of the signature peptide may compromise its accuracy and make it difficult to apply to multiple species. Here, we demonstrate that an alternative protease (chymotrypsin) can produce signature peptides suitable for a LC-MRM absolute quantification (AQUA) experiment. The new method offers several advantages, including: (1) a chymotryptic signature peptide lacking chemically active residues (Cys, Met) that can confound assay accuracy; (2) low attomole limits of detection and quantitation (LOD and LOQ); and (3) a signature peptide retaining the same amino acid sequence across most mammals naturally susceptible to prion infection as well as important laboratory models. To the authors’ knowledge, this is the first report of the use of a non-tryptic peptide in a LC-MRM AQUA workflow.
Introduction
Transmissible spongiform encephalopathies (TSEs) or prion diseases are a family of fatal neurodegenerative diseases that affect humans (Creutzfeldt-Jakob disease, CJD) and a variety of farmed and free-ranging mammals including cattle (bovine spongiform encephalopathy, BSE), sheep and goats (scrapie), North American members of the deer family (chronic wasting disease, CWD), and mink (transmissible mink encephalopathy, TME) [1–6]. Prion diseases are unique among protein misfolding disorders in that they are transmissible. Sheep scrapie and CWD appear unique among prion diseases in that epizootics can be sustained through horizontal transmission and that an environmental reservoir of infectivity can contribute to transmission [4,7–15]. Chronic wasting disease and BSE in domestic, captive, and free-ranging animals have caused substantial agricultural, economic, and social impacts. Bovine spongiform encephalopathy has transmitted to humans in the form of variant CJD, raising concern about the potential for CWD to cross the human species barrier [16–20]. The pathogenic prion protein, denoted as PrPTSE, can induce conformational changes in the normal, benign cellular form of the protein, PrPC, leading to disease propagation and accumulation of PrPTSE [21]. The cellular and pathogenic isoforms of the prion protein possess identical primary sequences and covalent posttranslational modifications, and differ only in their conformation [21–27]. Circular dichroism and infrared spectroscopy studies have indicated that conversion of PrPC to PrPTSE is accompanied by a dramatic increase β-sheet content [28,29]. The differences in secondary and higher structures of PrPC and PrPTSE confer on them profoundly different biophysical properties. PrPC is susceptible to proteolysis, is soluble in water and detergents, and exists as a monomer. In contrast, PrPTSE is partially resistant to proteolysis, insoluble in water and non-denaturing detergents, and has a propensity to aggregate. Differences in protease resistance allow discrimination between the two conformers. Treatment of PrPTSE with proteinase K (PK) yields the PK-resistant core of the molecule, denoted PrP27-30 due to the position it migrates to in SDS-PAGE. In contrast, similar treatment of PrPC leads to the generation of many small peptide fragments [21]. Therefore, PK-treatment of PrPC or PrPTSE yields distinct peptide products, enabling their detection and quantification.
Detection and quantification of PrPTSE remain challenging. An intracerebral infectious dose of PrPTSE for hamsters has been estimated to contain ~104 to 105 molecules (~0.02 to 0.2 amol) [30,31]. The most widely employed methods to detect prions and PrPTSE are animal bioassay and immunological assays. Animal bioassay is the most direct technique to measure prion infectivity and is considered the “gold standard” for prion detection, but is animal-intensive, time-consuming, expensive, and generally limited to rodent-adapted prion strains or requires the use of transgenic mice expressing the prion protein of interest [32]. Immunological assays such as Western blotting, enzyme-linked immunosorbent assays, and a conformation-dependent immunoassay are commonly used in prion research but have limits of detection (LOD) from 20 pmol to 0.1 pmol [33–40]. Other classes of prion detection methods are in vitro conversion assays and cell culture methods. Protein misfolding cyclic amplification (PMCA) is a powerful prion conversion assay that exploits the ability of PrPTSE to convert PrPC to a PK-resistant form in a manner conceptually similar to the polymerase chain reaction [41–43]. Many amplification/conversion cycles can be performed to increase PrPTSE levels 101- to 107-fold prior to detection [41]. A drawback of this method is that amplification of PrPTSE can take several days to achieve ultrasensitivity, stringent conditions are required to prevent false positive results [44], and levels of PrPTSE are not directly measured. Addition of polymeric beads to PMCA reactions has recently been shown to improve the robustness of the method [45,46]. Cell culture-based methods have recently been reported for several economically important species [47,48].
Mass spectrometry-based methods using stable isotope-labeled standards (SIS) for absolute quantification (AQUA) is an attractive alternative to the above prion detection methods. The AQUA method involves introducing a known concentration of a SIS peptide to a protein digest that yielded the same unlabeled peptide. Peptide abundances of the two peptides (SIS and native) are then measured by liquid-chromatography with multiple reaction monitoring (LC-MRM), and the concentration of the targeted protein is calculated using a calibration curve. This method allows for sub-femtomole quantification of proteins in complex mixtures [49,50].
A LC-MRM assay was recently reported for the detection and quantitation of PrPTSE in Syrian hamster brain homogenate [51,52]. A drawback to the reported method is that the signature tryptic peptide used for quantitation contains two chemically reactive residues (Cys, Met) that can compromise assay accuracy. In addition to concerns about assay accuracy, prion research is conducted on a wide variety of animal models. Although prion protein is observed in all these animal models, the primary structure differs slightly in each [53]. When the primary sequence differs in the target peptide region the assay must be redeveloped and revalidated. In the original report, the LC-MRM method was validated in the Syrian hamster using a signature peptide limited to this species (VVEQMCTTQYQK) [51]. Recently, the tryptic SIS peptide has been revalidated for additional species [54]. Application of this assay to additional animal models with different primary structures in the target region requires revalidation of the assay, adding expense to the lab.
This work explores the use of alternative proteases to produce a peptide more suitable for LC-MRM AQUA assays of prion protein. A suitable AQUA peptide is defined here as (1) lacking chemically active residues (Cys, Met) that can compromise the accuracy of the assay, (2) providing sub-femtomole LOD and LOQ, and (3) identical across multiple mammalian species susceptible to natural prion disease or used as laboratory models. Many LC-MRM AQUA publications mention in passing that proteases other than trypsin could in principle be used to produce a peptide suitable for a quantification assay [50,55], but to the authors’ knowledge, accounts of other enzymes being utilized for this type of assay have not been published.
Experimental
Chemicals
Iodoacetamide (IAA), beta-octyl glucopyranoside (BOG), α-cyano-4-hydroxycinnamic acid (CHCA), thermolysin, trifluoroacetic acid (TFA), and hydrofluoric acid (HF) were purchased from Sigma-Aldrich (St Louis, MO, USA). Ammonium bicarbonate, formic acid (FA), and heavy AQUA peptides RPV[13C5,15N]DQY, YRPV[13C5,15N]DQY, and FL[13C6,15N2]NPWEK were supplied by Thermo Fisher Scientific (Waltham, MA, USA). Dithiothreitol (DTT) and sequence grade modified trypsin were purchased from Promega (Madison, WI, USA). Bovine chymotrypsin and glutamic-C (glu-C) were purchased from Princeton Separations (Adelphia, NJ, USA). Bovine recombinant prion protein (bovine rPrP) was purchased from Abcam (Cambridge, MA, USA). Syrian hamster recombinant prion protein (hamster rPrP90-231) was supplied by Jena Bioscience (Jena, Germany). Sterile filtered mouse serum was purchased from Equitech-Bio, Inc (Kerrville, TX, USA). The 18 MΩ·cm water used in these experiments was obtained from a Milli-Q purification system (Millipore, Bedford, MA, USA). The LC-MS grade solvents acetonitrile (ACN) and water for the Waters nanoAcquity UPLC (Milford, MA, USA) were purchased from Burdick and Jackson (Muskegon, MI, USA). The Optima grade solvents ACN and water for the Eksigent Ultra 2D UPLC (Dublin, CA, USA) were purchased from Fisher Scientific (Fair Lawn, NJ, USA).
Chymotrypsin, Trypsin, Glu-C, Thermolysin Digestions for AQUA Peptide Identification
Bovine rPrP (5 μL, 1 μg·μL−1) was combined with 15 μL 0.01% BOG (w/v) and sonicated for 5 min in an ultrasonic bath. Cysteine residues in bovine rPrP were reduced with 10 μL 10 mM DTT prepared in buffer A (25 mM ammonium bicarbonate in 0.01% BOG (w/v)) for 1 h at 37 °C. After cooling to room temperature, cysteines were alkylated with 40 μL 10 mM IAA in darkness for 1 h at room temperature. Alkylation was quenched by incubation in 20 μL 10mM DTT for 15 min at room temperature. All digestions were performed under the manufacturer’s suggested conditions: chymotrypsin (1:50 enzyme:protein ratio, 30 °C, 9 h); Glu-C (1:20 enzyme:protein ratio, 25 °C, 16 h); thermolysin (1:25 enzyme:protein ratio, 37 °C, 24 h); and trypsin (1:50 ratio, 37 °C, 16 h). Each digest was quenched by addition of 2.5 μL 10 % (v/v) FA(aq), desalted with C18 OMIX pipette tip (Agilent Technologies, Santa Clara, CA, USA), and concentrated by speedvac (Thermo Scientific, Waltham, MA, USA). Dried eluent was resuspended in 60 μL 50 % ACN/0.1 % TFA(aq) (v/v) and stored at 4 °C until analysis on a Bruker amaZon ETD ion trap (Billerica, MA, USA) mass spectrometer.
Optimization of Chymotrypsin Digestion Time
Three 5 μL aliquots of hamster rPrP90-231 (1 μg·μL−1) were reduced, alkylated, and chymotrypsinized as described above except the total volume was diluted to 100 μL. At desired time points, 20 μL aliquots of each digest were removed, and 0.5 μL (0.1 μg·μL−1) fresh chymotrypsin was added to each digestion tube. The chymotryptic digestion in each aliquot was quenched by addition of 1 μL 10 % FA(aq) (v/v), and the reaction was desalted with C18 OMIX pipette tip and concentrated by speedvac. Concentrated samples were reconstituted in 100 μL 3%ACN/FA(aq) (v/v) containing SIS peptide YRPVDQY. Subsequent serial dilutions yielded final analyte and internal standard concentrations of 5 fmol·μL−1 and 10 fmol·μL−1, respectively. Each time point sample was analyzed in triplicate using a Bruker autoflex III TOF/TOF and AB Sciex 5500 QTRAP (Foster City, CA, USA) mass spectrometer.
Hamster rPrP90-231 Quantification
Three 2 μL aliquots of hamster rPrP90-231 (1 μg·μL−1) were reduced, alkylated and chymotrypsinized (vida supra). Chymotryptic digestion was allowed to proceed for 9.5 h before 2.5 μL 10 % FA(aq) (v/v) was added to quench the reaction. A 5 μL aliquot of the digest was placed in 45 μL 0.1% FA(aq) (v/v) containing 11.1 fmol·μL−1 SIS peptides RPVDQY and YRPVDQY, desalted with C18 OMIX pipette tip, and concentrated by speedvac. Concentrated samples were reconstituted in 200 μL 3% FA(aq) (v/v). Each digest was analyzed in triplicate on an AB Sciex 5500 QTRAP mass spectrometer.
Qualitative Mass Spectrometry
Bovine rPrP peptide fragments produced by tryptic, chymotryptic, thermolytic, and Glu-C digestions were identified by nanoESI-direct infusion (300 nL·min−1) into a Bruker amaZon ETD ion trap mass spectrometer. In addition, digestions were analyzed by nanoLC-MS/MS using an Eksigent Ultra 2D UPLC. Mobile phase A was water in 0.1% FA(aq) (v/v), and mobile phase B was ACN. Samples were introduced into the system by a 5 μL injection onto an Agilent Technologies Zorbax 300 SB-C18 5 μm, 5 × 0.3 mm trap cartridge (Santa Clara, CA) at a flow rate of 5 μL·min−1 of 95% mobile phase A for 5 min. Peptides were separated on a 3 μm Waters Atlantis dC18 75 μm × 150 mm column (Milford, MA) with a 40 min gradient from 5% to 35% mobile phase B at a flow rate of 250 nL·min−1 and column temperature of 30 °C. Electrospray emitter tips were prepared in house from 75 μm i.d., 360 μm o.d. capillary tubing (Polymicro Technologies, Phoenix, AZ) using a Sutter P-2000 laser capillary puller (Novator, CA).
Capillary voltage was set to −1300 V, end plate offset voltage was −500 V, dry gas flow was 4.0 L·min−1, and dry gas temperature was 125 °C. MS data were acquired from 300 to 1700 m/z in enhanced resolution mode (8100 m/z s−1) with a target mass of 700 m/z and trap drive level of 100%. CAD MS2 spectra were manually collected with a 1 V MS/MS fragmentation amplitude, and smart fragmentation was set at 30 to 300 %.
Bovine rPrP peptide fragments produced by chymotryptic digestion in the timed digestion experiment were identified using a Bruker MALDI autoflex III mass spectrometer. For this analysis, equal volumes (0.5 μL) of reconstituted digest and matrix solution, consisting of 5 mg CHCA dissolved in 1 mL 50 % ACN/0.1 % TFA(aq) (v/v), were mixed on a ground-steel plate and allowed to dry. Mass spectra were acquired in reflectron positive ion mode with a 200 Hz repetition rate averaging 500 laser shots.
In silico tryptic, chymotryptic, thermolytic, and Glu-C digestions of bovine and hamster rPrP were performed using MS-Digest tool from the Protein Prospector suite (http://prospector.ucsf.edu, v. 5.9.2) developed by UCSF. Carbamidomethylation was set as a static modification, methionine oxidation was selected as a variable modification, the minimum peptide length equaled five, peptide mass ranged from 300 to 2500 m/z, and multiple charged ions were reported in the algorithm’s output. Prion protein amino acid alignment was carried out with Jalview software (http://www.jalview.org/download.html) [56,57] and the clustalW alignment algorithm [58]. Protein sequences were from uniprot/swissprot knowledgebase (http://www.uniprot.org/help/uniprotkb).
Quantitative Mass Spectrometry
LC-MRM experiments were performed with a Waters nanoAcquity UPLC connected online with an AB Sciex 5500 QTRAP equipped with a nanospray III ion source. Initial experiments to manually optimize instrument response for YRPVDQY and FLNPWEK were done by direct infusion. The two peptides (2.5 pmol·μL−1 in 50 % ACN/0.1 % FA(aq) (v/v)) were infused into the mass spectrometer at 500 nL·min−1. A precursor scan was used to confirm the presence of analyte ions in the mass spectrometer, and a product ion scan was used to determine the dominant MS2 fragment peaks. Curtain gas, ionspray voltage, ion source gas 1, interface heater temperature, and ESI emitter position were optimized for the precursor signal, while the declustering potential (DP), entrance potential (EP), collision energy (CE), and collision cell exit potential were optimized for each precursor/product ion MRM transition to obtain maximum analyte/MRM intensity while maintaining stable electrospray intensity. Instrument settings for RPVDQY were optimized using Skyline software (https://skyline.gs.washington.edu, v. 0.7) developed by the MacCoss lab [59].
For LC-MRM experiments curtain gas was set to 20 psi, collision gas was medium, ionspray voltage was 2300 V, ion source gas 1 was set to 16 psi, and interface heater temperature was 150 °C. UPLC elution solvents were: A (0.1 % FA(aq) (v/v)) and B (ACN/0.1 % FA(aq) (v/v)). Samples resuspended in 3% ACN/0.1% FA(aq) (v/v) were loaded onto a self-packed C18 trap cartridge (Phenomenex Jupiter C18 5 μm, 100 μm × 70 mm;) at a flow rate of 2.00 μL·min−1 solvent A over 10.0 min. Samples were eluted from a self-packed reversed-phase column (Phenomenex Jupiter C18 5 μm, 75 μm × 120 mm) integrated with a HF etched 8 μm ESI emitter tip (Sutter Instrument P-2000, Novato, CA, USA) using an 11 min linear gradient from 3 % to 35 % solvent B at a flow rate of 500 nL·min−1. Solvent B was raised to 70 % to elute remaining peptides, and the column was re-equilibrated for 10 min in 3 % solvent B prior to injection of the next sample. The mass spectrometer was operated in MRM mode, alternating between detection of YRPVDQY (ion transitions m/z 470.7+2 to 759.3+1 (b6), 470.7+2 to 631.3+1 (b5), 470.7+2 to 182.1+1 (y1)), and YRPV[13C5,15N]DQY (ion transitions m/z 473.7+2 to 765.3+1 (b6), 473.7+2 to 637.3+1 (b5), 473.7+2 to 182.1+1 (y1)), RPVDQY (ion transitions m/z 389.2+2 to 596.3+1 (b5), 389.2+2 to 468.3+1(b4), 389.2+2 to 182.1+1 (y1)), RPV[13C5,15N]DQY (ion transitions m/z 392.2+2 to 602.3+1 (b5), 392.2+2 to 474.3+1 (b4), 392.2+2 to 182.1+1 (y1)). Q1 and Q3 were operated in unit resolution mode, and dwell time for each transition was set to 40 ms. Quantification was performed with the Intelliquan quantification algorithm of Analyst 1.5.1 software (AB Sciex) using default parameters. Calibration curves, proteolysis time, and digest accuracy graphs were prepared using Graphpad Prism (ver 5.0) software.
Results and Discussion
Recently, Silva and Onisko developed a mass spectrometric MRM assay using a tryptic peptide for AQUA quantification of PrPTSE in brain homogenate with an estimated limit of detection (LOD) in the low attomole range (20–30 amol) and limit of quantification (LOQ) below 100 amol [51,52,54,60]. In this method, a hamster PrPTSE tryptic peptide (VVEQMCTTQYQK, m/z 825.4) was subjected to collisionally activated dissociation (CAD) in quadrupole 2 of an AB Sciex 4400 QTRAP mass spectrometer, and the fragment a2 ion (m/z 171.1) was used for detection and quantification [51,52]. This work showcased the potential of mass spectrometry for prion quantification, but used a peptide containing chemically reactive amino acids that should be avoided in MRM assays (Cys and Met) [50,55,61–63]. Of the two amino acids, methionine has larger potential for affecting quantification accuracy since it can be present in three different oxidation states (normal, sulfide, sulfone) splitting the target peptide signal into three unique m/z values that need to be measured. In addition to the potential of the native peptide toward oxidation, the SIS peptide would be subject to the same quantitative challenges. We tested our concerns by analyzing a recombinant prion protein (rPrP) bovine tryptic digest and SIS peptide VVEQMCTTQYQK. We observed both normal and sulfoxide forms of the native and SIS peptides in each analysis (Supplemental Figure 1). The aforementioned researchers became aware of this complication in their most recent publication and now monitor both normal and sulfoxide forms of the native and SIS peptides for PrPTSE quantification [54,64]. Although normal and sulfoxide SIS peptides could be utilized, the possibility still exists for the normal SIS peptide to contribute to the sulfoxide SIS peak, and for the sulfoxide SIS peptide to further oxidize to the sulfone state, thus confounding quantification. Here, we aim to identify a peptide lacking Met and Cys residues to use as a signature peptide for an LC-MRM quantitation of prion protein.
Identifying a Non-tryptic Peptide for AQUA Quantification of PrPTSE
We simulated digestion of the proteinase K-resistant core of Syrian hamster and bovine PrPTSE by thirteen proteases in silico with Protein Prospector’s MS-Digest algorithm: trypsin, chymotrypsin, Lys C, Lys N, pepsin, Arg C, Asp N, Asp C, DE-N, Glu-C, Tyr-C, thermolysin, and Pro-C. Chymotrypsin, Glu-C, and thermolysin showed promise in that they produced peptides in silico that met our first criteria for a suitable AQUA peptide (Supplemental Table 1): lack of chemically active residues (Cys, Met) that can compromise assay accuracy (Supplemental Figure 1). Chymotrypsin cleaves peptide bonds predominantly on the carboxy side of Tyr, Phe, and Trp. Glu-C cleaves peptide bonds at the carboxy side of Glu residues. Thermolysin is predicted to cleave peptides at the N-terminus side of hydrophobic amino acid residues Leu, Ile, Phe, Val, Ala, and Met [65].
After identifying proteases that may potentially produce a peptide meeting our first criteria, digestions were performed according to manufacturers’ instructions. Desalted proteolytic digests were directly infused into a Bruker amaZon ETD ion trap and also subjected to nanoLC-MS/MS analysis to experimentally determine which peptides predicted in in silico digests warranted further consideration for a quantitative assay. Target peptides from the Glu-C and thermolysin digests were not observed at high intensities in either the direct infusion or online nanoLC analyses (data not shown). In contrast, chymotrypsin generated an intense ion peak at m/z 470.7 (2+) corresponding to the doubly charged peptide YRPVDQY. We originally targeted RPVDQY as a potential peptide for MRM quantification, but we did not observe ions for this peptide during direct infusion or nanoLC-MS/MS analyses (Supplemental Figure 2). Instead, the predominant ion observed was the doubly charged YRPVDQY ion (one missed cleavage). The reason that the missed cleavage form of RPVDQY is primarily produced over the fully digested form is unclear. However, the doubly charged YRPVDQY peptide met our first criteria for a suitable AQUA peptide since it lacks chemically reactive Cys and Met residues. We therefore further investigated this peptide.
The candidate AQUA peptide also met our third criterion of sharing homology with a large number of agriculturally important and research model species. Comparison of prion protein primary structure across 46 mammalian species showed that YRPVDQY was conserved in 35 of 46 species [53]. Of the remaining eleven species, six different isoforms of residue 163–169 peptide were observed and only one species did not contain this sequence due to C-terminal truncation. Figure 1 displays the amino acid sequence alignment of 20 prion proteins from mammalian species naturally susceptible to prion disease or serving as important research models. Peptide YRPVDQY resides at amino acid positions 163 to 169 (human PrP numbering). Figure 1 shows that YRPVDQY is common in sixteen of the twenty species including rodents (hamster, mouse, rat, voles), cervids (deer, elk, moose), cattle, and goats. Rodents (especially mice and Syrian hamsters) are important models in prion research because mouse genetics can be manipulated to test hypotheses and hamsters have relatively short incubation times and high infectious titers [66]. Cervid CWD is an emerging infectious disease in North America with a rapidly expanding range. BSE in cattle and scrapie in goats are TSEs of agricultural concern, and BSE has spread to humans in the form of variant CJD [16–20]. Discovery that an environmental reservoir of infectivity can contribute to horizontal (animal-to-animal) spread of CWD and scrapie has prompted research to identify sources of infectivity and mechanisms of transmission [4,7–15]. A LC-MRM assay using the signature peptide YRPVDQY could be implemented in research in these species without the need for assay revalidation. This is not the case for the previously reported peptide VVEQMCTTQYQK (PrP residues 209–220) that is unique to only Syrian hamster (Figure 1); revalidation would be required for species with alternative amino acid sequences in this region. Indeed, application of the previously reported LC-MRM assay using tryptic AQUA peptides to additional species required use of four additional 209 to 220 sequence isoforms (VVEQMCITQYQK, VVEQMCVTQYQK, VVEQMCITQYQR, VVEQMCITQYER) and the corresponding sulfoxide forms of each [54], with a coverage of 30 of the 46 species compared in [53]. This is due to the increased variation in amino acid residues located in the 209 to 220 domain. Additionally, peptide 209 to 220 is not observed in 4 of the 46 species due to C-terminal truncation. Therefore, the sequence YRPVDQY (163–169) appears to be a more attractive peptide for LC-MRM AQUA experiments because the investment in heavy isotope peptides is decreased compared to peptide 209–220 and the chymotryptic peptide lacks chemically reactive amino acids that could affect quantification accuracy.
Figure 1.
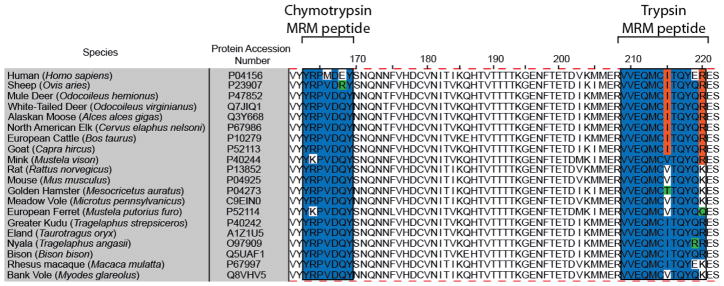
Amino acid alignment of PrP residues 161 to 222 (human PrP numbering) from 20 mammalian species either susceptible to natural prion infection or used as laboratory models. Note that the amino acid sequence for the chymotrypsin MRM peptide is more highly conserved than that for the trypsin-derived MRM peptide. The alignment was performed using the ClustalW algorithm within Jalview software.
We investigated whether known species-specific polymorphisms in prion protein could affect this LC-MRM assay. The sequence YRPVDQY is conserved across the cervid, bovid, and rodent species presented in Figure 1. For a polymorphism to affect this assay, the amino acid substitution would have to occur within residues 163 to 169. Substitution of amino acid 169 with a residue other than Phe or Trp would prevent production of the chymotryptic AQUA peptide and would interfere with analysis. Cervids have at least 20 known polymorphisms, but only seven of these alter the primary sequence of PrP [5,67], and none affect amino acids 163 to 169 or the generation of the chymotryptic peptide. Thirteen polymorphisms have been identified in the bovine Prnp gene with only three affecting the translated protein and none impacting this assay [67,68]. In the case of rodents, Syrian hamsters have no known polymorphisms and mice only have two polymorphisms (L108F and T189V), neither of which would interfere with the assay developed here [69]. Although voles have several known polymorphisms, none of them occur between residues 163 and 169 [70]. Therefore, polymorphisms in Prnp gene for these species should not hinder the efficacy of the LC-MRM method presented here.
Determining the Limit of Quantification (LOQ) and Detection (LOD) for YRPVDQY
The LOQ, LOD, and linear dynamic range for YRPVDQY were empirically determined by LC-MRM on an AB Sciex 5500 QTRAP. Figure 2 shows the CAD MS/MS spectra of YRPV[13C5,15N]DQY. Interestingly, b-ions dominate this MS/MS spectrum whereas y-ions dominate MS/MS spectra for tryptic peptides. The y-series dominate tryptic peptide MS/MS spectra because basic Arg and Lys residues reside on the C-terminus resulting in higher signal intensities. In the case of YRPVDQY, chymotryptic digestion produced a peptide with an Arg near the N-terminus giving rise to higher intensity b-ions [71]. Instrument and MRM transition settings for SIS peptide YRPVDQY y1, b2, b4, b5, and b6 ion transitions were manually optimized to provide reproducible and highly sensitive measurements (vide supra). After optimization, the b6 ion transition was chosen for quantification, and two transitions (b5, y1) were selected for peptide confirmation (Table 1).
Figure 2.

Collisionally activated dissociation (CAD) spectrum of stable isotope-labeled standard peptide YRPVDQY obtained at a collision energy of 20 V.
Table 1.
Optimized MRM transitions for selected chymotryptic peptides derived from bovine and hamster rPrPs.a
| Peptide | Precursor Ion (m/z; z=+2) | Declustering Potential (volts) | Collision Energy (volts) | Product Ion (m/z; z=+1) |
|---|---|---|---|---|
| RPVDQY (native) | 389.2 | 63.5 | 17.9 | 596.3 (b5) |
| 389.2 | 71.0 | 24.1 | 468.3 (b4) | |
| 389.2 | 71.0 | 17.9 | 182.1 (y1) | |
|
| ||||
| RPV* DQY (SIS) | 392.2 | 63.5 | 17.9 | 602.3 (b5) |
| 392.2 | 71.0 | 24.1 | 474.3 (b4) | |
| 392.2 | 71.0 | 17.9 | 182.1 (y1) | |
|
| ||||
| YRPVDQY (native) | 470.7 | 71.0 | 20.7 | 759.3 (b6) |
| 470.7 | 71.0 | 26.8 | 631.3 (b5) | |
| 470.7 | 71.0 | 20.8 | 182.1 (y1) | |
|
| ||||
| YRPV* DQY (SIS) | 473.7 | 71.0 | 20.7 | 765.3 (b6) |
| 473.7 | 71.0 | 26.8 | 637.3 (b5) | |
| 473.7 | 71.0 | 20.8 | 182.1 (y1) | |
The italicized/bolded transitions for each peptide indicate the transition used for quantification. The two non-italicized/bolded transitions were used to confirm the peptide’s identity.
indicates the location of the heavy amino acid (13C, 15N) for SIS peptides.
A calibration curve for the optimized m/z 473.7+2 to 765.3+1 transition was constructed (Figure 3a). The SIS peptide YRPV[13C5,15N]DQY was serially diluted from 1 pmol·μL−1 to 10 amol·μL−1 in 0.2 μg·μL−1 mouse serum tryptic digest to minimize non-specific adsorption to the sample vial walls. The SIS peptide FL[13C6,15N2]NPWEK served as internal standard and was added to each serial dilution sample at 2.5 fmol·μL−1. The mean MRM response value for quintuplicate injections of each calibration standard displayed a linear dynamic range spanning approximately 3.5 orders of magnitude and a LOQ of 80 amol. We collected data for the calibration curve using a mouse serum tryptic digest as sample matrix because we observed departures in linearity in the low attomole range (< 100 amol) when performing the experiment in a neat matrix (data not shown). We hypothesized that this was due to analyte adsorption to microcentrifuge tubes, pipette tips, and glass sample vials. Additionally, we hypothesized that the addition of a complex sample tryptic digest would mask these adsorption sites without interfering with MRM transitions. We were able to obtain a more accurate and linear calibration curve using this background matrix without affecting the purity of our MRM transitions. Figure 3a shows the MRM response of seven dilutions of the standards ranging from 80 amol to 200 fmol. The correlation coefficient for this curve was 0.996. Figure 3b and 3c display the chromatographic profiles for 40 fmol and 80 amol injections, respectively. The signal-to-noise ratio (S/N) was calculated using the standard deviation of a blank containing only background matrix ± 15 s of YRPVDQY elution time as an estimate of instrumental/chemical noise. For quintuplicate measurements of 80 amol, calculated S/N was ~11; for 40 amol, calculated S/N was ~6. By defining the LOD as the amount of analyte yielding a S/N = 3, we estimate this LC-MRM method can achieve a limit of detection between 20 and 40 amol, comparable to that estimated for the tryptic MRM peptide [51]. These results indicate that peptide YRPVDQY meets our second criterion for a suitable AQUA peptide by having a sub-femtomole LOD and LOQ.
Figure 3.

MRM response of chymotryptic stable isotope-labeled standard peptides YRPV*DQY and RPV*DQY spiked into a 0.2 μg·μL−1 mouse serum tryptic digest. (a) Linear calibration curve for YRPV*DQY spanning 3.5 orders of magnitude. (b) MRM response for 40 fmol YRPV*DQY and (c) 80 amol YRPV*DQY. (d) Linear calibration curve for RPVDQY spanning 3.5 orders of magnitude. (e) MRM response for 40 fmol RPV*DQY and (f) 80 amol RPV*DQY. For both peptides, the LOQ was less than 100 amol. Points on the calibration curve correspond to mean values; error bars represent the standard deviation (n = 5). The inset is an expansion of the MRM ratios from the three lowest amounts on the calibration curve. * indicates the location of the heavy amino acid (13C,15N) for SIS peptides.
Optimization of Chymotrypsin Digestion Time
After establishing that YRPVDQY has a LOQ < 100 amol and a LOD between 20 and 40 amol, we optimized digestion time using bovine rPrP. Although chymotrypsin cleaves peptide bonds predominantly on the carboxy side of Tyr, Phe, and Trp, non-specific hydrolysis can occur if digestion is conducted for too long [72,73], making the use of the shortest possible time for complete digestion of PrP advisable. Based on manufacturer recommendation and previous work by Arsene et al. [74] we determined that the digestion was complete by 9.5 h using the 5500 QTRAP to measure the native and SIS form of YRPVDQY (Figure 4). In addition, we monitored the digestion using a MALDI autoflex III TOF/TOF and surprisingly observed that after 3 h of digestion a low intensity doubly charged ion peak (m/z 389.2) appeared (Supplemental Figure 3). This ion peak corresponds with the m/z of the fully digested RPVDQY peptide. The MALDI spectra collected during the digestion time optimization experiment and the lack of detectable signal for RPVDQY during the enzyme selection experiment (Supplemental Figure 2) indicate that YRPVDQY is the predominant peptide form produced during chymotrypsin digestion. The reason that chymotrypsin digestion of rPrP favors production of YRPVDQY over RPVDQY remains unclear. To determine the extent to which YRPVDQY could form RPVDQY we digested 5 × 10−7 M SIS YRPVDQY with chymotrypsin for 9.5 h. The results indicate that that only a very small fraction (~0.1%) of YRPVDQY was converted to RPVDQY during digestion (Supplemental Figure 4). We hypothesize that during digestion a majority of rPrP forms the missed cleavage peptide YRPVDQY while a small portion of rPrP hydrolyzes as expected from in silico digestion to form the peptide RPVDQY.
Figure 4.
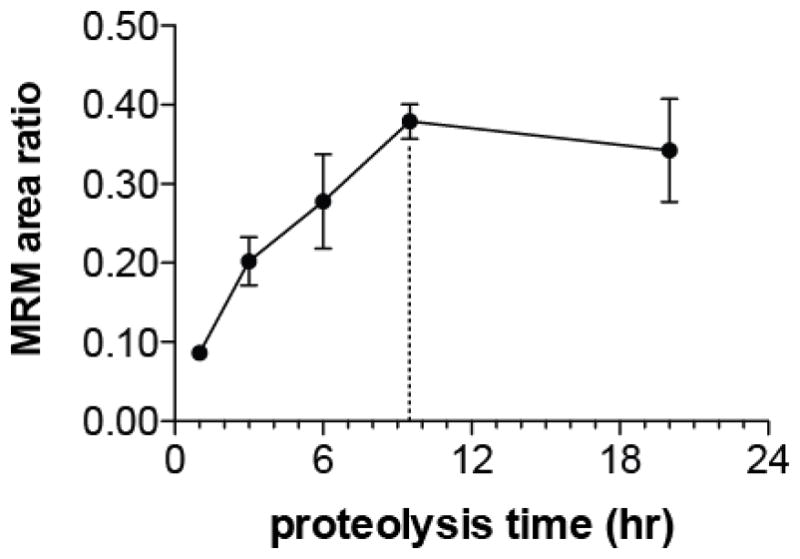
Formation of chymotryptic fragment YRPVDQY from digestion of bovine rPrP. Each point corresponds to the mean of three technical replicates from three separate digests; error bars represent one standard deviation. Native YRPVDQY MRM response was normalized to SIS YRPVDQY MRM response (10 fmol on-column). Dashed line indicates optimal digestion time (9.5 hours).
To increase the accuracy of the AQUA assay we monitored both RPVDQY and YRPVDQY with SIS peptides in the final assay. We optimized the b5, b4, and y1 transitions of RPVDQY using Skyline software to generate a range of DPs and CEs that could be tested in subsequent MRM experiments. The final optimized parameters for YRPVDQY and RPVDQY are listed in Table 1. We note that RPVDQY behaved similarly to YRPVDQY with respect to LOD, LOQ, and linear dynamic range (Figure 3d–f). The main difference between YRPVDQY and RPVDQY calibration curves is that the relative standard deviation (RSD) for points on RPVDQY curves was higher than that for YRPVDQY (Table 2). This may be attributed to optimizing RPVDQY by testing only the limited number of DPs and CEs suggested by Skyline software. For YRPVDQY, we optimized all transition voltages by directly infusing YRPVDQY into the mass spectrometer. In any case, the RSD for all points on the calibration curves for both SIS peptides were ≤ 15 % which is within suitable limits for a quantification method [75].
Table 2.
Calibration curve statistics for YRPVDQY and RPVDQY MRM quantification transitions.a
| YRPV*DQY, b6 transition
| |||||
|---|---|---|---|---|---|
| ExpectedAmount (fmol) | # of replicates | Calculated Amount (fmol) | Standard Deviation | RSD | Accuracy |
| 0.080 | 5 | 0.080 | 0.002 | 2.0 | 99.5 |
| 0.14 | 5 | 0.14 | 0.007 | 4.9 | 97.5 |
| 0.40 | 5 | 0.42 | 0.01 | 3.4 | 104.7 |
| 8.0 | 5 | 8.4 | 0.3 | 3.3 | 105.4 |
| 40 | 5 | 35 | 2 | 4.6 | 86.6 |
| 100 | 5 | 108 | 2 | 1.9 | 107.8 |
| 200 | 5 | 197 | 10 | 5.2 | 98.5 |
|
RPV*DQY, b5 transition
| |||||
| ExpectedAmount (fmol) | # of replicates | Calculated Amount (fmol) | Standard Deviation | RSD | Accuracy |
|
| |||||
| 0.080 | 5 | 0.069 | 0.007 | 10.6 | 86.5 |
| 0.14 | 5 | 0.15 | 0.02 | 11.1 | 107.6 |
| 0.40 | 5 | 0.40 | 0.04 | 10.3 | 99.7 |
| 40 | 5 | 45 | 5 | 10.3 | 111.7 |
| 100 | 5 | 94 | 12 | 13.0 | 93.5 |
| 200 | 5 | 202 | 24 | 11.6 | 100.9 |
The RSD and accuracy of each point on the calibration curve are < 15% of the real value.
Hamster rPrP90-231 Quantification by LC-MRM
We tested the performance of our LC-MRM AQUA method on a chymotryptic digest of known amount of Syrian hamster rPrP. Here, three 2 μg aliquots of hamster rPrP90-231 were digested with chymotrypsin. Sample preparation comprised reduction/alkylation, digestion, addition of SIS peptides, OMIX C18 pipette tip desalting, speedvac concentration, and sample re-suspension/dilution. Recovery experiments were performed to estimate the overall yield of RPVDQY and YRPVDQY after the sample preparation steps. Chymotryptic digests of PrP90-231 ranging from 1 fmol·μL−1 to 100 fmol·μL−1 rPrP90-231 were found to have recoveries of 60 % to 81 %, respectively. The 2 μg digests were diluted so that for a 2.0 μL injection the amounts of native RPVDQY and YRPVDQY would sum to approximately 62.7 fmol on-column when measured. SIS peptides for RPVDQY and RPVDQY were spiked into the digests so the total injected amount of each SIS would be 10 fmol on-column.
Figure 5a and 5b show the calibration curves for YRPVDQY and RPVDQY over the range 2 to 200 fmol on-column. Figure 5c shows the MRM responses for native and SIS AQUA peptides. Comparison of the SIS transitions to the native ion transitions demonstrates that YRPVDQY was the predominant chymotryptic fragment produced during digestion. The average amounts of YRPVDQY and RPVDQY were 44 ± 1 fmol and 18.6 ± 0.4 fmol on-column over all three digests, respectively. The total hamster rPrP90-231 amounts calculated on-column for each digest were the following: Digest 1 = 64.0 ± 1 fmol, Digest 2 = 63.7 ± 0.5 fmol, Digest 3 = 61.3 ± 0.5 fmol. These values correspond to errors of 2.15 %, 1.64 %, and 2.12 % (Figure 5d). These results underscore the suitability of YRPVDQY and RPVDQY to serve as AQUA peptides for the quantification of PrPTSE using a LC-MRM strategy.
Figure 5.

Quantification of hamster rPrP90-231 in a chymotryptic digest using the AQUA approach. (a,b) Three-point calibration curves of YRPVDQY and RPVDQY demonstrating a linear relationship over the range of quantification (2 fmol to 200 fmol on-column), respectively. (c) Extracted ion chromatogram of MRM responses for each of the four targeted peptides. Hamster rPrP90-231 (2 μg) was digested with chymotrypsin and diluted such that a 2 μL injection yielded 62.7 fmol digest on column. Both SIS internal standards were spiked into each digest to give an amount of 10 fmol on-column. Twelve MRM transitions are displayed (three for each peptide). (d) Quantitative accuracy of rPrP90-231 chymotryptic digests. All three rPrP digests were quantified accurately within 5 % of the native rPrP90-231 amount using the AQUA approach. Each digest was analyzed three times using the 5500 QTRAP. Solid line: amount of native rPrP90-231 digest injected on-column (62.7 fmol or 1.1 nmol/g). Dashed line: Indicates 5 % error boundary.
Conclusion
Mass spectrometry represents an attractive alternative to more commonly used assays to measure prions because it offers high-throughput, direct measurement of a protein’s signature peptide with sub-femtomole sensitivities. Using recombinant forms of the prion protein we demonstrated the utility of a signature peptide produced via chymotrypsin digestion for a LC-MRM AQUA assay. Our new peptides meet the following criteria: (1) lack of chemically active residues (Cys, Met) that can confound assay accuracy, (2) provision of sub-femtomole LOD and LOQ, and (3) identical amino acid sequences across multiple commercially and experimentally important mammalian species. Chymotryptic peptides YRPVDQY and RPVDQY do not contain residues susceptible to modification during sample handling and analysis that could hinder quantification experiments. Each provides low attomole LOD and LOQ sensitivities while maintaining a dynamic range over 3.5 orders of magnitude. In addition, both peptides show a high degree of conservation across mammalian species and should not be affected by known polymorphisms. When used to quantify recombinant prion protein, quantification accuracy error was minimal (< 3%). The LC-MRM approach presented here is expected to translate well into research studying pathogenic prion protein in a large number of mammalian species. In fact, we are currently applying this assay to a variety of mammalian species. In summary, we present an inexpensive, high-throughput, and accurate LC-MRM AQUA method for quantification of prion protein in a variety of economically and research important species. To the authors’ knowledge, this is the first report of an LC-MRM AQUA method utilizing signature peptides derived from a non-tryptic peptide.
Supplementary Material
Acknowledgments
The authors thank the Analytical Instrumentation Center at the UW School of Pharmacy for the access to the amaZon ETD ion trap, and Bruker Daltonics for graciously loaning the autoflex III MALDI TOF/TOF mass spectrometer. R.S. acknowledges the NIH-supported Clinical Neuroengineering Training Program Predoctoral Fellowship (NRSA T32 EB011434). G.K. was supported by an NHGRI training grant to the Genomic Sciences Training Program (5T32HG002760). L.S. acknowledges support from the Wisconsin Center of Excellence in Genomics Science through NIH/NHGRI grant 1P50HG004952. L.L. acknowledges an H.I. Romnes Faculty Research Fellowship. This work was supported by NSF CAREER grant CBET-0547484 (J.P.).
References
- 1.Prusiner SB. The Prion Diseases. Brain Pathol. 1998;8(3):499–513. doi: 10.1111/j.1750-3639.1998.tb00171.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baeten LA, Powers BE, Jewell JE, Spraker TR, Miller MW. A Natural Case of Chronic Wasting Disease in a Free-ranging Moose (Alces alces shirasi) J Wildl Dis. 2007;43(2):309. doi: 10.7589/0090-3558-43.2.309. [DOI] [PubMed] [Google Scholar]
- 3.Joly DO, Ribic CA, Langenberg JA, Beheler K, Batha CA, Dhuey BJ, Rolley RE, Bartelt G, Van Deelen TR, Samuel MD. Chronic Wasting Disease in Free-ranging Wisconsin White-tailed Deer. Emerging Infect Dis. 2003;9(5):599–601. doi: 10.3201/eid0905.020721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mathiason CK, Hays SA, Powers J, Hayes-Klug J, Langenberg J, Dahmes SJ, Osborn DA, Miller KV, Warren RJ, Mason GL, Hoover EA. Infectious Prions in Pre-Clinical Deer and Transmission of Chronic Wasting Disease Solely by Environmental Exposure. PloS ONE. 2009;4(6):e5916. doi: 10.1371/journal.pone.0005916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Williams ES. Chronic Wasting Disease. Vet Pathol. 2005;42(5):530–549. doi: 10.1354/vp.42-5-530. [DOI] [PubMed] [Google Scholar]
- 6.Williams ES, Young S. Chronic Wasting Disease of Captive Mule Deer: A Spongiform Encephalopathy. J Wildl Dis. 1980;16(1):89–98. doi: 10.7589/0090-3558-16.1.89. [DOI] [PubMed] [Google Scholar]
- 7.Miller M, Wild M, Williams E. Epidemiology of Chronic Wasting Disease in Captive Rocky Mountain Elk. J Wildl Dis. 1998;34(3):532. doi: 10.7589/0090-3558-34.3.532. [DOI] [PubMed] [Google Scholar]
- 8.Miller M, Williams E, McCarty C, Spraker T, Kreeger T, Larsen C, Thorne E. Epizootiology of Chronic Wasting Disease in Free-ranging Cervids in Colorado and Wyoming. J Wildl Dis. 2000;36(4):676–690. doi: 10.7589/0090-3558-36.4.676. [DOI] [PubMed] [Google Scholar]
- 9.Miller MW, Williams ES. Prion disease:Horizontal Prion Transmission in Mule Deer. Nature. 2003;425(6953):35–36. doi: 10.1038/425035a. [DOI] [PubMed] [Google Scholar]
- 10.Williams E, Miller M. Chronic Wasting Disease in Deer and Elk in North America. Rev Sci Tech. 2002;21(2):305–316. doi: 10.20506/rst.21.2.1340. [DOI] [PubMed] [Google Scholar]
- 11.Georgsson G, Sigurdarson S, Brown P. Infectious Agent of Sheep Scrapie May Persist in the Environment for at Least 16 Years. J Gen Virol. 2006;87:3737–3740. doi: 10.1099/vir.0.82011-0. [DOI] [PubMed] [Google Scholar]
- 12.Johnson CJ, Phillips KE, Schramm PT, McKenzie D, Aiken JM, Pedersen JA. Prions Adhere to Soil Minerals and Remain Infectious. PloS Pathog. 2006;2(4):296–302. doi: 10.1371/journal.ppat.0020032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miller MW, Williams ES, Hobbs NT, Wolfe LL. Environmental Sources of Prion Transmission in Mule Deer. Emerging Infect Dis. 2004;10(6):1003–1006. doi: 10.3201/eid1006.040010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pálsson P. Rida (scrapie) in Iceland and its Epidemiology: Slow transmissible diseases of the nervous system. In: Prusiner SB, Hadlow WJ, editors. Slow Transmissible Diseases of the Nervous System. Academic Press; New York: 1979. pp. 357–366. [Google Scholar]
- 15.Tamgüney G, Miller MW, Wolfe LL, Sirochman TM, Glidden DV, Palmer C, Lemus A, DeArmond SJ, Prusiner SB. Asymptomatic Deer Excrete Infectious Prions in Faeces. Nature. 2009;461(7263):529–532. doi: 10.1038/nature08289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Michigan Surveillance and Response Plan for Chronic Wasting Disease of Free-Ranging and Privately-Ownned/Captive Cervids. 2002:1–19. [Google Scholar]
- 17.Wisconsin’s Chronic Wasting Disease Response Plan: 2010–2025. 2010. pp. 1–44. [Google Scholar]
- 18.Belay ED, Schonberger LB. The Public Health Impact of Prion Diseases. Annu Rev Public Health. 2005;26:191–212. doi: 10.1146/annurev.publhealth.26.021304.144536. [DOI] [PubMed] [Google Scholar]
- 19.Joly DO, Samuel MD, Langenberg JA, Blanchong JA, Batha CA, Rolley RE, Keane DP, Ribic CA. Spatial Epidemiology of Chronic Wasting Disease in Wisconsin White-tailed Deer. J Wildl Dis. 2006;42(3):578–588. doi: 10.7589/0090-3558-42.3.578. [DOI] [PubMed] [Google Scholar]
- 20.Barria MA, Telling GC, Gambetti P, Mastrianni JA, Soto C. Generation of a New Form of Human PrPSc in vitro by Interspecies Transmission From Cervid Prions. J Biol Chem. 2011;286(9):7490–7495. doi: 10.1074/jbc.M110.198465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Prusiner SB. Prions. Proc Nat Acad Sci US A. 1998;95(23):13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baldwin MA, Stahl N, Reinders LG, Gibson BW, Prusiner SB, Burlingame AL. Permethylation and Tandem Mass Spectrometry of Oligosaccharides Having Free Hexosamine: Analysis of the Glycoinositol Phospholipid Anchor Glycan From the Scrapie Prion Protein. Anal Biochem. 1990;191(1):174–182. doi: 10.1016/0003-2697(90)90405-x. [DOI] [PubMed] [Google Scholar]
- 23.Stahl N, Baldwin M, Hecker R, Pan K, Burlingame A, Prusiner S. Glycosylinositol Phospholipid Anchors of the Scrapie and Cellular Prion Proteins Contain Sialic-Acid. Biochemistry. 1992;31(21):5043–5053. doi: 10.1021/bi00136a600. [DOI] [PubMed] [Google Scholar]
- 24.Stahl N, Baldwin M, Teplow D, Hood L, Gibson B, Burlingame A, Prusiner S. Structural Studies of the Scrapie Prion Protein Using Mass-Spectrometry and Amino-Acid Sequencing. Biochemistry. 1993;32(8):1991–2002. doi: 10.1021/bi00059a016. [DOI] [PubMed] [Google Scholar]
- 25.Stahl N, Baldwin MA, Burlingame AL, Prusiner SB. Identification of Glycoinositol Phospholipid Linked and Truncated Forms of the Scrapie Prion Protein. Biochemistry. 1990;29(38):8879–8884. doi: 10.1021/bi00490a001. [DOI] [PubMed] [Google Scholar]
- 26.Stahl N, Baldwin MA, Prusiner SB. Electrospray Mass Spectrometry of the Glycosylinositol Phospholipid of the Scrapie Prion Protein. Cell Biol Int Rep. 1991;15(9):853–862. doi: 10.1016/0309-1651(91)90037-j. [DOI] [PubMed] [Google Scholar]
- 27.Stahl N, Borchelt D, Hsiao K, Prusiner S. Scrapie Prion Protein Contains a Phosphatidylinositol Glycolipid. Cell. 1987;51(2):229–240. doi: 10.1016/0092-8674(87)90150-4. [DOI] [PubMed] [Google Scholar]
- 28.Caughey BW, Dong A, Bhat KS, Ernst D, Hayes SF, Caughey WS. Secondary Structure Analysis of the Scrapie-Associated Protein PrP 27–30 in Water by Infrared Spectroscopy. Biochemistry. 1991;30(31):7672–7680. doi: 10.1021/bi00245a003. [DOI] [PubMed] [Google Scholar]
- 29.Safar J, Roller PP, Gajdusek DC, Gibbs CJ. Conformational Transitions, Dissociation, and Unfolding of Scrapie Amyloid (Prion) Protein. J Biol Chem. 1993;268(27):20276–20284. [PubMed] [Google Scholar]
- 30.Prusiner SB, Bolton DC, Groth DF, Bowman KA, Cochran SP, McKinley MP. Further Purification and Characterization of Scrapie Prions. Biochemistry. 1982;21(26):6942–6950. doi: 10.1021/bi00269a050. [DOI] [PubMed] [Google Scholar]
- 31.Prusiner SB, McKinley MP, Bowman KA, Bolton DC, Bendheim PE, Groth DF, Glenner GG. Scrapie Prions Aggregate to Form Amyloid-like Birefringent Rods. Cell. 1983;35(2 Pt 1):349–358. doi: 10.1016/0092-8674(83)90168-x. [DOI] [PubMed] [Google Scholar]
- 32.Prusiner SB, Cochran SP, Groth DF, Downey DE, Bowman KA, Martinez HM. Measurement of the Scrapie Agent Using an Incubation Time Interval Assay. Ann Neurol. 1982;11(4):353–358. doi: 10.1002/ana.410110406. [DOI] [PubMed] [Google Scholar]
- 33.Biffiger K, Zwald D, Kaufmann L, Briner A, Nayki I, Pürro M, Bottcher S, Struckmeyer T, Schaller O, Meyer R, Fatzer R, Zurbriggen A, Stack M, Moser M, Oesch B, Kübler E. Validation of a Luminescence Immunoassay for the Detection of PrPSc in Brain Homogenate. J Virol Methods. 2002;101(1–2):79–84. doi: 10.1016/s0166-0934(01)00421-9. [DOI] [PubMed] [Google Scholar]
- 34.Deslys JP, Comoy E, Hawkins S, Simon S, Schimmel H, Wells G, Grassi J, Moynagh J. Screening Slaughtered Cattle for BSE. Nature. 2001;409(6819):476–478. doi: 10.1038/35054134. [DOI] [PubMed] [Google Scholar]
- 35.Grassi J, Comoy E, Simon S, Créminon C, Frobert Y, Trapmann S, Schimmel H, Hawkins SA, Moynagh J, Deslys JP, Wells GA. Rapid Test for the Preclinical Postmortem Diagnosis of BSE in Central Nervous System Tissue. Vet Rec. 2001;149(19):577–582. doi: 10.1136/vr.149.19.577. [DOI] [PubMed] [Google Scholar]
- 36.Lee DC, Stenland CJ, Hartwell RC, Ford EK, Cai K, Miller JL, Gilligan KJ, Rubenstein R, Fournel M, Petteway SR. Monitoring Plasma Processing Steps With a Sensitive Western Blot Assay for the Detection of the Prion Protein. J Virol Methods. 2000;84(1):77–89. doi: 10.1016/s0166-0934(99)00135-4. [DOI] [PubMed] [Google Scholar]
- 37.Safar J, Wille H, Itri V, Groth D, Serban H, Torchia M, Cohen FE, Prusiner SB. Eight Prion Strains Have PrPSc Molecules with Different Conformations. Nat Med. 1998;4(10):1157–1165. doi: 10.1038/2654. [DOI] [PubMed] [Google Scholar]
- 38.Wadsworth JD, Joiner S, Hill AF, Campbell TA, Desbruslais M, Luthert PJ, Collinge J. Tissue Distribution of Protease Resistant Prion Protein in Variant Creutzfeldt-Jakob Disease Using a Highly Sensitive Immunoblotting Assay. Lancet. 2001;358(9277):171–180. doi: 10.1016/s0140-6736(01)05403-4. [DOI] [PubMed] [Google Scholar]
- 39.Zanusso G, Righetti PG, Ferrari S, Terrin L, Farinazzo A, Cardone F, Pocchiari M, Rizzuto N, Monaco S. Two-Dimensional Mapping of Three Phenotype-Associated Isoforms of the Prion Protein in Sporadic Creutzfeldt-Jakob Disease. Electrophoresis. 2002;23(2):347–355. doi: 10.1002/1522-2683(200202)23:2<347::AID-ELPS347>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 40.Safar JG, Scott M, Monaghan J, Deering C, Didorenko S, Vergara J, Ball H, Legname G, Leclerc E, Solforosi L, Serban H, Groth D, Burton DR, Prusiner SB, Williamson RA. Measuring Prions Causing Bovine Spongiform Encephalopathy or Chronic Wasting Disease by Immunoassays and Transgenic Mice. Nature. 2002;20(11):1147–1150. doi: 10.1038/nbt748. [DOI] [PubMed] [Google Scholar]
- 41.Castilla J, Saa P, Soto C. Detection of Prions in Blood. Nat Med. 2005;11(9):982–985. doi: 10.1038/nm1286. [DOI] [PubMed] [Google Scholar]
- 42.Russo F, Johnson CJ, Johnson CJ, McKenzie D, Aiken JM, Pedersen JA. Pathogenic Prion Protein is Degraded by a Manganese Oxide Mineral Found in Soils. J Gen Virol. 2009;90:275–280. doi: 10.1099/vir.0.003251-0. [DOI] [PubMed] [Google Scholar]
- 43.Saborio GP, Permanne B, Soto C. Sensitive Detection of Pathological Prion Protein by Cyclic Amplification of Protein Misfolding. Nature. 2001;411(6839):810–813. doi: 10.1038/35081095. [DOI] [PubMed] [Google Scholar]
- 44.Cosseddu GM, Nonno R, Vaccari G, Bucalossi C, Fernandez-Borges N, Di Bari MA, Castilla J, Agrimi U. Ultra-Efficient PrPSc Amplification Highlights Potentialities and Pitfalls of PMCA Technology. PloS Pathog. 2011;7(11):e1002370. doi: 10.1371/journal.ppat.1002370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gonzalez-Montalban N, Makarava N, Ostapchenko VG, Savtchenk R, Alexeeva I, Rohwer RG, Baskakov IV. Highly Efficient Protein Misfolding Cyclic Amplification. PloS Pathog. 2011;7(2):e1001277. doi: 10.1371/journal.ppat.1001277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Johnson CJ, Aiken JM, McKenzie D, Samuel MD, Pedersen JA. Highly Efficient Amplification of Chronic Wasting Disease Agent by protein Misfolding Cyclic Amplificatoin with Beats (PMCAb) PLoS ONE. doi: 10.1371/journal.pone.0035383. (in review) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bian J, Napier D, Khaychuck V, Angers R. Cell-Based Quantification of Chronic Wasting Disease Prions. J Virol. 2010 doi: 10.1128/JVI.00633-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Neale M, Mountjoy S, Edwards J. Infection of Cell Lines with Experimental and Natural Ovine Scrapie Agents. J Virol. 2010 doi: 10.1128/JVI.01855-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gerber SA, Rush J, Stemman O, Kirschner MW, Gygi SP. Absolute Quantification of Proteins and Phosphoproteins from Cell Lysates by Tandem MS. Proc Nat Acad Sci US A. 2003;100(12):6940–6945. doi: 10.1073/pnas.0832254100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kirkpatrick DS, Gerber SA, Gygi SP. The Absolute Quantification Strategy: a General Procedure for the Quantification of Proteins and Post-translational Modifications. Methods. 2005;35(3):265–273. doi: 10.1016/j.ymeth.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 51.Onisko B, Dynin I, Requena JR, Silva CJ, Erickson M, Carter JM. Mass Spectrometric Detection of Attomole Amounts of the Prion Protein by nanoLC/MS/MS. J Am Soc Mass Spectrom. 2007;18(6):1070–1079. doi: 10.1016/j.jasms.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 52.Onisko BC, Silva CJ, Dynin I, Erickson M, Vensel WH, Hnasko R, Requena JR, Carter JM. Sensitive, Preclinical Detection of Prions in Brain by Nanospray Liquid Chromatography/Tandem Mass Spectrometry. Rapid Commun Mass Spectrom. 2007;21(24):4023–4026. doi: 10.1002/rcm.3310. [DOI] [PubMed] [Google Scholar]
- 53.Wopfner F, Weidenhöfer G, Schneider R, von Brunn A, Gilch S, Schwarz TF, Werner T, Schätzl HM. Analysis of 27 Mammalian and 9 Avian PrPs Reveals High Conservation of Flexible Regions of the Prion Protein. J Mol Biol. 1999;289(5):1163–1178. doi: 10.1006/jmbi.1999.2831. [DOI] [PubMed] [Google Scholar]
- 54.Silva CJ, Onisko BC, Dynin I, Erickson ML, Requena JR, Carter JM. Utility of Mass Spectrometry in the Diagnosis of Prion Diseases. Anal Chem. 2011;83(5):1609–1615. doi: 10.1021/ac102527w. [DOI] [PubMed] [Google Scholar]
- 55.Kettenbach AN, Rush J, Gerber SA. Absolute Quantification of Protein and Post-translational Modification Abundance with Stable Isotope-labeled Synthetic Peptides. Nat Protoc. 2011;6(2):175–186. doi: 10.1038/nprot.2010.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Clamp M, Cuff J, Searle SM, Barton GJ. The Jalview Java Alignment Editor. Bioinformatics. 2004;20(3):426–427. doi: 10.1093/bioinformatics/btg430. [DOI] [PubMed] [Google Scholar]
- 57.Waterhouse A, Procter J, Martin D. Jalview Version 2—A Multiple Sequence Alignment Editor and Analysis Workbench. Bioinformatics. 2009;25(9):1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: Improving the Sensitivity of Progressive Multiple Sequence Alignment Through Sequence Weighting, Position-specific Gap Penalties and Weight Matrix Choice. Nucleic Acids Res. 1994;22(22):4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, MacCoss MJ. Skyline: An Open Source Document Editor for Creating and Analyzing Targeted Proteomics Experiments. Bioinformatics. 2010;26(7):966–968. doi: 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Silva CJ, Onisko BC, Dynin I, Erickson ML, Vensel WH, Requena JR, Antaki EM, Carter JM. Assessing the Role of Oxidized Methionine at Position 213 in the Formation of Prions in Hamsters. Biochemistry. 2010;49(9):1854–1861. doi: 10.1021/bi901850n. [DOI] [PubMed] [Google Scholar]
- 61.Brun V, Masselon C, Garin J, Dupuis A. Isotope Dilution Strategies for Absolute Quantitative Proteomics. J Proteomics. 2009;72(5):740–749. doi: 10.1016/j.jprot.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 62.Lange V, Picotti P, Domon B, Aebersold R. Selected Reaction Monitoring for Quantitative Proteomics: A Tutorial. Molecular Systems Biology. 2008;4:222. doi: 10.1038/msb.2008.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pan S, Aebersold R, Chen R, Rush J, Goodlett DR, McIntosh MW, Zhang J, Brentnall TA. Mass Spectrometry Based Targeted Protein Quantification: Methods and Applications. J Proteome Res. 2009;8(2):787–797. doi: 10.1021/pr800538n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Canello T, Engelstein R, Moshel O, Xanthopoulos K, Juanes ME, Langeveld J, Sklaviadis T, Gasset M, Gabizon R. Methionine Sulfoxides on PrPSc: A Prion-Specific Covalent Signature. Biochemistry. 2008;47(34):8866–8873. doi: 10.1021/bi800801f. [DOI] [PubMed] [Google Scholar]
- 65.Arnold U, Rücknagel KP, Schierhorn A, Ulbrich-Hofmann R. Thermal Unfolding and Proteolytic Susceptibility of Ribonuclease A. Eur J Biochem. 1996;237(3):862–869. doi: 10.1111/j.1432-1033.1996.0862p.x. [DOI] [PubMed] [Google Scholar]
- 66.Prusiner SB, Safar J, DeArmond SJ. Bioassays of Prions. In: Prusiner SB, editor. Prion Biology and Diseases. Cold Spring Harbor Laboratory Press; 2003. pp. 143–186. [Google Scholar]
- 67.Heaton MP, Leymaster KA, Freking BA, Hawk DA, Smith TP, Keele JW, Snelling WM, Fox JM, Chitko-McKown CG, Laegreid WW. Prion Gene Sequence Variation Within Diverse Groups of U.S. Sheep, Beef Cattle, and Deer. Mamm Genome. 2003;14(11):765–777. doi: 10.1007/s00335-003-2283-y. [DOI] [PubMed] [Google Scholar]
- 68.Richt JA, Hall SM. BSE Case Associated With Prion Protein Gene Mutation. Plos Pathog. 2008;4(9):e1000156. doi: 10.1371/journal.ppat.1000156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Prusiner SB, Scott MR. Genetics of Prions. Annu Rev Genet. 1997;31:139–175. doi: 10.1146/annurev.genet.31.1.139. [DOI] [PubMed] [Google Scholar]
- 70.Agrimi U, Nonno R, Dell’Omo G, Di Bari MA, Conte M, Chiappini B, Esposito E, Di Guardo G, Windl O, Vaccari G, Lipp HP. Prion Protein Amino Acid Determinants of Differential Susceptibility and Molecular Feature of Prion Strains in Mice and Voles. PloS Pathog. 2008;4(7):e1000113. doi: 10.1371/journal.ppat.1000113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tabb D, Huang Y, Wysocki V, Yates J. Influence of Basic Residue Content On Fragment Ion Peak Intensities in Low-Energy -Collision-Induced Dissociation Spectra of Peptides. Anal Chem. 2004;76(5):1243–1248. doi: 10.1021/ac0351163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Blow DM. The Structure of Chymotrypsin. In: Boyer PD, editor. The Enzymes. Academic Press; New York: 1971. pp. 185–212. [Google Scholar]
- 73.Hess GP. Chymotrypsin-Chemical Properties and Catalysis. In: Boyer PD, editor. The Enzymes. Academic Press; New York: 1971. pp. 213–248. [Google Scholar]
- 74.Arsene CG, Ohlendorf R, Burkitt W, Pritchard C, Henrion A, O’Connor G, Bunk DM, Güttler B. Protein Quantification by Isotope Dilution Mass Spectrometry of Proteolytic Fragments: Cleavage Rate and Accuracy. Anal Chem. 2008;80(11):4154–4160. doi: 10.1021/ac7024738. [DOI] [PubMed] [Google Scholar]
- 75.USFDA. Guidance for Industry: Bioanalytical Method Validation. 2001. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.