

Abstract
We have identified naturally occurring 2-benzylidenebenzofuran-3-ones (aurones) as new templates for non-nucleoside hepatitis C virus (HCV) RNA-dependent RNA polymerase (RdRp) inhibitors. The aurone target site, identified by site-directed mutagenesis, is located in Thumb Pocket I of HCV RdRp. The RdRp inhibitory activity of 42 aurones was rationally explored in an enzyme assay. Molecular docking studies were used to determine how aurones bind to HCV RdRp and to predict their range of inhibitory activity. Seven aurone derivatives were found to have potent inhibitory effects on HCV RdRp, with IC50s below 5 μM and excellent selectivity. The most active aurone analogue was (Z)-2-((1-butyl-1H-indol-3-yl)methylene)-4,6-dihydroxybenzofuran-3(2H)-one (compound 51), with an IC50 of 2.2 μM. Their potent RdRp inhibitory activity, together with their low toxicity, make these molecules attractive candidate direct-acting anti-HCV agents.
Keywords: Antiviral Agents; chemical synthesis; pharmacology; Benzofurans; chemical synthesis; pharmacology; Hepacivirus; enzymology; Models, Molecular; RNA Replicase; antagonists & inhibitors; metabolism
Introduction
Hepatitis C virus (HCV) infection is a global public health problem. The World Health Organisation (WHO) estimates that 120–140 million people are infected worldwide (2–3% of the world population), while 3–4 million are newly infected each year. Chronic HCV infection is associated with chronic liver inflammation and fibrosis. In the absence of antiviral treatment, HCV liver disease progresses to cirrhosis in 20–30% of chronically infected patients, and patients with cirrhosis have a 1% to 4% annual risk of developing hepatocellular carcinoma.1 HCV infection is curable. The current standard of care is a combination of pegylated interferon alpha (peg-IFN) and ribavirin (RBV).2–5 Unfortunately, the response rate is low, especially among patients infected by HCV genotype 1, the most frequent genotype in the U.S., Europe, Asia, and Latin America (60 to 75% of cases).6 In 2012, a new treatment based on a combination of peg-IFN, ribavirin and a direct-acting antiviral drug that inhibits HCV serine protease activity (telaprevir or boceprevir) will be available for patients with HCV genotype 1 infection. Yet these new therapies will fail in 20–30% of treatment-naïve patients and in about 50% of patients in whom a first course of peg-IFN and ribavirin has failed to eradicate the infection. Thus, as a preventive vaccine will not be available for several years, there is an urgent need for more effective anti-HCV drugs.
The HCV RNA-dependent RNA polymerase (RdRp) is a particularly attractive target because of its key role in viral replication and the fact that this enzyme has no functional equivalent in mammalian cells, thus ensuring its selectivity. A number of HCV RdRp inhibitors have been identified, including nucleoside/nucleotide analogues that target the active site, and non-nucleoside allosteric inhibitors.7,8 The HCV RdRp structure resembles a right hand. Four allosteric binding sites have been identified so far, including “Thumb” Pockets I and II and “Palm” Pockets I and II.8,9 To date, two classes of molecules sharing a number of similarities have been reported to bind to Thumb Pocket I, including 2-aryl-1-cyclohexylbenzimidazole-5-carboxylic acid derivatives and their indole-based equivalents (Figure 1).10–16 Some of these compounds are currently being tested in HCV-infected patients. For example, a phase I clinical trial of MK-3281, an indole-based Thumb Pocket I inhibitor, has recently been completed,17 but this drug will not be further developed because of safety concerns.
Figure 1.

Two examples of known Thumb Pocket I inhibitors (top); general structure of the aurone backbone (bottom left); and structure of naturally occurring aureusidin (compound 1, bottom right).
Aurones (2-benzylidenebenzofuran-3-ones) are naturally occurring small molecules belonging to the flavonoid family, the pharmacological potential of which we reported for the first time several years ago.18 As part of an ongoing program investigating the therapeutic potential of aurones, we discovered that aureusidin (3′,4,4′,6-tetrahydroxyaurone, 1, Figure 1),19,20 a naturally occurring aurone, potently inhibits HCV RdRp, with an IC50 of 5.2 μM and a selectivity index of 90. Mutagenic studies and molecular modeling identified Thumb Pocket I as the aurone binding site. Based on these preliminary data, we used the aurone scaffold as a template for the design of aurone analogues with potent inhibitory activity on HCV RdRp.
Chemistry
The aurone derivatives described in this study were synthesized by means of aldol condensation of a substituted benzofuran-3(2H)-one with a benzaldehyde derivative, in basic KOH/MeOH conditions under reflux. Aurones bearing fluoro groups were obtained with a softer method using neutral alumina, as described by Varma et al., in order to avoid nucleophilic substitutions by in situ-formed potassium methoxide.21 4,6-dihydroxyaurones were synthesized by preparing the corresponding 4,6-dimethoxy analogues, followed by deprotection with boron tribromide. In some cases it was possible to obtain these derivatives directly by simple basic condensation in KOH/EtOH (Scheme 1).
Scheme 1.

Synthesis of aurone derivatives, starting from building blocks 2 – 9 a
aReagents and conditions: (a) for Procedure A: KOH/MeOH, 60°C, 1 – 18 h; for Procedure B: KOH/EtOH, 80°C, 2 – 5 h; for Procedure C: Al2O3, CH2Cl2, rt, 16 h. (b) BBr3, CH2Cl2, 0°C to rt, 24 – 72 h.
These pathways required the synthesis of benzofuran-3(2H)-ones 2 to 9 by various methods, as described elsewhere (Scheme 2). Compounds 2 and 3 were synthesized from resorcinol and phloroglucinol, respectively. Using chloroacetonitrile in HCl/Et2O, electrophilic addition in ortho position from one of the hydroxyl groups allowed the recovery of the chloroacetophenone. Cyclization then readily occurred in NaOMe/MeOH.22 Further reaction of 3 with methyl sulphate (Me2SO4) or benzylbromide (BnBr) in the presence of K2CO3 yielded compounds 6 and 7, respectively.23 Compounds 4 and 5 were obtained from the corresponding diacetoxyacetophenones after bromination of the α-position of the ketone with triphenylmethylammonium tribromide, and then one-pot deprotection and cyclization in KOH/MeOH.24 The same approach was used for compound 9, but with direct bromination of the 2-fluoro-6-hydroxyacetophenone with CuBr2, then cyclization in a K2CO3/DMF medium.25 Compound 8 was obtained from 3,5-difluorophenol, that was alkylated with chloroacetic acid. Subsequent chloration of the acid, followed by Friedel-Crafts cyclization, yielded the desired compound.26
Scheme 2.
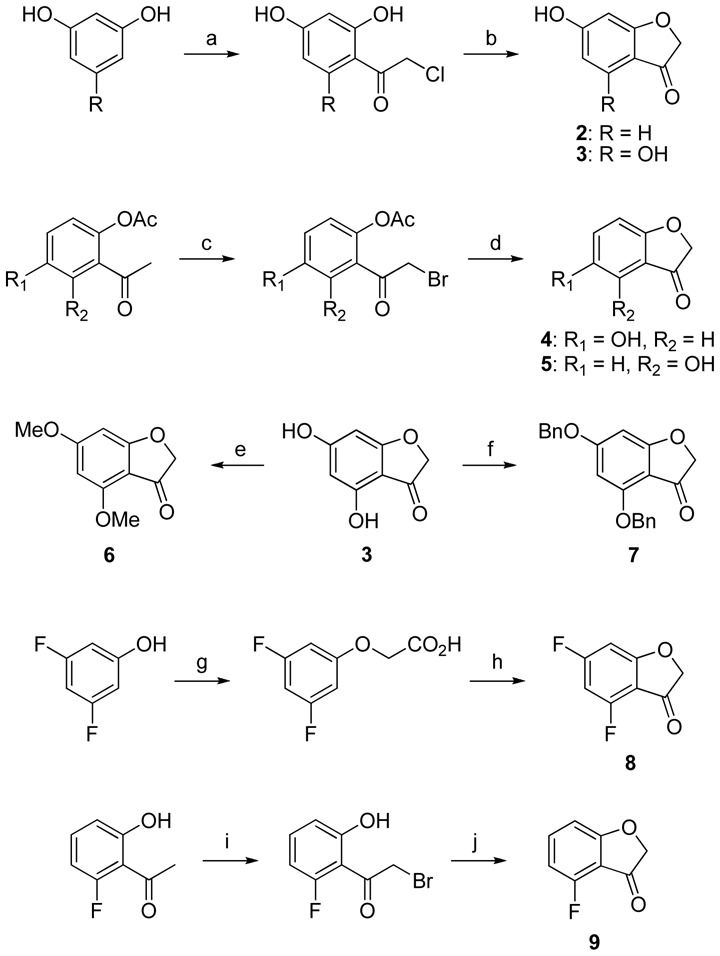
Synthesis of benzofuran-3(2H)-one derivatives 2 – 9 as the key starting blocksa
aReagents and conditions: (a) ClCH2CN, HCl, ZnCl2, Et2O, then HCl, H2O, 100°C, 1 h; (b) MeONa, MeOH, 60°C, 1.5 h; (c) trimethylphenylammonium tribromide, THF, rt, 6 h; (d) KOH, MeOH, 60°C, 3 h; (e) Me2SO4, K2CO3, dimethoxyethane, 80°C, 3 h; (f) BnBr, K2CO3, dimethoxyethane, rt, 3 days; (g) chloroacetic acid, NaOH, H2O, 90°C, 18 h; (h) SOCl2, PhMe, 110°C, 2 h, then AlCl3, CH2Cl2, 40°C, 18 h; (i) CuBr2, EtOAc, CHCl3, 60°C, 18 h; (j) K2CO3, DMF, 0°C, 1.5 h.
Benzaldehyde derivatives were purchased from commercial sources, or synthesized using published approaches. 4-Cyclohexylbenzaldehyde was synthesized from cyclohexylbenzene in a formylation reaction, using hexamethylenetetramine in TFA.27 1-Butyl-1H-indole-3-carbaldehyde was obtained from indole-3-carbaldehyde and butyl bromide, using NaH in DMF.
Results and Discussion
An enzyme assay was used to assess the inhibition of the polymerase activity of a purified RdRp (non-structural 5B [NS5B] protein), deleted of its 21 C-terminal amino acids in order to ensure solubility (HCV-NS5BΔ21). This assay measures the amount of double-stranded RNA synthesized in the presence of HCV-NS5BΔ21, a homopolymeric RNA template and ATP, as previously described.28 Initial screening was performed at an aurone concentration of 20 μM. The naturally occurring aureusidin (1, Figure 1) was found to be a potent inhibitor of HCV-NS5BΔ21 enzyme activity (85% inhibition at 20 μM, IC50 = 5.4±1.9 μM). This compound served as a “hit” for our subsequent investigations. As most naturally occurring aurones are hydroxylated at positions 4, 6, 2′, 3′ and 4′, we first investigated aurones bearing one, two, three or four hydroxyl groups at the above-mentioned positions (Table 1). The first structure-activity relationship study, undertaken to identify important substituents and their positions in the aurone core structure, showed that 4′ was the most important position for substitutions in the B-ring, whereas the most promising substituents in the A-ring were 4- and 6-hydroxyl groups (Table 1).
Table 1.
Initial array of NS5B inhibition by aureusidin analogues (compounds 10 – 20).
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|

|

|

|
|||||||
|
| |||||||||
| Cd | Inh. (20 μM, %) | IC50 (μM) | Cd | Inh. (20 μM, %) | IC50 (μM) | Cd | Inh. (20 μM, %) | IC50 (μM) | |

|
10 | 35.3 | n.d. | 14 | 10.3 | n.d. | 17 | 18.4 | n.d. |

|
11 | 60.3 | 5.9 | 15 | 37.7 | n.d. | 18 | 56.0 | 11.1 |

|
12 | 60.9 | 9.7 | 19 | 33.6 | n.d. | |||

|
13 | 37.6 | n.d. | 16 | 11.7 | n.d. | 20 | 0.7 | n.d. |
The next step was to identify the aurone binding site in HCV RdRp. Four sites, Thumb Pocket I and II and Palm Pockets I and II, have been identified as putative binding sites for non-nucleoside allosteric inhibitors of HCV RdRp (Figure 2). Amino acid substitutions at these sites have been shown to reduce susceptibility to the corresponding drugs. In order to determine if aurones target one of these sites, we examined whether these substitutions reduced HCV RdRp susceptibility to aurones in our cell-free enzyme assay. For this purpose we engineered HCV-NS5BΔ21 carrying the P495L, M423T, C316Y or H95Q substitutions, that are known to confer resistance to Thumb Pocket I, Thumb Pocket II, Palm Pocket I and Palm Pocket II inhibitors, respectively. The experiments were carried out with 4,4′-dihydroxyaurone (aurone 11). The IC50s of aurone 11 for each HCV-NS5BΔ21 mutant were compared with the IC50 for wild-type HCV-NS5BΔ21, and the results were recorded as fold differences (Figure 3). A benzimidazole inhibitor of HCV RdRp, known to target Thumb Pocket I, was used as a control. As expected, this latter compound was 10-fold less potent against HCV-NS5BΔ21-P495L than against HCV-NS5BΔ21-Wt, whereas its activity was not affected by the other amino acid substitutions. The inhibitory activity of aurone 11 was therefore only affected by the P495L substitution, indirectly suggesting that this compound binds to (or close to) Thumb Pocket I.
Figure 2.

Structure of HCV RdRp with its three subdomains (Fingers in red, Palm in blue and Thumb in green) and its four allosteric binding sites for known non-nucleoside inhibitors. Key substitution sites of adjacent amino acids are indicated in bold.
Figure 3.

Fold increase in IC50 relative to wild-type NS5BΔ21 obtained with aurone 11 and P495L, H95Q, C316Y and M423T mutants; these mutations are known to reduce susceptibility to Thumb Pocket I, Palm Pocket II, Palm Pocket I and Thumb Pocket II inhibitors, respectively.
We used the findings reported in Table 1 and mutagenesis experiments to examine the structure-activity relationship of aurones. Various substituents were used to determine the influence of physico-chemical parameters, including hydrogen bond donor potential, electronegativity, sterical effects and lipophilicity. Molecular docking was used throughout the decision process to identify the relevant substituents and their positions. The results are shown in Table 2. The presence of a hydroxyl group at position 4 appeared to be crucial. Replacement of the catechol moiety (B-ring) of 1 with a resorcinol in 34 did not affect inhibitory potency. Deletion of one of the B-ring hydroxyl groups of 34 yielded the less active derivative 12, while suppression of both B-ring hydroxyl groups led to even less active aurones. Table 2 also indicates that the presence of two hydroxyl groups, at positions 4 and 6, instead of only one at position 4, slightly increased the activity.
Table 2.
Inhibition activity of compounds 1 and 21 – 51.

| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compd | R4 | R5 | R6 | R2′ | R3′ | R4′ | R6′ | Inhibition (20 μM, %) | IC50 (μM) |
| 1 | OH | H | OH | H | OH | OH | H | 85.0 | 5.4 |
| 21 | OH | H | H | H | H | H | H | 57.8 | 14.6 |
| 22 | OH | H | H | OH | H | OH | H | 94.1 | 2.6 |
| 23 | OH | H | H | Me | H | Me | H | 64.2 | 5.8 |
| 24 | OH | H | H | OMe | H | OMe | H | 75.6 | 9.7 |
| 25 | OH | H | H | OMe | H | H | OMe | 47.2 | n.d. |
| 26 | OH | H | H | OMe | H | OMe | OMe | 51.8 | n.d. |
| 27 | OH | H | H | H | H | Et | H | 46.5 | n.d. |
| 28 | OH | H | H | H | H | i-Pr | H | 29.9 | n.d. |
| 29 | OH | H | H | H | H | n-Bu | H | 42.7 | n.d. |
| 30 | OH | H | H | H | H | t-Bu | H | 60.8 | n.d. |
| 31 | OH | H | H | H | H | N-Me-Ppz | H | 94.4 | 3.8 |
| 32 | OH | H | H | F | H | H | H | 58.5 | n.d. |
| 33 | OH | H | OH | H | H | H | H | 59.1 | 15.1 |
| 34 | OH | H | OH | OH | H | OH | H | 80.8 | 5.8 |
| 35 | OH | H | OH | OMe | OMe | H | H | 74.5 | n.d. |
| 36 | OH | H | OH | OMe | OMe | OMe | H | 50.2 | n.d. |
| 37 | OH | H | OH | H | H | Et | H | 79.7 | 3.6 |
| 38 | OH | H | OH | H | H | i-Pr | H | 78.3 | 3.1 |
| 39 | OH | H | OH | H | H | n-Bu | H | 93.4 | 2.5 |
| 40 | OH | H | OH | H | H | c-Hex | H | 91.8 | 2.3 |
| 41 | OH | H | OH | H | H | morpholinyl | H | 66.6 | 11.4 |
| 42 | OH | H | OH | H | H | F | H | 80.5 | 11.2 |
| 43 | OMe | H | OMe | Me | H | Me | H | 14.1 | n.d. |
| 44 | OMe | H | OMe | H | H | n-Bu | H | 3.3 | n.d. |
| 45 | OMe | H | OMe | H | H | c-Hex | H | 2.0 | n.d. |
| 46 | H | H | H | H | H | n-Bu | H | -33.8 | n.d. |
| 47 | F | H | F | H | H | n-Bu | H | -33.0 | n.d. |
| 48 | F | H | H | F | H | F | H | 10.6 | n.d. |
| 49 | H | OH | H | H | H | OH | H | 46.6 | n.d. |
| 50 | OBn | H | OBn | H | H | c-Hex | H | 3.6 | n.d. |
| 51 | 95.1 | 2.2 | |||||||
Based on the reported X-ray crystallographic structure of genotype 1b HCV RdRp complexed with a tetracyclic indole-based inhibitor binding to Thumb Pocket I (PDB ID: 2dxs),29 docking calculations were used to rationalize the observed activities. The best poses generated with Autodock software for compound 1 showed that Arginine 503 anchored the molecule, forming two hydrogen bonds with the 4-hydroxyl group and the ketone of the aurone. Glycine 493 allowed the formation of a third hydrogen bond with the 6-hydroxy group. These findings potentially explained the slightly better activities of 4,6-dihydroxyaurones compared with 4-hydroxyaurones. At the B-ring, the 3′- and 4′-hydroxyl groups of 1 allowed the formation of two additional chelating hydrogen bonds with the carbonyl group of Leucine 392 inside the hydrophobic pocket, as shown in Figure 4A. These findings explain the strong impact of hydroxy groups at these positions.
Figure 4.

Surface and ribbon views of predicted binding modes in RdRp Thumb Pocket I of a reported X-ray structure (PDB id: 2dxs) from docking studies with Autodock. (A) For compound 1, involved in five hydrogen bonds: two with Arginine 503 (1.9 Å and 2.5 Å), one with Glycine 493 (1.8 Å) and two with Leucine 392 (2.0 Å and 2.0 Å). (B) For compound 51, involved in three hydrogen bonds: two with Arginine 503 (1.8 Å and 2.0 Å) and one with Glycine 493 (2.2 Å). The pictures were built with Pymol software.
Replacement of the 4′-hydroxyl group by its isosteric, fluorine atom led to the slightly less active derivative 42, suggesting that hydrogen bonding plays a minor role in this region of the inhibitor. The introduction of methoxy groups instead of hydroxy groups at the B-ring led to compounds with similar (compounds 24, 35) or weaker (compounds 26, 36) activities, indicating that, in some cases, the insertion of more hydrophobic and bulky substituents is well tolerated. Conversely, methoxylation of the hydroxyl groups at the A-ring, or replacement of these groups by fluoro substituents, led to a marked fall in inhibitory activity (compounds 13, 20, 47, 48).
Given the strongly hydrophobic environment of Thumb Pocket I and the results of our initial docking studies, we tested the effect of adding hydrophobic substituents at the B-ring, especially at position 4′, on interactions with the pocket. The introduction of methyl and ethyl groups at the B-ring resulted in better inhibitory activity (compounds 23 and 37). Thus, alkyl substituents were grafted at position 4′. The introduction of a n-butyl or a cyclohexyl group provided the best inhibitory activity (compounds 39 and 40), indicating that both highly lipophilic and bulky substituents at the C-4′ position were associated with the strongest activities.
Overall, we identified the following pharmacophoric elements as crucial in the inhibitory activity of aurones on HCV RdRp: a) a hydroxyl group at position 4, or a dihydroxyl group at positions 4 and 6; and b) a hydrophobic and bulky substituent at position 4′ of the B-ring. We also found that replacement of the cyclohexyl by a 4-methylpiperazine in 31 was tolerated. Molecular docking of aurones 39 and 40, based on the known crystallographic structure of genotype 1b HCV RdRp complexed with a tetracyclic indole-based inhibitor binding to Thumb Pocket I (PDB ID: 2dxs),29 showed strong fitting of the 4′-n-butyl and 4′-cyclohexyl moieties into the pocket. Further virtual docking screening suggested that aurone analogues in which the B-ring is replaced by a substituted indole moiety are potential ligands.
In models built with GOLD and Autodock software, the best poses were obtained for N-alkylindole analogues (Z)-2-(N-alkylindolyl)methylene)-4,6-dihydroxybenzofuran-3(2H)-one). Arginine 503 was found to anchor the molecule in a similar way to compound 1, whereas hydrophobic residues (especially Leucine 492, Valine 37, Alanine 393, Leucine 392, Alanine 396, Alanine 395, Phenylalanine 429 and Isoleucine 424), forming two pockets, maintained both the indole part and the N-alkyl substituent of the ligand. This explains the positive effect of 4′-hydrophobic substituents. Compound 51, which belongs to this group of indole derivatives, had a highly discriminant Goldscore of 52.4 (36.9 to 43.6 for compounds 23, 24, 27, 28, 29, 39 and 40). The fixation mode of compound 51, which had the strongest inhibitory activity (IC50 = 2.2 μM), is shown in Figure 4B. These findings validated the docking results and confirmed the inhibitory potential of this group of indole derivatives.
Overall, the results obtained with weak inhibitors were, as expected, difficult to interpret, but the geometries of poses obtained with more efficient compounds were homogeneous and relevant. The strong activities of both 4′-hydroxyl and 4′-hydrophobic compounds could be explained by the good fit of these two subclasses of aurones in the pocket. Compounds 1 and 51 were representative of the hypothetic dual mode of aurone binding to Thumb Pocket I of HCV RdRp.
All the reported compounds were evaluated for cytotoxicity, based on the MTT assay with two human cell lines (HuH7 and HEK293) (data not shown). No cell death was observed at effective concentrations.
Conclusion
We investigated the capacity of aurones, non-toxic natural compounds, to inhibit HCV RdRp. Aureusidin potently inhibited the activity of this enzyme. By using classical medicinal chemistry, we identified positions and substituents that are crucial for the inhibitory activity of aurones on RdRp. A combination of site-directed mutagenesis and molecular modeling was successfully used to understand and optimize aurones’ anti-HCV activity. Our findings constitute a milestone in the development of new non-nucleoside inhibitors of HCV RdRp. Studies are underway to synthesize more active compounds warranting preclinical evaluation.
Experimental section
Chemistry
1H and 13C NMR spectra were recorded on a Brüker AC-400 instrument (400 MHz for 1H and 100 MHz for 13C). Chemical shifts (δ) are reported in ppm relative to Me4Si (internal standard). Electrospray ionization ESI mass spectra were acquired by the Analytical Department of Grenoble University on an Esquire 300 Plus Bruker Daltonis instrument with a nanospray inlet. Elemental analyses were performed by the Analytical Department of Grenoble University. Thin-layer chromatography (TLC) used Merck silica gel F-254 plates (thickness 0.25 mm). Flash chromatography used Merck silica gel 60, 200–400 mesh. All solvents were distilled prior to use. Unless otherwise stated, reagents were obtained from commercial sources and were used without further purification. Compounds 2 to 9 were prepared with published procedures.22–26
General procedure A for the preparation of (Z)-2-benzylidenebenzofuran-3(2H)-one derivatives
To a solution of a benzofuran-3(2H)-one derivative in methanol (15 mL/mmol) were added an aqueous solution of potassium hydroxide (50%, 1.5 mL/mmol) and a benzaldehyde derivative (1.5 equivalents). The solution was then refluxed until TLC showed complete disappearance of the starting materials (1 to 18 hours). After cooling, the mixture was concentrated under reduced pressure, then the residue was diluted in water (50 mL/mmol), and an aqueous solution of hydrochloric acid (1N) was added to adjust the pH to 2–3. The mixture was extracted with ethyl acetate or dichloromethane and washed with water and brine. The combined organic layers were dried over magnesium sulfate and filtered, and the filtrate was concentrated under reduced pressure to give the crude compound.
General procedure B for the preparation of (Z)-2-benzylidenebenzofuran-3(2H)-one derivatives
To a solution of a benzofuran-3(2H)-one derivative in ethanol (4 mL/mmol) were added an aqueous solution of potassium hydroxide (50%, 5 mL/mmol) and a benzaldehyde derivative (2 equivalents). The solution was then refluxed until TLC showed complete disappearance of the starting materials (2 to 5 hours). After cooling, the mixture was concentrated under reduced pressure, then the residue was diluted in water (50 mL/mmol), and an aqueous solution of hydrochloric acid (1N) was added to adjust the pH to 2–3. The mixture was extracted with ethyl acetate or dichloromethane and washed with water and brine. The combined organic layers were dried over magnesium sulfate and filtered, and the filtrate was concentrated under reduced pressure to give the crude compound.
General procedure C for the preparation of (Z)-2-benzylidenebenzofuran-3(2H)-one derivatives
To a solution of a benzofuran-3(2H)-one derivative in anhydrous dichloromethane (20 mL/mmol) were added a benzaldehyde derivative (1.5 equivalents) and aluminium oxide (4000 mg/mmol). The suspension was stirred under argon overnight, and the solid was removed by filtration. The filtrate was distilled under reduced pressure to give the crude compound.
General procedure D for the preparation of (Z)-2-benzylidenebenzofuran-3(2H)-one derivatives
To a solution of a (Z)-2-benzylidenebenzofuran-3(2H)-one derivative in anhydrous dichloromethane (10 mL/mmol) was added pure boron tribromide (20 equivalents) at 0°C. The solution was then stirred at room temperature until TLC showed complete disappearance of the starting materials (24 to 72 hours). Cold water was then added and the suspension was extracted three times with ethyl acetate, and washed with water and brine. The combined organic layers were dried over magnesium sulfate and filtered, and the filtrate was concentrated under reduced pressure to give the crude compound.
(Z)-2-(3,4-dihydroxybenzylidene)-4,6-dihydroxybenzofuran-3(2H)-one (1)
The crude product was prepared according to general procedure D starting from (Z)-2-(3,4-dimethoxy-benzylidene)-4,6-dimethoxybenzofuran-3(2H)-one (52), and was washed three times with distilled water to yield a yellow solid, which was analytically pure and used without further purification (61%). m.p. > 260°C (decomposition). 1H NMR (400 MHz, MeOD) δ 7.47 (d, 1H, J = 1.4 Hz, H2′), 7.18 (dd, 1H, J = 8.2 Hz, J = 1.4 Hz, H6′), 6.82 (d, 1H, J = 8.2 Hz, H5′), 6.56 (s, 1H, -CH=), 6.19 (s, 1H, H7), 6.01 (s, 1H, H5). 13C NMR (100 MHz, MeOD) δ 178.9, 167.4, 166.9, 158.1, 147.4, 145.8, 145.4, 123.9, 123.6, 117.5, 115.9, 109.5, 102.8, 97.5, 90.2. MS (ESI) m/z 287 (M+H)+, 309 (M+Na)+. Anal. Calcd for C15H10O6·1.3H2O: C, 57.57, H, 4.05. Found: C, 57.12, H, 3.81.
(Z)-2-(4-hydroxybenzylidene)-6-hydroxybenzofuran-3(2H)-one (10)
The crude product was prepared according to general procedure A starting from 6-hydroxybenzofuran-3(2H)-one (2) and 4-hydroxybenzaldehyde, and was recrystallized from acetonitrile to yield pure bright yellow crystals (77%). m.p. 294–296°C. 1H NMR (400 MHz, d6-DMSO) δ 11.14 (br s, 1H, OH), 10.14 (br s, 1H, OH), 7,82 (d, 2H, J = 8.7 Hz, H2′,6′), 7.60 (d, 1H, J = 8.4 Hz, H4), 6.88 (d, 2H, J = 8.7 Hz, H3′,5′), 6.78 (d, 1H, J = 1.9 Hz, H7), 6.73 (s, 1H, -CH=), 6.70 (dd, 1H, J1 = 8.4 Hz, J2 = 1.9 Hz, H5). 13C NMR (100 MHz, d6-DMSO) δ 181.2, 167.4, 166.0, 159.2, 145.6, 133.2, 125.6, 123.0, 116.0, 113.1, 112.8, 111.3, 98.4. MS (ESI) m/z 255 (M+H)+, 277 (M+Na)+. Anal. Calcd for C15H10O4·½H2O: C, 68.44, H, 4.18. Found: C, 68.28, H, 4.11.
(Z)-2-(4-hydroxybenzylidene)-4-hydroxybenzofuran-3(2H)-one (11)
The crude product was prepared according to general procedure A starting from 4-hydroxybenzofuran-3(2H)-one (5) and 4-hydroxybenzaldehyde, and was recrystallized from acetonitrile to yield pure orange crystals (37%). m.p. 245–247°C. 1H NMR (400 MHz, d6-DMSO) δ 11.05 (s, 1H, OH), 10.13 (s, 1H, OH), 7.81 (d, 2H, J = 8.4 Hz, H2′,6′), 7.51 (t, 1H, J = 8.1 Hz, H6), 6.88 (d, 2H, J = 8.4 Hz, H3′,5′), 6.84 (d, 1H, J = 8.1 Hz, H7), 6.70 (s, 1H, -CH=), 6.61 (d, 1H, J = 8.1 Hz, H5). 13C NMR (100 MHz, d6-DMSO) δ 181.1, 165.7, 159.2, 156.9, 144.9, 138.2, 133.2, 123.1, 116.0, 110.9, 110.3, 109.4, 102.4. MS (ESI) m/z 255 (M+H)+, 277 (M+Na)+. Anal. Calcd for C15H10O4·0.1H2O: C, 70.37, H, 3.99. Found: C, 70.10, H, 3.96.
(Z)-2-(4-hydroxybenzylidene)-4,6-dihydroxybenzofuran-3(2H)-one (12)
To a solution of 4-hydroxybenzaldehyde (147 mg, 1.21 mmol) in methanol (10 mL) was added concentrated sulfuric acid (64.2 μL, 1.21 mmol), and the solution was stirred at room temperature for 30 minutes. 4,6-Dihydroxybenzofuran-3(2H)-one (3, 200 mg, 1.21 mmol) was then added, and the solution was refluxed for 4 hours. After cooling, the mixture was concentrated under reduced pressure, then the residue was diluted in water (100 mL), extracted with ethyl acetate and washed with water and brine. The combined organic layers were dried over magnesium sulfate and filtered, and the filtrate was concentrated under reduced pressure to give the crude compound, which was purified by column chromatography on silica gel (eluent: ethyl acetate 3/cyclohexane 2) to yield a pure yellow solid (17%). m.p. > 295°C (decomposition). 1H NMR (400 MHz, d6-DMSO) δ 10.84 (br s, 1H, OH), 10.03 (br s, 1H, OH), 7.75 (d, 2H, J = 8.7 Hz, H2′,6′), 6.86 (d, 2H, J = 8.7 Hz, H3′,5′), 6.54 (s, 1H, -CH=), 6.20 (d, 1H, J = 1.6 Hz, H7), 6.06 (d, 1H, J = 1.6 Hz, H5). 13C NMR (100 MHz, d6-DMSO) δ 179.1, 167.6, 167.1, 158.8, 158.2, 146.0, 132.8, 123.4, 116.0, 109.1, 102.9, 97.7, 90.5. MS (ESI) m/z 271 (M+H)+, 293 (M+Na)+. Anal. Calcd for C15H10O5: C, 66.67, H, 3.73. Found: C, 66.43, H, 3.83.
(Z)-2-(4-hydroxybenzylidene)-4,6-dimethoxybenzofuran-3(2H)-one (13)
The crude product was prepared according to general procedure A starting from 4,6-dimethoxy-benzofuran-3(2H)-one (6) and 4-hydroxybenzaldehyde, and was recrystallized from methanol to yield pure yellow crystals (63%). m.p. > 275°C (decomposition). 1H NMR (400 MHz, d6-DMSO) δ 10.11 (s, 1H, OH), 7.79 (d, 2H, J = 8.6 Hz, H2′,6′), 6.87 (d, 2H, J = 8.6 Hz, H3′,5′), 6.68 (d, 1H, J = 1.1 Hz, H7), 6.65 (s, 1H, -CH=), 6.33 (d, 1H, J = 1.1 Hz, H5), 3.91 (s, 3H, OMe), 3.88 (s, 3H, OMe). 13C NMR (100 MHz, d6-DMSO) δ 178.9, 168.6, 167.9, 159.1, 158.8, 145.6, 133.0, 123.1, 116.0, 110.4, 104.3, 94.3, 89.7, 56.4, 56.1. MS (ESI) m/z 299 (M+H)+, 321 (M+Na)+. Anal. Calcd for C17H14O5: C, 68.46, H, 4.74. Found: C, 68.13, H, 4.78.
(Z)-2-(3-hydroxybenzylidene)-6-hydroxybenzofuran-3(2H)-one (14)
The crude product was prepared according to general procedure A starting from 6-hydroxybenzofuran-3(2H)-one (2) and 3-hydroxybenzaldehyde, and was recrystallized from acetonitrile to yield pure yellow crystals (86%). m.p. 272–274°C. 1H NMR (400 MHz, d6-DMSO) δ 11.27 (br s, 1H, OH), 9.69 (br s, 1H, OH), 7.63 (d, 1H, J = 8.4 Hz, H4), 7.36 (m, 2H, H2′,6′), 7.28 (t, 1H, J = 7.6 Hz, H5′), 6.84 (d, 1H, J = 7.6 Hz, H4′), 6.78 (s, 1H, H7), 6.72 (d, 1H, J = 8,4 Hz, H5), 6.69 (s, 1H, -CH=). 13C NMR (100 MHz, d6-DMSO) δ 181.5, 167.9, 166.6, 157.6, 147.3, 133.1, 129.9, 126.1, 122.4, 117.3, 117.1, 113.1, 112.8, 110.7, 98.5. MS (ESI) m/z 255 (M+H)+, 277 (M+Na)+. Anal. Calcd for C15H10O4·¼H2O: C, 69.63, H, 4.06. Found: C, 69.47, H, 3.94.
(Z)-2-(3-hydroxybenzylidene)-4-hydroxybenzofuran-3(2H)-one (15)
The crude product was prepared according to general procedure A starting from 4-hydroxybenzofuran-3(2H)-one (5) and 3-hydroxybenzaldehyde, and was recrystallized from acetonitrile to yield pure yellow crystals (55%). m.p. 207–209°C. 11.21 (br s, 1H, OH), 9.69 (br s, 1H, OH), 7.54 (t, 1H, J = 8.2 Hz, H6), 7.36 (m, 2H, H2′,6′), 7.28 (t, 1H, J = 7.8 Hz, H5′), 6.84 (m, 2H, H4′,7), 6.66 (s, 1H, -CH=), 6.64 (d, 1H, J = 8.2 Hz, H5). 13C NMR (100 MHz, d6-DMSO) δ 181.4, 166.0, 157.6, 157.2, 146.5, 138.7, 133.2, 129.9, 122.3, 117.2, 117.0, 110.7, 110.2, 109.0, 102.3. MS (ESI) m/z 255 (M+H)+, 277 (M+Na)+. Anal. Calcd for C15H10O4: C, 70.87, H, 3.97. Found: C, 70.84, H, 4.74.
(Z)-2-(3-hydroxybenzylidene)-4,6-dimethoxybenzofuran-3(2H)-one (16)
The crude product was prepared according to general procedure A starting from 4,6-dimethoxy-benzofuran-3(2H)-one (6) and 3-hydroxybenzaldehyde, and was recrystallized from methanol to yield pure yellow crystals (76%). m.p. > 250°C (decomposition). 1H NMR (400 MHz, d6-DMSO) δ 9.65 (s, 1H, OH), 7.34 (m, 2H, H2′,6′), 7.27 (t, 1H, J = 8.0 Hz, H5′), 6.83 (d, 1H, J = 8.0 Hz, H4′), 6.67 (d, 1H, J = 1.2 Hz, H7), 6.61 (s, 1H, -CH=), 6.35 (d, 1H, J = 1.2 Hz, H5), 3.92 (s, 3H, OMe), 3.89 (s, 3H, OMe). 13C NMR (100 MHz, d6-DMSO) δ 179.0, 169.0, 168.3, 159.0, 157.6, 147.2, 133.2, 129.9, 122.1, 117.3, 116.9, 109.8, 104.0, 94.4, 89.8, 56.5, 56.2. MS (ESI) m/z 299 (M+H)+, 321 (M+Na)+. Anal. Calcd for C17H14O5·H2O: C, 64.56, H, 5.06. Found: C, 64.31, H, 5.06.
(Z)-2-(2-hydroxybenzylidene)-6-hydroxybenzofuran-3(2H)-one (17)
The crude product was prepared according to general procedure A starting from 6-hydroxybenzofuran-3(2H)-one (2) and 2-hydroxybenzaldehyde, and was recrystallized from acetonitrile to yield pure yellow crystals (25%). m.p. > 250°C (decomposition). 1H NMR (400 MHz, d6-DMSO) δ 11.21 (br s, 1H, OH), 10.35 (br s, 1H, OH), 8.09 (d, 1H, J = 7.0 Hz, H6′), 7.62 (d, 1H, J = 8.4 Hz, H4), 7.26 (t, 1H, J = 7.0 Hz, H4′), 7.09 (s, 1H, -CH=), 6.94 (m, 2H, H3′,5′), 6.79 (d, 1H, J = 1.5 Hz, H7), 6.72 (dd, 1H, J = 8.4 Hz, J = 1.5 Hz, H5). 13C NMR (100 MHz, d6-DMSO) δ 181.4, 167.7, 166.3, 157.0, 146.8, 131.3, 130.9, 125.8, 119.6, 118.8, 115.6, 112.9, 104.6, 98.5. MS (ESI) m/z 255 (M+H)+, 277 (M+Na)+. Anal. Calcd for C15H10O4·¼ H2O: C, 69.63, H, 4.06. Found: C, 69.81, H, 4.05.
(Z)-2-(2-hydroxybenzylidene)-4-hydroxybenzofuran-3(2H)-one (18)
The crude product was prepared according to general procedure A starting from 4-hydroxybenzofuran-3(2H)-one (5) and 2-hydroxybenzaldehyde, and was recrystallized from acetonitrile to yield pure yellow crystals (28%). m.p. > 250°C. 1H NMR (400 MHz, d6-DMSO) δ 11.10 (br s, 1H, OH), 10.35 (s, 1H, OH), 8.08 (d, 1H, J = 7.2 Hz, H6′), 7.52 (t, 1H, J = 7.6 Hz, H6), 7.24 (t, 1H, J = 7.2 Hz, H4′), 7.06 (s, 1H, -CH=), 6.94 (m, 2H, H3′,5′), 6.86 (d, 1H, J = 7.6 Hz, H7), 6.62 (d, 1H, J = 7.6 Hz, H5). 13C NMR (100 MHz, d6-DMSO) δ 181.2, 165.8, 157.0, 157.0, 146.0, 138.4, 131.2, 130.8, 119.6, 118.9, 115.6, 110.4, 109.1, 104.1, 102.4. MS (ESI) m/z 255 (M+H)+, 277 (M+Na)+. Anal. Calcd for C15H10O4·¼H2O: C, 69.63, H, 4.06. Found: C, 69.19, H, 3.89.
(Z)-2-(2-hydroxybenzylidene)-4,6-dihydroxybenzofuran-3(2H)-one (19)
The crude product was prepared according to general procedure D starting from (Z)-2-(2-hydroxybenzylidene)-4,6-dimethoxybenzofuran-3(2H)-one (16), and was purified by column chromatography on silica gel (eluent: ethyl acetate 1/cyclohexane 1) to yield a pure yellow solid (66%). m.p. 262–264°C. 1H NMR (400 MHz, d6-acetone) δ 8.12 (d, 1H, J = 7.2 Hz, H6′), 7.17 (m, 1H, H4′), 7.12 (s, 1H, -CH=), 6.90 (m, 2H, H3′, H5′), 6.29 (d, 1H, J = 2.0 Hz, H7), 6.08 (d, 1H, J = 2.0 Hz, H5). 13C NMR (100 MHz, d6-acetone) δ 180.9, 167.6, 167.5, 157.9, 156.7, 147.5, 131.2, 130.9, 120.0, 119.6, 115.5, 103.6, 103.4, 97.7, 91.2. MS (ESI) m/z 271 (M+H)+. Anal. Calcd for C15H10O5·⅔H2O: C, 65.22, H, 3.86. Found: C, 65.37, H, 4.61.
(Z)-2-(2-hydroxybenzylidene)-4,6-dimethoxybenzofuran-3(2H)-one (20)
The crude product was prepared according to general procedure A starting from 4,6-dimethoxy-benzofuran-3(2H)-one (6) and 2-hydroxybenzaldehyde, and was recrystallized from methanol to yield pure yellow crystals (51%). m.p. > 250°C (decomposition). 1H NMR (400 MHz, d6-DMSO) δ 10.31 (s, 1H, OH), 8.06 (d, 1H, J = 7.8 Hz, H6′), 7.24 (t, 1H, J = 7.8 Hz, H4′), 7.01 (s, 1H, -CH=), 6.94 (d, 1H, J = 7.8 Hz, H3′), 6.92 (t, 1H, J = 7.8 Hz, H5′), 6.71 (d, 1H, J = 1.4 Hz, H7), 6.34 (d, 1H, J = 1.4 Hz, H5), 3.91 (s, 3H, OMe), 3.89 (s, 3H, OMe). 13C NMR (100 MHz, d6-DMSO) δ 179.0, 168.7, 168.0, 158.8, 156.9, 146.7, 131.1, 130.6, 119.4, 118.8, 115.6, 104.0, 103.6, 94.3, 89.8, 56.4, 56.0. MS (ESI) m/z 299 (M+H)+, 321 (M+Na)+. Anal. Calcd for C17H14O4: C, 68.46, H, 4.74. Found: C, 68.39, H, 4.77.
(Z)-2-benzylidene-4-hydroxybenzofuran-3(2H)-one (21)
The crude product was prepared according to general procedure A starting from 4-hydroxybenzofuran-3(2H)-one (5) and benzaldehyde, and was purified by column chromatography on silica gel (eluent: dichloromethane) to yield a pure yellow solid (72%). m.p. 148°C. 1H NMR (400 MHz, d6-DMSO) δ 11.19 (s, 1H, OH), 7.95 (d, 2H, J = 7.2 Hz, H2′,6′), 7.54 (t, 1H, J = 8.1 Hz, H6), 7.46 (m, 3H, H3′,4′,5′), 6.87 (d, 1H, J = 8.1 Hz, H7), 6.77 (s, 1H, -CH=), 6.64 (d, 1H, J = 8.1 Hz, H5). 13C NMR (100 MHz, d6-DMSO) δ 181.4, 166.1, 157.2, 146.6, 138.8, 132.2, 131.0, 129.6, 129.0, 110.7, 109.9, 109.0, 102.5. MS (ESI) m/z 239 (M+H)+, 261 (M+Na)+. Anal. Calcd for C15H10O3·⅔H2O: C, 72.00, H, 4.53. Found: C, 71.99, H, 4.15.
(Z)-2-(2,4-dihydroxybenzylidene)-4-hydroxybenzofuran-3(2H)-one (22)
The crude product was prepared according to general procedure D starting from (Z)-2-(2,4-dimethoxybenzylidene)-4-hydroxybenzofuran-3(2H)-one (24), and was washed three times with distilled water to yield a yellow solid, which was analytically pure and used without further purification (61%). m.p. > 230°C (decomposition). 1H NMR (400 MHz, d6-DMSO) δ 10.94 (s, 1H, OH), 10.29 (s, 1H, OH), 10.02 (s, 1H, OH), 7.96 (d, 1H, J = 8.2 Hz, H6′), 7.48 (t, 1H, J = 8.0 Hz, H6), 7.03 (s, 1H, -CH=), 6.83 (d, 1H, J = 8.0 Hz, H7), 6.59 (d, 1H, J = 8.0 Hz, H5), 6.40 (m, 2H, H3′,5′). 13C NMR (400 MHz, d6-DMSO) δ 181.0, 165.6, 160.9, 159.1, 156.8, 144.3, 137.8, 132.4, 110.7, 110.1, 109.7, 108.3, 105.6, 102.5, 102.3. MS (ESI) m/z 271 (M+H)+, 293 (M+Na)+. Anal. Calcd for C15H10O5·H2O: C, 62.50, H, 4.17. Found: C, 62.64, H, 3.70.
(Z)-2-(2,4-dimethylbenzylidene)-4-hydroxybenzofuran-3(2H)-one (23)
The crude product was prepared according to general procedure A starting from 4-hydroxybenzofuran-3(2H)-one (5) and 2,4-dimethylbenzaldehyde, and was recrystallized from methanol to yield pure yellow crystals (76%). m.p. 193–194°C. 1H NMR (400 MHz, d6-DMSO) δ 11.14 (s, 1H, OH), 8.01 (d, 1H, J = 7.9 Hz, H6′), 7.52 (t, 1H, J = 8.1 Hz, H6), 7.12 (m, 2H, H3′,5′), 6.84 (d, 1H, J = 8.1 Hz, H7), 6.78 (s, 1H, -CH=), 6.63 (d, 1H, J = 8.1 Hz, H5), 2.40 (s, 3H, Me), 2.29 (s, 3H, Me). 13C NMR (100 MHz, d6-DMSO) δ 181.2, 166.0, 157.1, 146.2, 139.4, 138.5, 138.4, 131.3, 130.3, 127.6, 127.1, 110.6, 109.1, 106.5, 102.4, 21.0, 19.6. MS (ESI) m/z 267 (M+H)+, 289 (M+Na)+. Anal. Calcd for C17H14O3·⅓H2O: C, 75.00, H, 5.39. Found: C, 74.96, H, 5.35.
(Z)-2-(2,4-dimethoxybenzylidene)-4-hydroxybenzofuran-3(2H)-one (24)
The crude product was prepared according to general procedure A starting from 4-hydroxybenzofuran-3(2H)-one (5) and 2,4-dimethoxybenzaldehyde, and was recrystallized from methanol to yield pure yellow crystals (76%). m.p. 234–235°C. 1H NMR (400 MHz, d6-DMSO) δ 11.05 (s, 1H, OH), 8.11 (d, 1H, J = 8.6 Hz, H6′), 7.51 (t, 1H, J = 8.0 Hz, H6), 6.99 (s, 1H, -CH=), 6.84 (d, 1H, J = 8.0 Hz, H7), 6.70 (d, 1H, J = 8.6 Hz, H5′), 6.66 (s, 1H, H3′), 6.62 (d, 1H, J = 8.0 Hz, H5), 3.90 (s, 3H, OMe), 3.84 (s, 3H, OMe). 13C NMR (100 MHz, d6-DMSO) δ 181.2, 165.8, 162.4, 159.8, 157.0, 145.3, 138.3, 132.2, 113.2, 110.5, 109.4, 106.7, 103.9, 102.6, 98.3, 56.0, 55.6. MS (ESI) m/z 299 (M+H)+, 321 (M+Na)+, 619 (2M+Na)+. Anal. Calcd for C17H14O5·½H2O: C, 66.44, H, 4.92. Found: C, 66.60, H, 4.88.
(Z)-2-(2,6-dimethoxybenzylidene)-4-hydroxybenzofuran-3(2H)-one (25)
The crude product was prepared according to general procedure A starting from 4-hydroxybenzofuran-3(2H)-one (5) and 2,6-dimethoxybenzaldehyde, and was purified by column chromatography on silica gel (eluent: dichloromethane) to yield pure yellow solid (34%). m.p. 152–153°C. 1H NMR (400 MHz, CDCl3) δ 7.95 (br s, 1H, OH), 7.45 (t, 1H, J = 8.1 Hz, H6), 7.34 (t, 1H, J = 8.4 Hz, H4′), 7.09 (s, 1H, -CH=), 6.66 (d, 1H, J = 8.1 Hz, H7), 6.61 (d, 2H, J = 8.4 Hz, H3′,5′), 6.58 (d, 1H, J = 8.1 Hz, H5), 3.88 (s, 6H, OMe). 13C NMR (100 MHz, CDCl3) δ 185.5, 165.1, 159.1, 156.7, 147.5, 139.2, 131.5, 110.1, 109.9, 109.3, 106.5, 104.0, 103.5, 56.0. MS (ESI) m/z 299 (M+H)+, 321 (M+Na)+, 619 (2M+Na)+. Anal. Calcd for C17H14O5·½H2O: C, 66.45, H, 4.88. Found: C, 66.45, H, 4.78.
(Z)-2-(2,4,6-trimethoxybenzylidene)-4-hydroxybenzofuran-3(2H)-one (26)
The crude product was prepared according to general procedure A starting from 4-hydroxybenzofuran-3(2H)-one (5) and 2,4,6-trimethoxybenzaldehyde, and was recrystallized from methanol to yield pure yellow crystals (71%). m.p. 139–140°C. 1H NMR (400 MHz, d6-DMSO) δ 11.00 (s, 1H, OH), 7.46 (t, 1H, J = 8.1 Hz, H6), 6.68 (d, 1H, J = 8.1 Hz, H7), 6.66 (s, 1H, -CH=), 6.57 (d, 1H, J = 8.1 Hz, H5), 6.31 (s, 2H, H3,5), 3.84 (s, 3H, OMe), 3.83 (s, 6H, OMe). 13C NMR (100 MHz, d6-DMSO) δ 181.1, 165.9, 162.8, 159.5, 156.9, 146.1, 138.4, 109.9, 109.5, 103.4, 102.2, 91.1, 55.9, 55.5. MS (ESI) m/z 329 (M+H)+, 351 (M+Na)+, 679 (2M+Na)+. Anal Calcd for C18H16O6·⅓H2O: C, 64.67, H, 4.99. Found: C, 64.58, H, 5.03.
(Z)-2-(4-ethylbenzylidene)-4-hydroxybenzofuran-3(2H)-one (27)
The crude product was prepared according to general procedure A starting from 4-hydroxybenzofuran-3(2H)-one (5) and 4-ethylbenzaldehyde, and was recrystallized from methanol to yield pure yellow crystals (50%). m.p. 141–142°C. 1H NMR (400 MHz, d6-DMSO) δ 11.16 (s, 1H, OH), 7.86 (d, 2H, J = 8.0 Hz, H2′,6′), 7.53 (t, 1H, J = 8.1 Hz, H6), 7.32 (d, 2H, J = 8.0 Hz, H3′,5′), 6.86 (d, 1H, J = 8.1 Hz, H7), 6.74 (s, 1H, -CH=), 6.64 (d, 1H, J = 8.1 Hz, H5), 2.64 (q, 2H, J = 7.5 Hz, PhCH 2CH3), 1.19 (t, 3H, J = 7.5 Hz, PhCH2CH 3). 13C NMR (100 MHz, d6-DMSO) δ 181.3, 166.0, 157.1, 146.2, 145.8, 138.6, 131.1, 129.6, 128.4, 110.6, 110.1, 109.1, 102.4, 28.2, 15.3. MS (ESI) m/z 267 (M+H)+, 289 (M+Na)+. Anal. Calcd for C17H14O3·⅛H2O: C, 76.04, H, 5.31. Found: C, 75.93, H, 5.26.
(Z)-2-(4-isopropylbenzylidene)-4-hydroxybenzofuran-3(2H)-one (28)
The crude product was prepared according to general procedure A starting from 4-hydroxybenzofuran-3(2H)-one (5) and 4-isopropylbenzaldehyde, and was recrystallized from methanol to yield pure yellow crystals (44%). m.p. 126–127°C. 1H NMR (400 MHz, d6-DMSO) δ 11.16 (s, 1H, OH), 7.86 (d, 2H, J = 7.9 Hz, H2′,6′), 7.54 (t, 1H, J = 8.1 Hz, H6), 7.36 (d, 2H, J = 7.9 Hz, H3′,5′), 6.86 (d, 1H, J = 8.1 Hz, H7), 6.74 (s, 1H, -CH=), 6.64 (d, 1H, J = 8.1 Hz, H5), 2.92 (sept., 1H, J = 6.8 Hz, PhCH(CH3)2), 1.21 (d, 6H, J = 6.8 Hz, PhCH(CH 3)2). 13C NMR (100 MHz, d6-DMSO) δ 181.3, 166.0, 157.1, 150.4, 146.2, 138.6, 131.2, 129.8, 127.0, 110.6, 110.1, 109.1, 102.4, 33.5, 23.6. MS (ESI) m/z 281 (M+H)+, 303 (M+Na)+. Anal. Calcd for C18H16O3·¼H2O: C, 75.92, H, 5.79. Found: C, 75.55, H, 5.62.
(Z)-2-(4-butylbenzylidene)-4-hydroxybenzofuran-3(2H)-one (29)
The crude product was prepared according to general procedure A starting from 4-hydroxybenzofuran-3(2H)-one (5) and 4-butylbenzaldehyde, and was recrystallized from methanol to yield pure yellow crystals (56%). m.p. 121–122°C. 1H NMR (400 MHz, d6-DMSO) δ 11.15 (s, 1H, OH), 7.85 (d, 2H, J = 7.9 Hz, H2′,6′), 7.53 (t, 1H, J = 8.1 Hz, H6), 7.30 (d, 2H, J = 7.9 Hz, H3′,5′), 6.86 (d, 1H, J = 8.1 Hz, H7), 6.74 (s, 1H, -CH=), 6.63 (d, 1H, J = 8.0 Hz, H5), 2.61 (t, 2H, J = 7.4 Hz, PhCH 2CH2CH2CH3), 1.56 (quint., 2H, J = 7.4 Hz, PhCH2CH 2CH2CH3), 1.30 (sext., 2H, J = 7.4 Hz, PhCH2CH2CH 2CH3), 0.89 (t, 3H, J = 7.4 Hz, PhCH2CH2CH2CH 3). 13C NMR (100 MHz, d6-DMSO) δ 181.3, 166.0, 157.1, 146.2, 144.5, 138.6, 131.1, 129.6, 129.0, 110.6, 110.2, 109.1, 102.4, 34.8, 32.9, 21.8, 13.8. MS (ESI) m/z 295 (M+H)+, 317 (M+Na)+. Anal. Calcd for C19H18O3: C, 77.53, H, 6.17. Found: C, 77.32, H, 6.33.
(Z)-2-(4-tertiobutylbenzylidene)-4-hydroxybenzofuran-3(2H)-one (30)
The crude product was prepared according to general procedure A starting from 4-hydroxybenzofuran-3(2H)-one (5) and 4-tertiobutylbenzaldehyde, and was recrystallized from methanol to yield pure yellow crystals (61%). m.p. 167–168°C. 1H NMR (400 MHz, d6-DMSO) δ 11.16 (s, 1H, OH), 7.86 (d, 2H, J = 8.0 Hz, H2′,6′), 7.54 (t, 1H, J = 8.1 Hz, H6), 7.49 (d, 2H, J = 8.0 Hz, H3′,5′), 6.85 (d, 1H, J = 8.1 Hz, H7), 6.74 (s, 1H, -CH=), 6.64 (d, 1H, J = 8.1 Hz, H5), 1.28 (s, 9H, Ph(CH 3)3). 13C NMR (100 MHz, d6-DMSO) δ 181.3, 166.0, 157.2, 152.6, 146.3, 138.6, 130.9, 129.4, 125.8, 110.6, 110.0, 109.2, 102.4, 34.6, 30.9. MS (ESI) m/z 295 (M+H)+, 317 (M+Na)+. Anal. Calcd for C19H18O3: C, 77.53, H, 6.17. Found: C, 77.22, H, 6.24.
(Z)-2-(4-(4-methylpiperazin-1-yl)benzylidene)-4-hydroxybenzofuran-3(2H)-one (31)
The crude product was prepared according to general procedure A starting from 4-hydroxybenzofuran-3(2H)-one (5) and 4-(4-methylpiperazin-1-yl)benzaldehyde, and was washed three times with distilled water to yield an orange solid, which was the analytically pure hydrochloride salt, used without further purification (71%). m.p. > 260°C (decomposition). 1H NMR (400 MHz, d6-DMSO) δ 11.14 (s, 1H, OH), 11.14 (br s, 1H, NH+) 7.85 (d, 2H, J = 8.4 Hz, H2′,6′), 7.51 (t, 1H, J = 8.1 Hz, H6), 7.10 (d, 2H, J = 8.4 Hz, H3′,5′), 6.84 (d, 1H, J = 8.1 Hz, H7), 6.72 (s, 1H, -CH=), 6.66 (d, 1H, J = 8.1 Hz, H5), 4.02 (m, 2H, CH2), 3.48 (m, 2H, CH2), 3.24 (m, 2H, CH2), 3.13 (m, 2H, CH2), 2.80 (s, 3H, NMe). 13C NMR (100 MHz, d6-DMSO) δ 180.8, 165.5, 156.9, 149.9, 145.0, 137.9, 132.4, 122.8, 115.1, 110.6, 110.3, 109.3, 102.2, 51.7, 44.2, 41.8. MS (ESI) m/z 337 (M+H)+. Anal. Calcd for C15H10O4·HCl·1.75H2O: C, 57.14, H, 5.83, N, 6.67. Found: C, 57.22, H, 5.95, N, 6.76.
(Z)-2-(2-fluorobenzylidene)-4-hydroxybenzofuran-3(2H)-one (32)
The crude product was prepared according to general procedure A starting from 4-hydroxybenzofuran-3(2H)-one (5) and 2-fluorobenzaldehyde, and was recrystallized from methanol to yield pure yellow crystals (30%). m.p. 162–163°C. 1H NMR (400 MHz, d6-DMSO) δ 11.29 (s, 1H, OH), 8.21 (m, 1H, H6′), 7.56 (t, 1H, J = 8.1 Hz, H6), 7.50 (m, 1H, H3′), 7.35 (m, 2H, H4′,5′), 6.87 (d, 1H, J = 8.1 Hz, H7), 6.75 (s, 1H, -CH=), 6.66 (d, 1H, J = 8.1 Hz, H5). 13C NMR (100 MHz, d6-DMSO) δ 181.1, 166.1, 160.6 (d, J = 251.6 Hz), 157.4, 147.6, 139.1, 131.7 (d, J = 8.0 Hz), 131.1, 125.2, 119.9 (d, J = 11.5 Hz), 115.8 (d, J = 21.5 Hz), 111.0, 108.8, 102.5, 99.8 (d, J = 7.3 Hz). MS (ESI) m/z 257 (M+H)+, 279 (M+Na)+. Anal. Calcd for C15H9FO3·⅔H2O: C, 67.16, H, 3.85. Found: C, 66.80, H, 3.58.
(Z)-2-benzylidene-4,6-dihydroxybenzofuran-3(2H)-one (33)
The crude product was prepared according to general procedure D starting from (Z)-2-benzylidene-4,6-dimethoxy-benzofuran-3(2H)-one (54), and was purified by column chromatography on silica gel (eluent: toluene 9/methanol 1) to yield a pure yellow solid (33%). m.p. > 225°C (decomposition). 1H NMR (400 MHz, d6-DMSO) δ 10.99 (br s, 2H, OH), 7.89 (d, 2H, J = 7.4 Hz, H3′,5′), 7.47 (m, 2H, H2′,6′), 7.39 (m, 1H, H4′), 6.61 (s, 1H, -CH=), 6.23 (s, 1H, H7), 6.08 (s, 1H, H5). 13C NMR (100 MHz, d6-DMSO) δ 180.0, 167.8, 158.4, 147.7, 132.3, 130.6, 129.1, 128.9, 108.0, 102.4, 97.7, 90.6. MS (ESI) m/z 255 (M+H)+, 277 (M+Na)+. Anal. Calcd for C15H10O4·⅔H2O: C, 67.67, H, 4.26. Found: C, 67.67, H, 4.15.
(Z)-2-(2,4-dihydroxybenzylidene)-4,6-dihydroxybenzofuran-3(2H)-one (34)
The crude product was prepared according to general procedure D starting from (Z)-2-(2,4-dimethoxy-benzylidene)-4,6-dimethoxybenzo-furan-3(2H)-one (53), and was washed three times with distilled water to yield an orange solid, which was analytically pure and used without further purification (58%). m.p. > 270°C (decomposition). 1H NMR (400 MHz, d6-DMSO) δ 10.77 (s, 1H, OH), 10.74 (s, 1H, OH), 10.16 (s, 1H, OH), 9.90 (s, 1H, OH), 7.89 (d, 1H, J = 8.4 Hz, H6′), 6.88 (s, 1H, -CH=), 6.37 (m, 2H, H3′,5′), 6.18 (d, 1H, J = 1.6 Hz, H7), 6.05 (d, 1H, J = 1.6 Hz, H5). 13C NMR (100 MHz, d6-DMSO) δ 179.1, 167.4, 166.7, 160.3, 158.6, 158.0, 145.4, 132.0, 110.8, 108.1, 103.6, 103.1, 102.3, 97.5, 90.4. MS (ESI) m/z 287 (M+H)+, 309 (M+Na)+. Anal. Calcd for C15H10O6·1.1H2O: C, 58.86, H, 3.99. Found: C, 58.78, H, 3.98.
(Z)-2-(2,3-dimethoxybenzylidene)-4,6-dihydroxybenzofuran-3(2H)-one (35)
The crude product was prepared according to general procedure B starting from 4,6-dihydroxybenzofuran-3(2H)-one (3) and 2,3-dimethoxybenzaldehyde, and was purified by column chromatography on silica gel (eluent: cyclohexane 2/ethyl acetate 1/acetic acid 0.005) to yield a pure yellow solid (72%). m.p. 272–273°C. 1H NMR (400 MHz, d6-DMSO) δ 11.00 (s, 1H, OH), 10.94 (s, 1H, OH), 7.73 (dd, 1H, J1 = 8.0 Hz, J2 = 0.8 Hz, H6′), 7.19 (t, 1H, J = 8.0 Hz, H5′), 7.12 (dd, 1H, J1 = 8.0 Hz, J2 = 0.8 Hz, H4′), 6.80 (s, 1H, -CH=), 6.21 (d, J = 1.6 Hz, 1H, H7), 6.08 (d, J = 1.6 Hz, 1H, H5), 3.84 (s, 3H, OMe), 3.80 (s, 3H, OMe). 13C NMR (100 MHz, d6-DMSO) δ 178.9, 167.7, 167.4, 158.5, 152.5, 148.3, 147.8, 125.9, 124.4, 122.0, 114.0, 102.4, 101.4, 97.8, 90.6, 60.9, 55.8.
(Z)-2-(2,3,4-trimethoxybenzylidene)-4,6-dihydroxybenzofuran-3(2H)-one (36)
The crude product was prepared according to general procedure B starting from 4,6-dihydroxybenzofuran-3(2H)-one (3) and 2,3,4-trimethoxybenzaldehyde, and was purified by column chromatography on silica gel (eluent: cyclohexane 2/ethyl acetate 1/acetic acid 0.005) to yield a pure yellow solid (80%). m.p. 245–246°C. 1H NMR (400 MHz, d6-DMSO) δ 11.13 (s, 2H, OH), 7.87 (d, 1H, J = 7.6 Hz, H6′), 6.97 (d, 1H, J = 7.6 Hz, H5′), 6.72 (s, 1H, -CH=), 6.27 (s, 1H, H7), 6.23 (s, 1H, H5), 3.86 (s, 3H, OMe), 3.85 (s, 3H, OMe), 3.76 (s, 3H, OMe). 13C NMR (100 MHz, d6-DMSO) δ 181.0, 169.5, 169.5, 160.4, 156.6, 154.6, 149.3, 143.7, 127.9, 120.6, 110.6, 104.5, 103.7, 99.9, 92.5, 63.6, 62.5, 58.0.
(Z)-2-(4-ethylbenzylidene)-4,6-dihydroxybenzofuran-3(2H)-one (37)
The crude product was prepared according to general procedure B starting from 4,6-dihydroxybenzofuran-3(2H)-one (3) and 4-ethylbenzaldehyde, and was purified by column chromatography on silica gel (eluent: cyclohexane 2/ethyl acetate 1/acetic acid 0.005) to yield a pure yellow solid (83%). m.p. 237–238°C. 1H NMR (400 MHz, d6-DMSO) δ 10.94 (s, 2H, OH), 7.81 (d, 2H, J = 8.0 Hz, H2′,6′), 7.31 (d, 2H, J = 8.0 Hz, H3′,5′), 6.58 (s, 1H, -CH=), 6.22 (s, 1H, H7), 6.08 (s, 1H, H5), 2.64 (q, 2H, J = 7.6 Hz, CH 2CH3), 1.18 (t, 3H, J = 7.6 Hz, CH2CH 3). 13C NMR (100 MHz, d6-DMSO) δ 181.1, 169.8, 169.4, 160.4, 149.3, 147.3, 132.8, 131.8, 130.4, 110.3, 104.6, 99.7, 92.6, 30.1, 17.4. Anal. Calcd for C17H14O4·¼H2O: C, 71.20, H, 5.06. Found: C, 71.47, H, 5.19.
(Z)-2-(4-isopropylbenzylidene)-4,6-dihydroxybenzofuran-3(2H)-one (38)
The crude product was prepared according to general procedure B starting from 4,6-dihydroxybenzofuran-3(2H)-one (3) and 4-isopropylbenzaldehyde, and was purified by column chromatography on silica gel (eluent: cyclohexane 2/ethyl acetate 1/acetic acid 0.005) to yield a pure yellow solid (88%). m.p. 229–230°C. 1H NMR (400 MHz, d6-DMSO) δ 10.94 (s, 2H, OH), 7.80 (d, 2H, J = 8.0 Hz, H2′,6′), 7.32 (d, 2H, J = 8.0 Hz, H3′,5′), 6.58 (s, 1H, -CH=), 6.23 (d, 1H, J = 2.0 Hz, H7), 6.09 (d, 1H, J = 2.0 Hz, H5), 2.89 (sept, 1H, J = 6.8 Hz, CH(CH3)2), 1.19 (d, 6H, J = 6.8 Hz, CH(CH 3)2). 13C NMR (100 MHz, d6-DMSO) δ 181.1, 169.8, 169.4, 160.4, 151.8, 149.4, 132.8, 132.0, 128.9, 110.3, 104.6, 99.7, 92.6, 35.4, 25.6. Anal. Calcd for C18H16O4·¼H2O: C, 71.88, H, 5.49. Found: C, 71.99, H, 5.52.
(Z)-2-(4-butylbenzylidene)-4,6-dihydroxybenzofuran-3(2H)-one (39)
The crude product was prepared according general procedure D starting from (Z)-2-(4-butylbenzylidene)-4,6-dimethoxybenzofuran-3(2H)-one (44), and was purified by column chromatography on silica gel (eluent: ethyl acetate 1/cyclohexane 1) to yield a pure yellow solid (52%). m.p. 221–222°C. 1H NMR (400 MHz, d6-DMSO) δ 10.95 (br s, 2H, OH), 7.79 (d, 2H, J = 8.1 Hz, H2′,6′), 7.29 (d, 2H, J = 8.1 Hz, H3′,5′), 6.57 (s, 1H, -CH=), 6.21 (s, 1H, H7), 6.07 (s, 1H, H5), 2.61 (t, 2H, J = 7.5 Hz, PhCH 2CH2CH2CH3), 1.56 (quint., 2H, J = 7.5 Hz, PhCH2CH 2CH2CH3), 1.31 (sext., 2H, J = 7.5 Hz, PhCH2CH2CH 2CH3), 0.89 (t, 3H, J = 7.5 Hz, PhCH2CH2CH2CH 3). 13C NMR (100 MHz, d6-DMSO) δ 179.0, 167.8, 167.6, 158.5, 147.4, 143.9, 130.7, 129.9, 128.9, 108.3, 102.6, 97.8, 90.5, 34.8, 32.9, 21.8, 13.8. MS (ESI) m/z 311 (M+H)+, 333 (M+Na)+, 643 (2M+Na)+. Anal. Calcd for C19H18O4·⅓H2O: C, 72.15, H, 5.91. Found: C, 72.00, H, 5.83.
(Z)-2-(4-cyclohexylbenzylidene)-4,6-dihydroxybenzofuran-3(2H)-one (40)
The crude product was prepared according to general procedure D starting from (Z)-2-(4-cyclohexylbenzylidene)-4,6-dimethoxybenzofuran-3(2H)-one (45), and was purified by column chromatography on silica gel (eluent: ethyl acetate 1/cyclohexane 1) to yield a pure yellow solid (64%). m.p. > 215°C (decomposition). 1H NMR (400 MHz, d6-DMSO) δ 10.98 (br s, 2H, OH), 7.79 (d, 2H, J = 8.3 Hz, H2′,6′), 7.31 (d, 2H, J = 8.3 Hz, H3′,5′), 6.56 (s, 1H, -CH=), 6.20 (d, 1H, J = 1.5 Hz, H7), 6.07 (d, 1H, J = 1.5 Hz, H5), 2.54 (m, 1H, c-Hex), 1.79 (m, 4H, c-Hex), 1.70 (m, 1H, c-Hex), 1.39 (m, 4H, c-Hex), 1.31 (m, 1H, c-Hex). 13C NMR (100 MHz, d6-DMSO) δ 179.0, 167.8, 167.5, 158.5, 149.0, 147.4, 130.7, 130.0, 127.3, 108.2, 102.5, 97.7, 90.5, 43.7, 33.7, 26.3, 25.5. MS (ESI) m/z 337 (M+H)+, 359 (M+Na)+, 695 (2M+Na)+. Anal. Calcd for C21H20O4·½H2O: C, 73.03, H, 6.13. Found: C, 72.95, H, 6.01.
(Z)-2-(4-morpholinobenzylidene)-4,6-dihydroxybenzofuran-3(2H)-one (41)
The crude product was prepared according to general procedure B starting from 4,6-dihydroxybenzofuran-3(2H)-one (3) and 4-morpholinobenzaldehyde, and was purified by column chromatography on silica gel (eluent: cyclohexane 2/ethyl acetate 1/acetic acid 0.005) to yield a pure red solid (32%). m.p. 273–273°C. 1H NMR (400 MHz, d6-DMSO) δ 10.83 (s, 1H, OH), 10.79 (s, 1H, OH), 7.77 (d, 2H, J = 8.6 Hz, H2′,6′), 7.02 (d, 2H, J = 8.6 Hz, H3′,5′), 6.55 (s, 1H, -CH=), 6.20 (d, 1H, J = 0.8 Hz, H7), 6.06 (d, 1H, J = 0.8 Hz, H5), 3.74 (t, 4H, J = 4.8 Hz, CH2O), 3.23 (t, 4H, J = 4.8 Hz, CH2N). 13C NMR (100 MHz, d6-DMSO) δ 180.9, 169.4, 168.9, 160.1, 153.3, 147.9, 134.2, 124.4, 116.4, 111.3, 104.9, 99.6, 92.4, 67.9, 49.2. Anal. Calcd for C19H17NO5·⅓H2O: C, 66.08, H, 5.12, N, 4.05. Found: C, 66.44, H, 5.54, N, 3.37.
(Z)-2-(4-fluorobenzylidene)-4,6-dihydroxybenzofuran-3(2H)-one (42)
The crude product was prepared according to general procedure D starting from (Z)-2-(4-fluorobenzylidene)-4,6-dimethoxybenzofuran-3(2H)-one (55), and was purified by column chromatography on silica gel (eluent: dichloromethane 9/methanol 1) to yield a pure yellow solid (50%). m.p. > 270°C (decomposition). 1H NMR (400 MHz, d6-DMSO) δ 11.00 (br s, 1H, OH), 10.97 (br s, 1H, OH), 7.96 (m, 2H, H2′,6′), 7.31 (m, 2H, H3′,5′), 6.64 (s, 1H, -CH=), 6.22 (d, 1H, J = 1.6 Hz, H7), 6.08 (d, 1H, J = 1.6 Hz, H5). 13C NMR (100 MHz, d6-DMSO) δ 179.0, 167.9, 167.5, 162.3 (d, J = 248.4 Hz), 158.5, 147.5, 132.9 (d, J = 8.2 Hz), 129.1, 116.0 (d, J = 21.7 Hz), 107.1, 102.5, 97.8, 90.7. MS (ESI) m/z 273 (M+H)+, 295 (M+Na)+. Anal. Calcd for C15H9FO4·⅓H2O: C, 64.74, H, 3.48. Found: C, 64.58, H, 3.67.
(Z)-2-(2,4-dimethylbenzylidene)-4,6-dimethoxybenzofuran-3(2H)-one (43)
The crude product was prepared according to general procedure A starting from 4,6-dimethoxy-benzofuran-3(2H)-one (6) and 2,4-dimethoxybenzaldehyde, and was recrystallized from methanol to yield pure pale yellow crystals (86%). m.p. 216°C. 1H NMR (400 MHz, CDCl3) δ 8.07 (d, 1H, J = 8.0 Hz, H6′), 7.10 (d, 1H, J = 8.0 Hz, H5′), 7.06 (s, 1H, H3′), 6.97 (s, 1H, -CH=), 6.37 (d, 1H, J = 1.7 Hz, H7), 6.12 (d, 1H, J = 1.7 Hz, H5), 3.95 (s, 3H, OMe), 3.90 (s, 3H, OMe), 2.46 (s, 3H, Me), 2.35 (s, 3H, Me). 13C NMR (100 MHz, CDCl3) δ 180.9, 169.2, 169.0, 159.6, 147.8, 139.8, 139.0, 131.6, 131.0, 128.4, 127.2, 108.1, 105.6, 94.2, 89.4, 56.4, 56.3, 21.6, 20.4. MS (ESI) m/z 311 (M+H)+, 333 (M+Na)+, 643 (2M+Na)+. Anal. Calcd for C19H18O4: C, 73.54, H, 5.85. Found: C, 73.04, H, 5.92.
(Z)-2-(4-butylbenzylidene)-4,6-dimethoxybenzofuran-3(2H)-one (44)
The crude product was prepared according to general procedure A starting from 4,6-dimethoxybenzofuran-3(2H)-one (6) and 4-butylbenzaldehyde, and was recrystallized from methanol to yield pure white crystals (75%). m.p. 153°C. 1H NMR (400 MHz, CDCl3) δ 7.78 (d, 2H, J = 8.1 Hz, H2′,6′), 7.24 (d, 2H, J = 8.1 Hz, H3′,5′), 6.77 (s, 1H, -CH=), 6.38 (d, 1H, J = 1.7 Hz, H7), 6.12 (d, 1H, J = 1.7 Hz, H5), 3.95 (s, 3H, OMe), 3.91 (s, 3H, OMe), 2.65 (t, 2H, J = 7.6 Hz, PhCH 2CH2CH2CH3), 1.61 (quint., 2H, J = 7.6 Hz, PhCH2CH 2CH2CH3), 1.37 (sext., 2H, J = 7.6 Hz, PhCH2CH2CH 2CH3), 0.96 (t, 3H, J = 7.6 Hz, PhCH2CH2CH2CH 3). 13C NMR (100 MHz, CDCl3) δ 181.0, 169.2, 169.1, 159.6, 147.7, 145.0, 131.3, 130.2, 129.1, 111.3, 105.5, 94.2, 89.4, 56.4, 56.3, 35.8, 33.6, 22.5, 14.1. MS (ESI) m/z 339 (M+H)+, 361 (M+Na)+, 699 (2M+Na)+. Anal. Calcd for C21H22O4: C, 74.54, H, 6.56. Found: C, 74.46, H, 6.33.
(Z)-2-(4-cyclohexylbenzylidene)-4,6-dimethoxybenzofuran-3(2H)-one (45)
The crude product was prepared according to general procedure A starting from 4,6-dimethoxybenzofuran-3(2H)-one (6) and 4-cyclohexylbenzaldehyde, and was recrystallized from methanol to yield pure white crystals (78%). m.p. 174°C. 1H NMR (400 MHz, CDCl3) δ 7.78 (d, 2H, J = 8.2 Hz, H2′,6′), 7.27 (d, 2H, J = 8.2 Hz, H3′,5′), 6.76 (s, 1H, -CH=), 6.37 (d, 1H, J = 1.6 Hz, H7), 6.11 (d, 1H, J = 1.6 Hz, H5), 3.94 (s, 3H, OMe), 3.90 (s, 3H, OMe), 2.53 (m, 1H, c-Hex), 1.86 (m, 4H, c-Hex), 1.76 (m, 1H, c-Hex), 1.42 (m, 4H, c-Hex), 1.35 (m, 1H, c-Hex). 13C NMR (100 MHz, CDCl3) δ 180.9, 169.1, 169.0, 159.5, 150.0, 147.7, 131.4, 130.3, 127.5, 111.2, 105.5, 94.1, 89.4, 56.4, 56.3, 44.7, 34.4, 27.0, 26.2. MS (ESI) m/z 365 (M+H)+, 751 (2M+Na)+. Anal. Calcd for C23H24O4: C, 75.81, H, 6.64. Found: C, 75.40, H, 6.78.
(Z)-2-(4-butylbenzylidene)-benzofuran-3(2H)-one (46)
The crude product was prepared according to general procedure C starting from benzofuran-3(2H)-one and 4-butylbenzaldehyde, and was purified by column chromatography on silica gel (eluent: dichloromethane 1/cyclohexane 1) to yield a pure pale yellow solid (44%). m.p. 77–78°C. 1H NMR (400 MHz, CDCl3) δ 7.87 (d, 2H, J = 8.3 Hz, H2′,6′), 7.83 (d, 1H, J = 8.3 Hz, H4), 7.67 (t, 1H, J = 8.3 Hz, H6), 7.35 (d, 1H, J = 8.3 Hz, H7), 7.30 (d, 2H, J = 8.3 Hz, H3′,5′), 7.24 (t, 1H, J = 8.3 Hz, H5), 6.92 (s, 1H, -CH=), 2.68 (t, 2H, J = 7.5 Hz, PhCH 2CH2CH2CH3), 1.65 (quint., 2H, J = 7.5 Hz, PhCH2CH 2CH2CH3), 1.40 (sext., 2H, J = 7.5 Hz, PhCH2CH2CH 2CH3), 0.96 (t, 3H, J = 7.5 Hz, PhCH2CH2CH2CH 3). 13C NMR (100 MHz, CDCl3) δ 185.0, 166.2, 146.7, 145.7, 136.9, 131.8, 129.9, 129.3, 124.8, 123.6, 122.0, 113.7, 113.1, 35.9, 33.6, 22.6, 14.2. MS (ESI) m/z 279 (M+H)+. Anal. Calcd for C15H10O4·¼H2O: C, 80.71, H, 6.55. Found: C, 80.96, H, 6.77.
(Z)-2-(4-butylbenzylidene)-4,6-difluorobenzofuran-3(2H)-one (47)
The crude product was prepared according to general procedure C starting from 4,6-difluorobenzofuran-3(2H)-one (8) and 4-butylbenzaldehyde, and was purified by column chromatography on silica gel (eluent: dichloromethane 1/cyclohexane 1) to yield a pure white solid (56%). m.p. 93–94°C. 1H NMR (400 MHz, CDCl3) δ 7.80 (d, 2H, J = 8.1 Hz, H2′,6′), 7.29 (d, 2H, J = 8.1 Hz, H3′,5′), 6.91 (s, 1H, -CH=), 6.88 (m, 1H, H7), 6.63 (m, 1H, H5), 2.68 (t, 2H, J = 7.5 Hz, PhCH 2CH2CH2CH3), 1.65 (quint., 2H, J = 7.5 Hz, PhCH2CH 2CH2CH3), 1.39 (sext., 2H, J = 7.5 Hz, PhCH2CH2CH 2CH3), 0.96 (t, 3H, J = 7.5 Hz, PhCH2CH2CH2CH 3). 13C NMR (100 MHz, CDCl3) δ 179.5, 170.1 (J = 13.0 Hz), 167.5 (J = 13.0 Hz), 160.8 (J = 16.2 Hz), 158.2 (J = 16.2 Hz), 146.4, 146.3, 131.9, 129.4, 129.2, 114.5, 100.0 (J1 = 27.0 Hz, J2 = 23.4 Hz), 97.5 (J1 = 27.0 Hz, J2 = 4.5 Hz). MS (ESI) m/z 315 (M+H)+. Anal. Calcd for C19H16F2O2: C, 72.61, H, 5.14. Found: C, 72.86, H, 5.47.
(Z)-2-(2,4-difluorobenzylidene)-4-fluorobenzofuran-3(2H)-one (48)
The crude product was prepared according to general procedure C starting from 4-fluorobenzofuran-3(2H)-one (9) and 2,4-difluorobenzaldehyde, and was purified by column chromatography on silica gel (eluent: dichloromethane 1/cyclohexane 1) to yield a pure white solid (83%). m.p. 164–165°C. 1H NMR (400 MHz, CDCl3) δ 8.29 (dt, 1H, J1 = 8.6 Hz, J2 = 6.5 Hz, H6′), 7.65 (dt, 1H, J1 = 8.3 Hz, J2 = 5.5 Hz, H6), 7.13 (d, 1H, J = 8.3 Hz, H7), 7.11 (s, 1H, -CH=), 7.01 (m, 1H, H5′), 6.90 (dt, 1H, J1 = 8.7 Hz, J2 = 2.6 Hz, H3′), 6.88 (t, 1H, J = 8.3 Hz, H5). 13C NMR (100 MHz, CDCl3) δ 180.6, 166.3 (J = 6.0 Hz), 164.4 (J1 = 171.8 Hz, J2 = 12.3 Hz), 161.9 (J1 = 174.6 Hz, J2 = 12.2 Hz), 160.1, 157.5, 146.9, 138.7 (J = 9.4 Hz), 133.2 (J = 9.5 Hz), 117.0 (J1 = 12.0 Hz, J2 = 4.1 Hz), 112.4 (J1 = 21.2 Hz, J2 = 2.9 Hz), 110.7 (J = 19.0 Hz), 109.0 (J = 4.1 Hz), 104.5 (J = 25.6 Hz), 103.9 (J = 5.7 Hz). MS (ESI) m/z 277 (M+H)+. Anal. Calcd for C15H7F3O2: C, 65.23, H, 2.56. Found: C, 65.36, H, 2.57.
(Z)-2-(4-hydroxybenzylidene)-5-hydroxybenzofuran-3(2H)-one (49)
The crude product was prepared according to general procedure A starting from 5-hydroxybenzofuran-3(2H)-one (4) and 4-hydroxybenzaldehyde, and was recrystallized from acetonitrile to yield pure yellow crystals (17%). m.p. > 300°C (decomposition). 1H NMR (400 MHz, d6-DMSO) δ 10.21 (br s, 1H, OH), 9.76 (br s, 1H, OH), 7.85 (d, 2H, J = 7.8 Hz, H2′,6′), 7.38 (d, 1H, J = 8.5 Hz, H6), 7.20 (d, 1H, J = 8.5 Hz, H7), 7.01 (s, 1H, H4), 6.89 (d, 2H, J = 7.8 Hz, H3′,5′), 6.83 (s, 1H, -CH=). 13C NMR (100 MHz, d6-DMSO) δ 183.4, 159.5, 158.8, 153.6, 145.5, 133.5, 125.2, 122.9, 121.5, 116.0, 113.7, 112.8, 107.4. MS (ESI) m/z 255 (M+H)+, 277 (M+Na)+. Anal. Calcd for C15H10O4·⅔H2O: C, 67.67, H, 4.29. Found: C, 67.56, H, 4.10.
(Z)-2-(4-cyclohexylbenzylidene)-4,6-dibenzyloxybenzofuran-3(2H)-one (50)
The crude product was prepared according to general procedure C starting from 4,6-dibenzyloxybenzofuran-3(2H)-one (7) and 4-cyclohexylbenzaldehyde, and was purified by column chromatography on silica gel (eluent: dichloromethane 2/cyclohexane 1) to yield a pure pale yellow solid (43%). m.p. 155–157°C. 1H NMR (400 MHz, CDCl3) δ 7.81 (d, 2H, J = 8.2 Hz, H2′,6′), 7.32–7.50 (m, 10H, OCH2 Ph), 7.29 (d, 2H, J = 8.2 Hz, H3′,5′), 6.78 (s, 1H, -CH=), 6.46 (d, 1H, J = 1.6 Hz, H7), 6.24 (d, J = 1.6 Hz, H5), 5.28 (s, 2H, OCH 2Ph), 5.11 (s, 2H, OCH 2Ph), 2.53 (m, 1H, c-Hex), 1.89 (m, 4H, c-Hex), 1.78 (m, 1H, c-Hex), 1.43 (m, 4H, c-Hex), 1.30 (m, 1H, c-Hex). 13C NMR (100 MHz, CDCl3) δ 180.8, 169.0, 167.8, 158.5, 150.0, 147.7, 136.2, 135.7, 131.4, 130.3, 129.0, 128.9, 128.7, 128.1, 127.8, 127.5, 127.0, 111.2, 106.1, 96.6, 90.7, 71.0, 70.9, 44.8, 34.4, 27.1, 26.3. MS (ESI) m/z 517 (M+H)+. Anal. Calcd for C35H32O4·⅓H2O: C, 80.46, H, 6.26. Found: C, 80.30, H, 6.67.
(Z)-2-((1-butyl-1H-indol-3-yl)methylene)-4,6-dihydroxybenzofuran-3(2H)-one (51)
The crude product was prepared according to general procedure B starting from 4,6-dihydroxybenzofuran-3(2H)-one (3) and 1-butyl-1H-indole-3-carbaldehyde, and was purified by column chromatography on silica gel (eluent: cyclohexane 2/ethyl acetate 1/acetic acid 0.005) to yield a pure red solid (65%). m.p. 248–249°C. 1H NMR (400 MHz, d6-DMSO) δ 10.75 (s, 2H, OH), 8.13 (s, 1H, H2′), 7.97 (d, 1H, J = 7.7 Hz, H4′), 7.56 (d, 1H, J = 7.7 Hz, H7′), 7.25 (t, 1H, J = 7.7 Hz, H6′), 7.18 (t, 1H, J = 7.7 Hz, H5′), 6.97 (s, 1H, -CH=), 6.27 (d, 1H, J = 1.2 Hz, H7), 6.08 (d, 1H, J = 1.2 Hz, H5), 4.28 (t, 2H, J = 7.2 Hz, CH 2CH2CH2CH3), 1.76 (quint., 2H, J = 7.2 Hz, CH2CH 2CH2CH3), 1.27 (sext., 2H, J = 7.2 Hz, CH2CH2CH 2CH3), 0.88 (t, 3H, J = 7.2 Hz, CH2CH2CH2CH 3). 13C NMR (100 MHz, d6-DMSO) δ 180.3, 168.9, 168.5, 159.9, 147.3, 138.0, 134.9, 129.2, 124.5, 122.7, 121.1, 112.6, 109.4, 105.7, 104.2, 99.5, 92.5, 47.9, 33.9, 21.5, 15.6. Anal. Calcd for C21H19NO4: C, 72.19, H, 5.48, N, 4.01. Found: C, 71.81, H, 5.79, N, 3.79.
(Z)-2-(3,4-dimethoxybenzylidene)-4,6-dimethoxybenzofuran-3(2H)-one (52)
The crude product was prepared according to general procedure A starting from 4,6-dimethoxybenzofuran-3(2H)-one (6) and 3,4-dimethoxybenzaldehyde, and was recrystallized from methanol to yield pure pale yellow crystals (83%). m.p. 173–174°C. 1H NMR (400 MHz, CDCl3) δ 7.44 (m, 2H, H2′,6′), 6.91 (d, 1H, J = 8.1 Hz, H5′), 6.73 (s, 1H, -CH=), 6.34 (s, 1H, H7), 6.12 (s, 1H, H5), 3.96 (s, 3H, OMe), 3.95 (s, 3H, OMe), 3.93 (s, 3H, OMe), 3.91 (s, 3H, OMe). 13C NMR (100 MHz, CDCl3) δ 180.5, 168.7, 159.3, 150.3, 148.9, 146.8, 125.5, 125.3, 113.5, 111.3, 111.1, 105.4, 93.9, 89.1, 56.2, 56.1, 55.9, 56.0. MS (ESI) m/z 343 (M+H)+, 707 (2M+Na)+.
(Z)-2-(2,4-dimethoxybenzylidene)-4,6-dimethoxybenzofuran-3(2H)-one (53)
The crude product was prepared according to general procedure A starting from 4,6-dimethoxybenzofuran-3(2H)-one (6) and 2,4-dimethoxybenzaldehyde, and was recrystallized from methanol to yield pure pale yellow crystals (91%). m.p. 213–214°C. 1H NMR (400 MHz, CDCl3) δ 8.19 (d, 1H, J = 8.7 Hz, H6′), 7.27 (s, 1H, -CH=), 6.57 (dd, 1H, J = 8.7 Hz, J = 2.3 Hz, H5′), 6.45 (d, 1H, J = 2.3 Hz, H3′), 6.36 (d, 1H, J = 1.7 Hz, H7), 6.11 (d, 1H, J = 1.7 Hz, H5), 3.94 (s, 3H, OMe), 3.90 (s, 3H, OMe), 3.87 (s, 3H, OMe), 3.86 (s, 3H, OMe). 13C NMR (100 MHz, CDCl3) δ 180.6, 168.5, 168.4, 162.2, 160.1, 159.2, 146.8, 132.8, 114.6, 105.6, 105.5, 105.3, 97.9, 93.7, 89.0, 56.1, 56.0, 55.5, 55.4. MS (ESI) m/z 343 (M+H)+, 707 (2M+Na)+.
(Z)-2-benzylidene-4,6-dimethoxybenzofuran-3(2H)-one (54)
The crude product was prepared according to general procedure A starting from 4,6-dimethoxybenzofuran-3(2H)-one (6) and benzaldehyde, and was recrystallized from methanol to yield pure pale yellow crystals (61%). m.p. 156–157°C. 1H NMR (400 MHz, CDCl3) δ 7.91 (d, 1H, J = 7.3 Hz, H2′,6′), 7.39–7.48 (m, 3H, H3′,4′,5′), 6.68 (s, 1H, -CH=), 6.66 (d, 1H, J = 1.5 Hz, H7), 6.31 (d, 1H, J = 1.5 Hz, H5), 3.91 (s, 3H, OMe), 3.89 (s, 3H, OMe). 13C NMR (100 MHz, CDCl3) δ 179.0, 168.9, 168.3, 158.9, 147.3, 132.1, 130.8, 129.3, 128.8, 109.3, 104.0, 94.3, 89.8, 56.4, 56.1. MS (ESI) m/z 283 (M+H)+, 587 (2M+H)+.
(Z)-2-(4-fluorobenzylidene)-4,6-dimethoxybenzofuran-3(2H)-one (55)
The crude product was prepared according to general procedure A starting from 4,6-dimethoxybenzofuran-3(2H)-one (6) and 4-fluorobenzaldehyde, and was recrystallized from methanol to yield pure pale yellow crystals (64%). m.p. 186–187°C. 1H NMR (400 MHz, CDCl3) δ 7.87 (m, 2H, H2′,6′), 7.12 (m, 2H, H3′,5′), 6.74 (s, 1H, -CH=), 6.40 (d, 1H, J = 1.8 Hz, H7), 6.15 (d, 1H, J = 1.8 Hz, H5), 3.97 (s, 3H, OMe), 3.93 (s, 3H, OMe). 13C NMR (100 MHz, CDCl3) δ 180.8, 168.2, 163.2 (d, J = 251.4 Hz), 159.6, 147.7, 133.2 (d, J = 8.2 Hz), 129.1, 116.2 (d, J = 21.7 Hz), 109.8, 105.4, 94.3, 89.4, 56.4, 56.4. MS (ESI) m/z 301 (M+H)+, 623 (2M+Na)+.
Molecular Modeling
Docking studies used the reported X-ray crystallographic structure of HCV genotype 1b RdRp complexed with a known inhibitor binding to Thumb Pocket I of the enzyme (PDB ID: 2dxs).29 The protein structure was extracted and water molecules were deleted. The complexed ligand was used to define the binding site for docking. The genetic algorithms of GOLD and Autodock4 docking softwares performed flexible docking with small molecules while keeping the protein structure rigid. All compounds were built and their energies minimized with Sybyl, using a Conjugate Gradient method and MMFF94 force field and charges. For each compound, 20 poses were generated by GOLD and ranked according to the Goldscore scoring function, while 100 poses were generated by Autodock and clustered according to their spatial proximity and calculated free energy of binding. Molecular surfaces were generated with MOLCAD and the docking solutions were analyzed with Sybyl.
Biology
Expression and purification of recombinant HCV NS5BΔ21
RdRp (NS5B protein) from the HCV J4 genotype 1b reference strain, truncated of its 21 C-terminal amino acids to ensure solubility (NS5BΔ21) and carrying a hexahistidine tag (His-Tag) at its N-terminus, was expressed in Escherichia coli C41(DE3) and purified. Chromatography was performed on Ni-NTA columns (Qiagen, Valencia, California). The columns were washed with a buffer containing 50 mM NaH2PO4 (pH 8.0), 500 mM NaCl, and 20 mM imidazole. The bound protein was eluted in 1-mL fractions with a buffer containing 50 mM NaH2PO4 (pH 8.0), 500 mM NaCl, and 250 mM imidazole. NS5BΔ21-enriched fractions were selected with a Bradford colorimetric assay, and NS5BΔ21 purity was determined by SDS-PAGE analysis with Coomassie staining. Purified NS5BΔ21 fractions were pooled and dialyzed against a buffer containing 5 mM Tris (pH 7.5), 0.2 M sodium acetate, 1 mM DTT, 1 mM EDTA, and 10% glycerol. NS5BΔ21 purity was >98%.
HCV-NS5BΔ21 polymerase assay
An enzyme assay was used to measure inhibition of HCV-NS5BΔ21 polymerase activity. The cell-free HCV-NS5BΔ21 polymerase assay is based on the amount of double-stranded RNA synthesized in the presence of HCV-NS5BΔ21, a homopolymeric RNA template (Poly U, GE Healthcare, Chalfont St. Giles, UK) and ATP, measured with an intercalating agent (SYBR® Green, Applied Biosystems), as previously described.30 The Quick Site-Directed Mutagenesis kit (Stratagene, La Jolla, California) was used to assess the effect of amino acid substitutions conferring resistance to other specific HCV RdRp inhibitors. The tested substitutions included M423T, C316Y, H95Q, and P495L. The constructs were verified by DNA sequencing and the corresponding proteins were purified as described above.
Cytotoxicity assay
Exponentially growing HuH7 and HEK293 cells were trypsinized and plated (2000 and 1000 cells per well, respectively) in 96-well microplates and allowed to attach for 96 hours at 37°C under 5% CO2. A 200-μL aliquot of drug solution, diluted in medium with 1% DMSO (final concentration) was added and the cells were incubated for 72 hours at 37°C under 5% CO2. Cytotoxicity was evaluated with a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide colorimetric assay (MTT assay).
Acknowledgments
R. H. is a recipient of a fellowship grant from the Ministère de l’Enseignement et de la Recherche to whom he is very thankful. A part of the project related to this article is funded by l’Agence Nationale de la Recherche (ANR). W.Y is a recipient of a fellowship from the University of Sun Yat-Sen (China) to whom he is thankful.
References
- 1.Wong T, Lee SS. Hepatitis C: A review for primary care physicians. CMAJ. 2006;174:649–659. doi: 10.1503/cmaj.1030034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pawlotsky JM, McHutchison JG. Hepatitis C. Development of new drugs and clinical trials: Promises and pitfalls. Hepatology. 2004;39:554–567. doi: 10.1002/hep.20065. [DOI] [PubMed] [Google Scholar]
- 3.Strader DB, Wright T, Thomas DL, Seeff LB. Diagnosis, management, and treatment of hepatitis C. Hepatology. 2004;39:1147–1171. doi: 10.1002/hep.20119. [DOI] [PubMed] [Google Scholar]
- 4.Dixit NM, Layden-Almer JE, Layden TJ, Perelson AS. Modelling how ribavirin improves interferon response rate in hepatitis C virus infection. Nature. 2004;432:922–924. doi: 10.1038/nature03153. [DOI] [PubMed] [Google Scholar]
- 5.Feld JJ, Hoofnagle JH. Mechanism of action of interferon and ribavirin treatment of hepatitis C. Nature. 2005;436:967–972. doi: 10.1038/nature04082. [DOI] [PubMed] [Google Scholar]
- 6.Zein NN. Clinical significance of hepatitis C virus genotypes. Clin Microbiol Rev. 2000:223–225. doi: 10.1128/cmr.13.2.223-235.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carroll SS, Olsen DB. Nucleoside analog inhibitors of hepatitis C virus replication. Infect Disord Drug Targets. 2006;6:17–29. doi: 10.2174/187152606776056698. [DOI] [PubMed] [Google Scholar]
- 8.Beaulieu PL. Recent advances in the development of NS5B polymerase inhibitors for the treatment of hepatitis C virus infection. Expert Opin Ther Patents. 2009;19:145–164. doi: 10.1517/13543770802672598. [DOI] [PubMed] [Google Scholar]
- 9.Pauwels F, Mostmans W, Quirynen LMM, van der Helm L, Boutton CW, Rueff AS, Cleiren E, Raboisson P, Surleraux D, Nyanguile O, Simmen KA. Binding-site identification and genotype profiling of hepatitis C virus polymerase inhibitors. J Virol. 2007;81:6909–6919. doi: 10.1128/JVI.01543-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beaulieu PL, Bös M, Bousquet Y, Fazal G, Gauthier J, Gillard J, Goulet S, LaPlante S, Poupart MA, Lebebvre S, McKercher G, Pellerin C, Austel V, Kukolj G. Non-nucleoside inhibitors of the hepatitis C virus NS5B polymerase: Discovery and preliminary SAR of benzimidazole derivatives. Bioorg Med Chem Lett. 2004;14:119–124. doi: 10.1016/j.bmcl.2003.10.023. [DOI] [PubMed] [Google Scholar]
- 11.Harper S, Pacini B, Avolio S, Di Filippo M, Migliaccio G, Laufer R, De Francesco R, Rowley M, Narjes F. Development and preliminary optimization of indole-N-acetamide inhibitors of hepatitis C virus NS5B polymerase. J Med Chem. 2005;48:1314–1317. doi: 10.1021/jm049122i. [DOI] [PubMed] [Google Scholar]
- 12.Stansfield I, Ercolani C, Mackay A, Conte I, Pompei M, Koch U, Gennari N, Giuliano C, Rowley M, Narjes F. Tetracyclic indole inhibitors of hepatitis C virus NS5B-polymerase. Bioorg Med Chem Lett. 2009;19:627–632. doi: 10.1016/j.bmcl.2008.12.068. [DOI] [PubMed] [Google Scholar]
- 13.Habermann J, Capito E, del Rosario Rico Ferreira M, Koch U, Narjes F. Discovery of pentacyclic compounds as potent inhibitors of hepatitis C virus NS5B RNA polymerase. Bioorg Med Chem Lett. 2009;19:633–638. doi: 10.1016/j.bmcl.2008.12.039. [DOI] [PubMed] [Google Scholar]
- 14.Goulet S, Poupart MA, Gillard J, Poirier M, Kukolj G, Beaulieu PL. Discovery of benzimidazole-diamide finger loop (Thumb Pocket I) allosteric inhibitors of HCV NS5B polymerase: Implementing parallel synthesis for rapid linker optimization. Bioorg Med Chem Lett. 2010;20:196–200. doi: 10.1016/j.bmcl.2009.10.136. [DOI] [PubMed] [Google Scholar]
- 15.Beaulieu PL, Jolicoeur E, Gillard J, Brochu C, Coulombe R, Dansereau N, Duan J, Garneau M, Jakalian A, Kühn P, Lagacé L, LaPlante S, McKercher G, Perrault S, Poirier M, Poupart MA, Stammers T, Thauvette L, Thavonekham B, Kukolj G. N-Acetamideindolecarboxylic acid allosteric ‘finger-loop’ inhibitors of the hepatitis C virus NS5B polymerase: discovery and initial optimization studies. Bioorg Med Chem Lett. 2010;20:857–861. doi: 10.1016/j.bmcl.2009.12.101. [DOI] [PubMed] [Google Scholar]
- 16.Beaulieu PL, Dansereau N, Duan J, Garneau M, Gillard J, McKercher G, LaPlante S, Lagacée L, Thauvette L, Kukolj G. Benzimidazole Thumb Pocket I finger-loop inhibitors of HCV NS5B polymerase: Improved drug-like properties through C-2 SAR in three sub-series. Bioorg Med Chem Lett. 2010;20:1825–1829. doi: 10.1016/j.bmcl.2010.02.003. [DOI] [PubMed] [Google Scholar]
- 17.Study to evaluate the safety, tolerability and pharmacokinetics of MK-3281 in healthy and hepatitis C infected male patients ( NCT00635804) National Institute of Health; Bethesda, MD, USA: 2008. http://clinicaltrials.gov/ct2/show/NCT00635804. [Google Scholar]
- 18.Boumendjel A. Aurones: A subclass of flavones with promising biological potential. Curr Med Chem. 2003;10:2621–2630. doi: 10.2174/0929867033456468. [DOI] [PubMed] [Google Scholar]
- 19.Seikel MK, Geissman TA. Anthochlor pigments. VII. The pigments of yellow Antirrhinum Majus . J Am Chem Soc. 1950;72:5725–5730. [Google Scholar]
- 20.Nakayama T, Yonekura-Sakakibara K, Sato T, Kikuchi S, Fukui Y, Fukuchi-Mizutani M, Ueda T, Nakao M, Tanaka Y. Aureusidin synthase: A polyphenol oxidase homolog responsible for flower coloration. Science. 2000;290:1163–1166. doi: 10.1126/science.290.5494.1163. [DOI] [PubMed] [Google Scholar]
- 21.Varma S, Varma M. Alumina-mediated condensation. A simple synthesis of aurones. Tetrahedron Lett. 1992;33:5937–5940. [Google Scholar]
- 22.Beney C, Mariotte AM, Boumendjel A. An efficient synthesis of 4,6-dimethoxy aurones. Heterocycles. 2001;55:967–972. [Google Scholar]
- 23.Büchi G, Weinreb M. Total syntheses of aflatoxins M1 and G1 and an improved synthesis of aflatoxin B1 . J Am Chem Soc. 1971;93:746–752. doi: 10.1021/ja00732a032. [DOI] [PubMed] [Google Scholar]
- 24.Okombi S, Rival D, Bonnet S, Mariotte AM, Perrier E, Boumendjel A. Discovery of benzylidenebenzofuran-3(2H)-one (aurones) as inhibitors of tyrosinase derived from human melanocytes. J Med Chem. 2006;49:329–333. doi: 10.1021/jm050715i. [DOI] [PubMed] [Google Scholar]
- 25.Brehm C, Zimmermann PJ, Zemolka S, Palmer A, Buhr W, Postius S, Simon W-A, Herrmann M. International Patent Application WO 2008/084067. Pharmaceutically active dihydrobenzofurane-substituted benzimidazole derivatives. 2008 Jul 17;
- 26.Brehm C, Zimmermann PJ, Zemolka S, Palmer A, Buhr W, Postius S, Simon W-A, Herrmann M. International Patent Application WO 2008/084067. Pharmaceutically active dihydrobenzofurane-substituted benzimidazole derivatives. 2008 Jul 17;
- 27.Kodra JT, Madsen P, Lau J, Jorgensen AS, Christensen IT. International Patent Application WO 2003/048109. Novel glucagon antagonists. 2003 Jun 12;
- 28.Ahmed-Belkacem A, Ahnou N, Barbotte L, Wychowski C, Pallier C, Brillet R, Pohl RT, Pawlotsky JM. Silibinin and related compounds are direct inhibitors of hepatitis C virus RNA-dependant RNA polymerase. Gastroenterology. 2010;138:1112–1122. doi: 10.1053/j.gastro.2009.11.053. [DOI] [PubMed] [Google Scholar]
- 29.Ikegashira K, Oka T, Hirashima S, Noji S, Yamanaka H, Hara Y, Adachi T, Tsuruha JI, Doi S, Hase Y, Noguchi T, Ando I, Ogura N, Ikeda S, Hashimoto H. Discovery of conformationally constrained tetracyclic compounds as potent hepatitis C virus NS5B RNA polymerase inhibitors. J Med Chem. 2006;49:6950–6953. doi: 10.1021/jm0610245. [DOI] [PubMed] [Google Scholar]
- 30.Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Krausslich HG, Mizokami M, Bartenschlager R, Liang TJ. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med. 2005;11:791–796. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]