

Abstract
Sialyl Lewis antigens are selectin ligands involved in leukocyte trafficking and cancer metastasis. Biosynthesis of these selectin ligands occurs by the sequential actions of several glycosyltransferases in the Golgi apparatus following synthesis of the protein backbone in the endoplasmic reticulum. In this study, we examine how the synthesis of sialyl Lewis a (sLea) is regulated in prostatic cells and identify a mucin that carries this glycotope. We treat human prostatic cells including one normal and three cancerous cells with histone deacetylase inhibitors, valproic acid, tricostatin A (TSA), and suberoylanilide hydroxamic acid (SAHA), and then monitor the expression of sLea. We have found that SAHA enhances the production of sLea in normal prostatic RWPE-1 cells but not prostatic cancer cells. Employing siRNA technology and co-immunoprecipitation, we show that the sLea is associated with MUC1, which is confirmed by confocal immunofluorescence microscopy and proximity ligation assay. The SAHA-induced production of sLea in RWPE-1 cells is resulted from upregulation of B3GALT1 gene via enhancement of acetylated histone-3 and histone-4. Interestingly, PC3 and LNCaP C-81 cells do not produce detectable amounts of sLea despite expressing high levels of B3GALT1. However, the MUC1-associated sLea is generated in these cells after introduction of MUC1 cDNA. We conclude that the synthesis of sLea is controlled by not only peptide backbone of the glycoprotein but also glycoprotein-specific glycosyltransferases involved in the synthesis of sLea. Further, the SAHA induction of this selectin ligand in normal prostatic cells may pose a potentially serious side effect of this drug recently approved by the US Food and Drug Administration.
Introduction
Tumor metastasis is the primary cause of the mortality of cancer patients. The tumor invasion and metastasis properties acquired during cancer progression include increased invasion of surrounding tissues, escape from primary site, and establishment of tumors at distant sites. This process is driven by different families of adhesion molecules including integrins, members of the immunoglobulin superfamily, selectins and carbohydrate ligands, such as sialyl Lewis x (sLex) and sialyl Lewis a (sLea) [1]. SLex, NeuAcα2,3Galβ1,4(Fucα1,3)GlcNAcβ1→R, is a carbohydrate antigen expressed on neutrophils, monocytes, certain T lymphocytes, and advanced cancers, and plays a key role in leukocyte trafficking and cancer metastasis [2], [3]. This antigen has been used as a diagnosis and prognosis marker for cancer [4], [5], [6]. Similar to sLex, sLea, NeuAcα2,3Galβ1,3(Fucα1,4)GlcNAc→R, also known as CA 19.9, is widely expressed on tumors in the gastrointestinal tract and has been used as a marker for pancreatic and colon cancer [7], [8]. SLea is also a ligand for endothelial leukocyte adhesion molecule and is associated with metastasis [9], [10], [11] of human colon cancer [12], [13] and pancreatic adenocarcinoma [14]. Both sialyl Lewis antigens are found on various glycoproteins and mucins, including MUC1, which serve as selectin ligands to mediate leukocyte adhesion and hematogenous metastasis of cancer cells [15], [16], [17]. Biosynthesis of these ligands occurs by sequential actions of several glycosyltransferases with the final reactions completed by α2,3-sialyltransferases and then α1,3/1,4-fucosyltransferases [18]. Four α2,3-sialyltransferases (ST3GAL3-6) [19], [20] and four α3/4 fucosyltransferases (α3/4 FUT3-5 and -7) [21] can act on type I (Galβ1,3GlcNAcβ1-R) structure to generate sLea and on type II (Galβ1,4GlcNAcβ1-R) structure to produce sLex oligosaccharides despite different degrees of preference for their glycan substrates. The biosynthetic pathway for core 2-associated sLea is shown in figure 1. The major difference between sLea and sLex lies in the fucose linked to terminal GlcNAc through α1-4 linkage in sLea and α1-3 in sLex on type-1 and type-2 backbone structures, respectively. The type-1 backbone structure is synthesized by β1,3-galactosyltransferases (B3GALTs) while type-2 by β1,4-galactosyltransferases (B4GALTs).
Figure 1. Biosynthetic pathway of nucin core 2-associated sialyl Lewis a (sLea) antigen.

Biosynthesis of mucin O-glycans is initiated by the addition of GalNAc to ser or thr of the peptide as catalyzed by polypeptide N-acetylgalactosaminyltransferases (GALNTs). This is followed by the addition of Gal in β1-3 linkage to GalNAc as catalyzed by C1GALT1 enzyme and produces core-1. Core 2 is generated by adding GlcNAc in β1-6 linkage to GalNAc by GCNTs. To synthesize sLea, Gal is transferred to GlcNAc in β1-3 linkage to form type 1 chain as catalyzed by B3GALTs, followed by the addition of α2-3NeuAc to Gal as catalyzed by ST3GALTs and then α4-fucose (Fuc) to GlcNAc as catalyzed by α3/4FUTs.
The expression of glycosyltransferase genes can be regulated epigenetically. Epigenetics is a gene regulation mechanism without involvement of alterations in the DNA sequence [22]. The epigenetic changes such as DNA methylation and histone modification [23] have been shown to contribute to the development of malignancy-associated phenotypes such as growth, invasion, metastasis, or angiogenesis. DNA methylation is involved in the silencing of many glycosyltransferase genes, including heparin sulfate 3-O-sulfotransferase, 3-OST-2 [24], ABO [25], B4GALNT2 [26], and ST3GAL6 [19]. Histone modification also is involved in the silencing of many genes in colon cancer [27]. These results and our previous studies demonstrating the upregulation of sLex upon sodium butyrate or 5-Aza-2’-deoxycytidine (5-Aza-dC) treatment [19], [28] prompted us to examine the effect of other potential chemotherapeutic agents on the biosynthesis of sLea in prostatic cells. We treated immortalized normal and cancerous prostatic cells with various histone deacetylase inhibitors (HDACi) [29], including valproic acid, tricostatin A (TSA), and suberoylanilide hydroxamic acid (SAHA), and then monitored changes of sLea antigen. We found that normal prostatic cells were the only cells that were induced to produce sLea and SAHA was the only inhibitor that was capable of doing that. We showed that sLea was associated with MUC1, which was resulted from enhancement of B3GALT1 gene expression through elevated levels of acetylated histone-3 (H3) and histone-4 (H4). We also found that PC3 and LNCaP C-81 cells were induced to produce MUC1-associated sLea after MUC1 cDNA was delivered to these cells. The results indicate that the ability of different prostatic cells to produce MUC1-associated sLea is controlled by the expression of either MUC1 or one of the glycosyltransferase genes involved in the synthesis of sLea.
Materials and Methods
Materials
Keratinocyte serum-free medium (KSFM), bovine pituitary extract (BPE), epidermal growth factor (EGF) supplement, RPMI, penicillin, streptomycin, fetal bovine serum (FBS) and trypsin/EDTA were procured from Life Technologies Inc. (Grand Island, NY). Acrylamide and protein estimation kit were purchased from Bio-Rad (Hercules, CA). A mixture of pre-stained protein molecular weight standard marker was from Fermentas (Glen Burnie, MD). ECL reagent kit was obtained from Pierce Biotechnology (Rockford, IL). HDACi valproic acid and SAHA were from Cayman Chemical Company (Ann Arbar, MI) while TSA was obtained from Sigma (St. Louis, MO). Nuclear stain DAPI was from Life Technologies Inc. (Grand Island, NY). KM231 mouse monoclonal Abs to sLea was from EMD Millipore Corporation (Billerica, MA). Mouse monoclonal Abs to β-actin was from Sigma (St. Louis, MO). VU-4H5 mouse monoclonal Ab to MUC1 was obtained from Life Span Biosciences (Seattle, WA) and rabbit polyclonal Abs to MUC1 was from Abcam (Cambridge, MA). Abs to acetylated-H3, acetylated-H4 and for total histone proteins were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Abs against acetylated-H2A and H2B were purchased from Cell Signaling Technology (Beverly, MA). Horseradish peroxidase, DyLight® 488 (green) and DyLight® 595 (red) conjugated secondary antibodies (donkey anti-rabbit, donkey anti-mouse) were obtained from Jackson ImmunoResearch (West Grove, PA).
Methods
Cell cultures and HDACi treatment
Immortalized normal human prostatic RWPE-1 cells were obtained from ATCC and grown in keratinocyte serum-free medium containing 0.05 mg/ml bovine pituitary extract and 5 ng/ml EGF. LNCaP C-81 cells derived from left supraclavicular lymph node metastasis were obtained from Dr. Ming-Fong Lin, prostatic cancer PC3 cells derived from bone metastasis (stage IV) and DU145 derived from brain metastasis were obtained from ATCC. These cells were cultured at 37 °C under 5% CO2 in RPMI-1640 medium containing 10% FBS and 100 U/ml penicillin and 100 µg streptomycin. For drug treatment, stock solutions of SAHA (1 mM) and valproic acid (1 M) were prepared fresh in sterilized PBS and TSA (50 µM) in DMSO within 1 h of the treatment. Final concentrations of HDACi were prepared by adding an appropriate amount of the stock solution directly to the culture medium. Drug and PBS (control) treatments of the cells were initiated at 50% confluence and then continued for 72 h.
Western blotting
Aliquots of the cell lysates from PBS or HDACi-treated cells were boiled for 5 min and then run on 4% SDS-PAGE (7.5×8.5 cm) under reducing conditions. After electrophoresis, the separated proteins were electro-transferred onto a PVDF membrane (Immobilon-P, 0.2 µ, Millipore, Bedford, MA), which was then blocked with tris-buffered saline containing 0.001% Tween 20 (TBST) and 5% (w/v) non-fat dried milk for 60 min. The membranes were then incubated with anti-sLea (KM231) (1∶500) or anti-MUC1 (VU-4H5) (1∶1,000) Abs overnight at 4 °C in same buffer. After five washings in same buffer, the membranes were treated for 1 h at room temperature (r.t.) with HRP-conjugated donkey anti-mouse IgG. The membranes were then washed five times (5 min each) with TBST and once with milli-Q water. Then, the blots were developed with ECL-sensitive film. For β-actin and acetylation analysis, proteins were separated on 10 and 15% SDS-PAGE, respectively and probed with respective primary and secondary Abs.
Immunoprecipitation (IP)
To isolate glycoproteins containing sLea ligand for characterization of conjugated glycans, cells were lysed in IP lysis buffer (Boston BioProducts, Worcester, MA) and precleared with protein G-agarose (Pierce). The precleared cell lysate (500 µg) was incubated with 5 µg of mouse anti-sLea (or anti-MUC1) mAb for 12 h at 4 °C. The immuno-complexes isolated with protein G-agarose for 1 h at 4 °C were analyzed for sLea by western blotting. The immunoprecipitates obtained with non-specific mouse IgG antibodies served as the negative control. Protein concentration was measured by Coomassie blue dye (Pierce).
Mixed glycosidases or Peptide-N-glycosidase (PNGase) F treatments
The anti-sLea immunoprecipitate was boiled (100 °C) for 10 min in 0.5% SDS and 1% β-mercaptoethanol and then treated with PNGase F (Sigma, USA) (5 U/mg protein) in 50 mM phosphate, pH 7.5 containing 1% NP-40 at 37 °C for 16 h. The immunoprecipitate was also treated with mixed glycosidases including neuraminidase (Glyko®; GK80040), β(1-4) galactosidase/β-N-acetylglucosaminidase (PRO-LINK Extender; GK80115), and O-glycanase (Glyko®; GK 80110) according to the manufacturer's instruction. The samples were analyzed by anti-sLea western blotting before and after treatment with glycosidases.
Transient transfection of RWPE-1 cells with MUC1 or B3GALT1 siRNA
Pool of three different siRNAs targeting B3GALT1 (sc-105001; SantaCrutz Biotechnology), one custom synthesized siRNA targeting MUC1 RNA and scrambled siRNA were purchased from Dharmacon (Waltman, MA) (See Table S1 for the nucleotide sequences of all three siRNAs). After treatment with 5 µM SAHA for 12 h, RWPE-1 cells at 50% confluence were transfected for 8 h in OPTI-MEM® reduced serum medium containing 100 nM siRNA and Lipofectamine RNAi MAX reagent (Life Technologies). The medium was then replaced with a fresh RPMI medium containing 10% FBS and 5 µM SAHA. After cultured for 52 h (total 72 h), the transfected cells were analyzed for MUC1 and B3GALT1 mRNA by quantitative PCR and sLea by western blotting as described above.
Confocal immunofluorescence microscopy
RWPE-1 cells were grown on cover slips in either presence or absence of 5 µM SAHA for 72 h. The cells were then fixed in 4% paraformaldehyde/PBS at r. t. for 30 min, washed thrice with PBS and blocked for 1 h in 3% normal donkey serum. After treated with mouse anti-sLea and rabbit anti-MUC1 Abs (1∶100) at 37 °C for 1 h, the cells were stained with DyLight 488 conjugated donkey anti-mouse Ab (green), and DyLight 594 conjugated donkey anti-rabbit Ab (red) (1∶200) and mounted in ProLong Gold antifade reagent with DAPI nuclear stain. Stained cells were examined on a Zeiss 510 Meta Confocal Laser Scanning Microscope and analyzed using Zeiss 510 software.
Flow cytometric analysis
Cell surface expression of sLea was quantitatively assessed by flow cytometry using anti-sLea antibodies [19]. Briefly, SAHA or PBS-treated RWPE-1 cells were dislodged, washed with PBS and washing buffer. The cells (0.2×106) were blocked for non-specific staining in blocking buffer as described above and then incubated with anti-sLea Abs (10 µg/ml in washing buffer) on ice for 1 h. After thorough washing, secondary Ab staining was performed with DyLight 488 conjugated donkey anti-mouse IgG (1∶200) for 30 min on ice and excessive Abs were washed with washing buffer. Finally, cells were resuspended in 500 µl of PBS containing 0.5% paraformaldehyde and analyzed using a FACS Vantage (Becton Dickson, San Jose, CA) equipped with 488 nm argon laser and with Cell Quest-pro software. The unstained cells and cells incubated with secondary Ab alone served as negative and Ab controls, respectively.
In situ Proximity Ligation Assay (PLA)
In situ PLA was performed in RWPE-1 and PC3 cells treated with PBS or SAHA. These two cells were grown on cover slip in the presence or absence of 5 µM SAHA for 72 h. The cells were then fixed in 4% paraformaldehyde/PBS at r. t. for 30 min, washed thrice with PBS and blocked for 1 h in 3% BSA. After treatment with mouse anti-sLea and rabbit anti-MUC1 Abs (1∶100 in PBS with 3% BSA) at 37 °C for 1 h, cells were washed thrice (5 min each) with PBST. Oligonucleotide-conjugated anti-mouse minus and anti-rabbit plus PLA secondary probes were added at appropriate dilutions prepared in PBS with 3% BSA and the cells were incubated in a humidified chamber for 1 h at 37 °C. The PLA assay was performed using the Duolink PLA kit (Olink Bioscience cat# LNK-92101-KI01, Uppsala, Sweden) according to the manufacture's instruction. Briefly, connector oligonucleotides were hybridized and circularized by ligation for 30 min at 37 °C. After thorough washing, the cells were incubated with DNA polymerase for 100 min at 37 °C to produce rolling circle amplification products tagged with a red fluorescence probe. The nuclei were counterstained with DAPI, and the PLA signals were visualized at 60 x magnifications under Zeiss 510 Meta Confocal Laser Scanning Microscope equipped with DAPI/Texas Red filters and analyzed using Zeiss 510 software.
Quantitative real-time PCR analysis of glycosyltransferase and mucin genes
Quantitative real-time PCR analyses of various glycogenes were performed as described earlier [19]. In brief, all four cell lines cultured in T-25 flask were lysed with 1 ml TRI-REAGENT (Molecular Research Center, Inc, Cincinnati, OH) and mRNA was isolated according to the manufacturer's instruction. For cDNA synthesis, 2 µg RNA was used in a 20 µl reaction mixture using a Verso reverse transcriptase kit (Thermo scientific) as follows: 5 min at r.t., 60 min at 42 °C, and 2 min at 95 °C. Quantitative real-time PCR was performed in 10 µl reaction volume in a 96-well plate using 2 µl of diluted cDNA with SYBR® Premix Ex Taq™ (TAKARA BIO INC.) on a Mastercycler Epgradient realplex (Eppendorf AG, Hamburg, Germany). The PCR conditions included 1 cycle at 95 °C for 2 min followed by 45 cycles at 95 °C for 15 s, 60 °C for 15 s, and 72 °C for 45 s. The data were analyzed using Eppendorf realplex software, version 1.5 (Eppendorf). The amounts of various glycogene transcripts were normalized to the amount of GAPDH transcript in same cDNA sample. Relative fold differences in transcript expression of each target gene were determined using the comparative CT method: 2-[ΔCt (SAHA)-ΔCt (PBS)] = 2-ΔΔCt, where ΔCt (in PBS or SAHA-treated sample) = Ct (in PBS or SAHA-treated sample)-Ct (GAPDH in PBS or SAHA-treated sample) as described by Tassone et al. [30]. The results were expressed as % of the target gene relative to that (100%) of GAPDH and plotted as mean fold changes ± standard error of the mean (SEM). Primer sequences used for expression analysis of all genes including GAPDH are summarized in Table S2.
Transient transfection of PC3 and LNCaP C-81 cells with MUC1 cDNA
Full length MUC1 cDNA containing N-terminal FLAG tag was prepared as previously described [31], [32]. Transient transfections of PC3 and LNCaP C-81 cells were carried out using the Lipofectamine 2000 (Life science technologies) following the manufacturer's protocol. Briefly, 1×106 cells/well in 6 well culture plates were incubated with 10 µg of MUC1F cDNA for 8 h in OPTI-MEM medium containing 15 µl of Lipofectamine 2000. After 8 h, the medium was replaced with complete PRMI containing 10% FBS and antibiotics. Cells were cultured for 72 h and harvested for western blot analysis.
Statistical analysis
All statistical analyses were carried out using the Student's t-test. SigmaPlot software (Systat Software, San Jose, CA) was used for all the graphing and statistical analyses. Data are expressed as mean ± SEM. p < 0.05 was considered statistically significant.
Results
SAHA treatment enhances the synthesis of sLea in RWPE-1 cells
To study the effect of histone acetylation on the synthesis of sLea, a ligand that mediates hematogenous metastasis of cancer cells, we treated three different prostatic cancer cell lines and one immortalized normal prostatic epithelial cell line with three different HDACi, valproic acid, SAHA, and TSA. We found that SAHA was the only one that enhanced the production of sLea in normal RWPE-1 cells (Fig. 2A). SLea was found to be associated primarily with a 250 kDa band. The SAHA effect was dose dependent (Fig. 2B). Because 5 µM SAHA was the lowest concentration that did not exhibit apparent cytotoxicity, it was used for subsequent experiments. It was noted that valproic acid showed no effect on all these cell lines tested. On the other hand, TSA inhibited the production of sLea in DU145 cells but did not show any effect on other cell lines. The cause for this inhibition is not known and will be a subject of future investigation.
Figure 2. Effects of HDACi treatment on production of sLea in one normal and three cancerous prostatic cells.

(A) RWPE-1, PC3, DU145, and LNCaP C-81 cells were treated with 1 mM valproic acid, 20 µM SAHA or 250 nM TSA for 72 h followed by western analysis of the cell lysates for sLea using KM231 Abs. SAHA was the only HDACi which induces marked production of sLea in RWPE-1 cells. (B) Dose-dependent (0-20 µM) effect of SAHA on the production of sLea in RWPE-1 cells. Minimum concentration of SAHA required to induce the production of sLea without causing cytotoxicity is 5 µM. β-actin was used as a loading control on 10% gels and probed with anti-β-actin Abs. Experiments were performed (3x) with similar results.
SLea is associated with O-glycans on MUC1 in RWPE-1 cells
To determine the nature of the SAHA-induced sLea in RWPE-1 cells, we treated the anti-sLea Ab pull-down with mixed glycosidases or N-glycanase. As shown in Fig. 3A, mixed glycosidases removed sLea epitope while N-glycanase did not, suggesting that the sLea is associated with O- and not N-glycans. Because the size of the sLea band (250 kDa) in the immunoprecipitate resembles that of MUC1, we predicted that the sLea might be associated with MUC1. This prediction was confirmed by the detection of sLea in anti-MUC1 Ab pull-down (Fig. 3A). Further, the sLea signal in SAHA-treated cells was greatly reduced after the MUC1 mRNA had been knocked down by 86.6 ± 2.2% (Fig. 3B &C) confirming the above prediction. The results suggest that the sLea induced by SAHA treatment in RWPE-1 cells is associated with O-glycans on MUC1.
Figure 3. SLea induced by SAHA in RWPE-1 cells is associated with O-glycans of MUC1.

Anti-sLea immunoprecipitates from the lysates of RWPE-1 cells treated with SAHA were digested with either mixed glycosidases (to remove sialic acid and O-glycans) or N-glycanase (which removes N-glycans) prior to western analysis for sLea. (A) SLea band in mixed glycosidase-treated samples were completely abolished while sLea staining was retained after N-glycanase treatment. SLea band was also detected in anti-MUC1 immunoprecipitate (MUC1 IP), indicating that sLea is associated with MUC1. IgG lane served as a background control. (B) Anti-sLea western blot of the lysates of cells treated with PBS, SAHA, SAHA plus scrambled RNAi, or SAHA plus MUC1 RNAi. (C) Relative expression level of MUC1 gene was determined according to ΔCt method (see materials and methods) after normalization with GAPDH ( = 100%) and then expressed as % of MUC1 mRNA in the control ( = 100%) (n = 3).
The SAHA-induced sLea is on the surface of RWPE-1 cells
By confocal immunofluorescence microscopy, we found that MUC1 co-localized with sLea induced by SAHA at the plasma membrane of RWPE-1 cells (Fig. 4A), validating the observation that MUC1 is the carrier of sLea. These results were further confirmed by flow cytometric analysis performed on RWPE-1 cells with or without SAHA treatment. SAHA treatment increased the mean fluorescence intensity, an arbitrary unit for measuring the fluorescence intensity, from 3 in PBS-untreated cells to 132.2 ± 11 in SAHA-treated cells (Fig. 4B). The result is consistent with the acquisition of MUC1-associated sLea in SAHA-treated cells. Further, SAHA treatment increased the % of sLea-positive cells from <1 to 82 ± 6% (Fig. 4C).
Figure 4. Confocal and flow cytometric analysis of RWPE-1 cells.

(A) Confocal immunofluorescence images of MUC1 (red) and sLea (green) on RWPE-1 cells treated with PBS (control) or SAHA (5 µM) for 72 h. SLea induced by SAHA is colocalized with MUC1. (B) SLea mean fluorescence intensity in RWPE-1 cells treated with PBS or SAHA as measured by flow cytometry analysis. (p < 0.05) (C) % of sLea-positive cells in PBS- or SAHA-treated cells. The data expressed as mean ± SEM were obtained from three independent experiments. (p < 0.05) Bar = 20 µm.
In situ proximity ligation assay (PLA) of the colocalization of MUC1 and sLea
The in situ PLA was performed to show that the newly synthesized sLea induced by SAHA is associated with MUC1. As shown in Fig. 5, after SAHA treatment, RWPE-1 cells produced sLea on MUC1 as shown by positive red dots in these cells while no red dots were detected in PBS-treated control cells. Similarly, as expected, both PBS-treated and SAHA-treated PC3 cells did not show any red dots. The SAHA-treated cells exposed to PLA probe, mouse anti-sLea Ab, or rabbit anti-MUC1 Ab, which served as negative controls, did not show any red dots, indicating clean background signals (Fig. S1).
Figure 5. Proximity ligation assay of MUC1 and sLea in RWPE-1 and PC3 cells as analyzed by confocal microscopy.

RWPE-1 and PC3 cells grown on cover slips were treated with PBS or SAHA and then fixed in paraformaldehyde. These cells were processed for PLA for MUC1 and sLea using an Olink Bioscience PLA kit. Briefly, these cells were treated with mouse anti-sLea and rabbit anti-MUC1 antibodies, and then with oligonucleotide-conjugated anti-mouse minus and anti-rabbit plus proximity ligation assay secondary probes. The oligonucleotides conjugated with the plus and minus antibodies were used to generate circular DNA, which was then amplified and tagged with a red fluorescence dye. Following DAPI staining of the nuclei, the cells were examined by confocal fluorescence microscopy. The expression as well as association of sLea with MUC1, significantly increased only in SAHA treated RWPE-1 cells as indicated by red foci all over the cells. Bar = 20 µm.
Enhanced B3GALT1 gene expression is responsible for the synthesis of sLea in RWPE-1 cells treated with SAHA
To examine how SAHA treatment enhanced the synthesis of MUC1-associated sLea in RWPE-1 cells but did not affect the expression of sLea in the prostatic cancer cells, we performed real time PCR analyses to determine the expression levels of MUC1 gene and glycosyltransferase genes involved in the synthesis of sLea in these cells treated with PBS or SAHA. As shown in Fig. S2 & S3, the glycogenes we analyzed include C1GALT1, three GCNTs, three ST3GALs, six FUTs, four B3GALTs, fourteen GALNTs and four membrane-bound MUCs. We found that the expression level of the B3GALT1 gene was increased 26.6-fold from 0.006% to 0.173% of the expression level of GAPDH gene (Fig. 6), suggesting that this is the key enzyme involved in the synthesis of sLea induced by SAHA in RWPE-1 cells. The basal expression level (0.09% of GAPDH) of MUC1 gene was much higher than the expression levels (< 0.0007% of GAPDH) of the other three MUC genes (MUC4, MUC16 and MUC17)(Fig. S2). SAHA treatment did not show an appreciable effect on the expression of all these MUC genes. Although MUC1 gene was not enhanced in the SAHA-treated RWPE-1 cells, the basal expression level of this gene was sufficiently high (0.09% of GAPDH) that it was not considered a limiting factor for the synthesis of MUC1-associated sLea in these cells. However, in the prostatic cancer cells, the basal expression level of the B3GALT1 gene was from moderate (0.05% of GAPDH) in DU145 cells to very high in the other two prostatic cancer cells, i.e. 0.324% of GAPDH in PC3 cells and 0.334% of GAPDH in LNCaP C-81 cells (Fig. 6), suggesting that this enzyme is not a limiting factor for the synthesis of sLea in these cells. Further, the MUC1 gene expression level was very high in DU145 cells, i.e. 1.27% of GAPDH, and very low in PC3 cells, i.e. 0.0002% of GAPDH, and LNCaP C-81 cells, i.e. 0.008%, suggesting that the low expression level of MUC1 gene is a limiting factor for the synthesis of sLea in these two prostatic cancer cells.
Figure 6. SAHA treatment stimulates the expression of B3GALT1 gene in RWPE-1 cells.

Quantitative real-time PCR analysis was carried out on RWPE-1, PC3, DU145 and LNCaP C-81 cells treated with PBS or 5 µM SAHA for 72 h. Relative expression levels of B3GALT1 and MUC1 genes were sorted according to ΔCt (see Materials and Methods) method, normalized with GAPDH in same cell preparation and expressed as fold changes ± SEM determined by calculating the ratio of the expression level of each gene in SAHA-treated vs. that in PBS-treated control cells. Relative amount of each gene versus that of GAPDH (100%) in PBS-treated control cells was given in the parenthesis (n = 3). The numbers shown in parenthesis represent % basal expression level relative to GAPDH ( = 100%).
B3GALT1 gene participates in the synthesis of sLea on MUC1
To prove that B3GALT1 is involved in the synthesis of sLea on MUC1, we performed a B3GALT1 mRNA knockdown experiment. As shown in Fig. 7A & B, knockdown of the B3GALT1 mRNA by 77% greatly reduced the sLea signal. The reduction of sLea signal in B3GALT1 gene knocked down cells did not affect the amount of MUC1 (Fig. 7C), indicating that the reduction of sLea signal in these cells is due to reduction of the synthesis of this epitope and not reduction of MUC1 protein.
Figure 7. Knockdown of B3GALT1 mRNA reduces sLea in SAHA-treated RWPE-1 cells.

(A) Anti-sLea western blot of the lysates of control cells, SAHA plus scrambled siRNA, or SAHA plus B3GALT1 siRNA. (B) Knockdown of B3GALT1 gene in RWPE-1 cells was confirmed by 77% reduction of B3GALT1 mRNA measured by quantitative real-time PCR analysis. Relative expression level of B3GALT1 gene was determined according to ΔCt method (see materials and methods) after normalization with GAPDH ( = 100%) and then expressed as % of B3GALT1 mRNA relative to same in the control ( = 100%) (n = 3). (C) Anti-MUC1 western blot of the lysates of cells treated with SAHA, SAHA plus scrambled siRNA, or SAHA plus B3GALT1 siRNA. Knockdown of 77% of B3GALT1 mRNA reduces sLea in SAHA-treated RWPE-1 cells without affecting MUC1 glycoprotein. Experiments were performed (3x) with similar results.
Transfection of PC3 and LNCaP C-81 cells with MUC1 cDNA results in the production of sLea
To prove that the failure of PC3 and LNCaP C-81 cells to produce MUC1-associated sLea is the result of the lack of MUC1 expression, we monitored MUC1 as well as sLea generated in these cells after transfection of both cells with a plasmid containing MUC1 cDNA expressing a FLAG tag as described previously [31]. As shown in Fig. 8, sLea epitope was detected in a 250 kDa band and a 130 kDa band in MUC1 cDNA transfected PC3 cells (Fig. 8A & B). This 130 kDa band likely represents a truncated MUC1. Similar expression of sLea was also observed in LNCaP C-81 cells after introduction of MUC1 cDNA (Fig. 8C). The results indicate that the low level of MUC1 gene expression is the limiting factor for the synthesis of MUC1-associated sLea in PC3 and LNCaP C-81 cells under basal conditions.
Figure 8. Transfection of PC3 and LNCaP C-81 cells with MUC1 cDNA results in production of sLea.
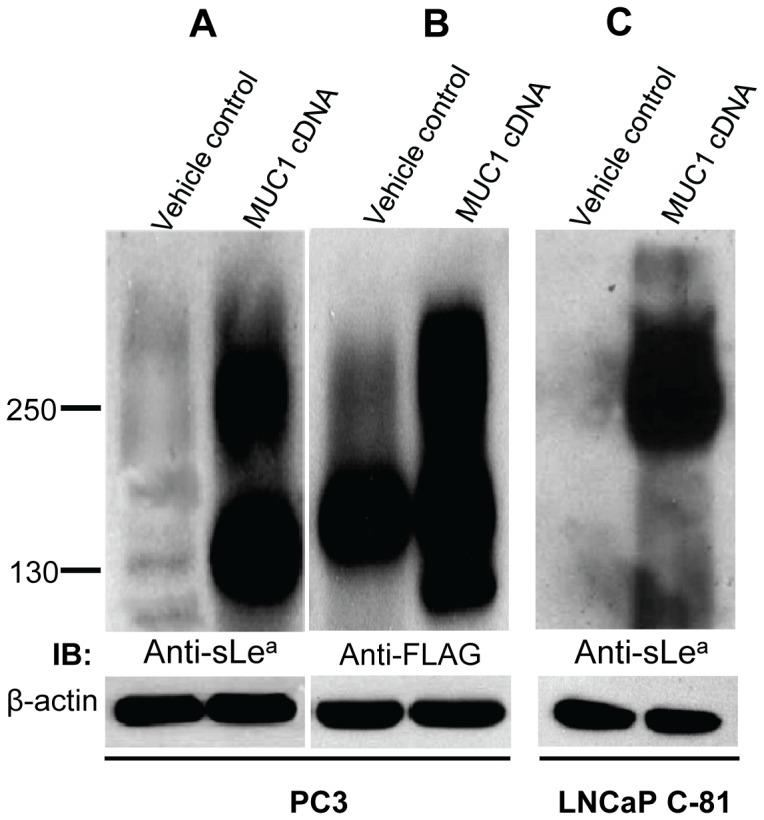
Full length MUC1 cDNA composed of tandem repeats and N-terminal FLAG epitope was used to transfect PC3 and LNCaP C-81 cells. After 72 h, cell lysates from both vehicle control and MUC1 cDNA-transfected cells were analyzed by sLea (KM231) western blotting. The MUC1 band corresponding to 250 kDa appeared after MUC1 cDNA transfection in both cell lines. The 130 kDa band in frame A probably represents truncated MUC1 in MUC1 cDNA-transfected cells. Antibodies did not detect any sLea band in vehicle-treated cells. For equal protein loading, β-actin was used and run on 12% SDS-PAGE.
H3 and H4 histone deacetylation is involved in the regulation of the expression of sLea in RWPE-1 cells
To assess the role of histone acetylation in the B3GALT1-mediated sLea synthesis, we compared the levels of acetylated histones in these prostatic cells treated with PBS or SAHA. As shown in Fig. 9, all these prostatic cells showed up-regulation of the levels of acetylated H3 histone although the basal level of acetylated H3 histone in RWPE-1 cells was lower than those of the other three prostatic cells. Similarly, the basal level of acetylated H4 histone in RWPE-1 cells was lower than those in other prostatic cells. But, SAHA treatment greatly up-regulated the levels of acetylated H4 histone in RWPE-1 cells but not the other prostatic cells. However, we did not observe any increase in acetylation of H2A and H2B in RWPE-1 and DU145 cells but a small increase in the acetylation of H2A and H2B was observed in LNCaP C-81 and PC3 cells, respectively (Fig. S4). The results suggest that SAHA treatment inhibits histone deacetylase in RWPE-1 cells, which results in elevation of acetylated H3 and H4 histones, activation of the B3GALT1 gene, and then induction of sLea decorated on MUC1.
Figure 9. SAHA treatment of RWPE-1 cells increases acetylated H3 and H4 histones.

Cell lysates were prepared from RWPE-1, PC3, LNCaP C-81 and DU145 cells treated with PBS or 5 µM SAHA for 72 h. Proteins (100 µg) were separated on 15% SDS-PAGE and blotted onto a PVDF membrane. The acetylated histone-3 and histone-4 proteins were detected with H3 and H4-specific antibodies along with total histone protein antibodies. The β-actin from same samples was used as a protein loading control.
Discussion
SLea and sLex are carbohydrate antigens serving as ligands for selectins to mediate adhesion of cancer cells to the endothelium, thereby facilitating hematogenous metastasis [33], [34]. Synthesis of these antigens requires a tight coordination of several glycosyltransferases. Some of these glycosyltransferase genes are subject to epigenetic regulation. For example, we previously showed that the synthesis of MUC1-associated sLex in HCT15 colon cancer cells was controlled by methylation of the ST3GAL6 promoter [19]. The expression of many other glycosyltransferase genes is also under the control of DNA methylation [26], [35], [36], [37], [38], [39]. Apart from glycosyltransferases, many other glycogenes, including enzymes involved in the biosynthesis of sugar nucleotides [40], [41], galectins [42], and some transporters [27], also are regulated by epigenetics. Although these studies demonstrate epigenetic regulation of glycogenes and their phenotypes in cancer cells, no attempts have been made to investigate normal counterparts. Our study provides evidence that the sLea induced by SAHA decorates MUC1 as demonstrated by proximity ligation, confocal microscopy, and flow cytometry assays. Current study also shows that the synthesis of MUC1-associated sLea in human normal prostatic RWPE-1 cells is regulated by the B3GALT1 gene expression via histone acetylation.
Biosynthesis of a given mucin-associated glycan requires not only the mucin protein backbone but also all the glycosyltransferases needed for the synthesis of this glycan. Missing either the mucin protein backbone or any one of these glycosyltransferases would make the cells incapable of producing either this mucin at all or this mucin without the specific glycan, respectively. In our study, we show that one normal prostatic epithelial cell line can produce MUC1 protein and all the glycosyltransferases needed for the synthesis of sLea except B3GALT1. Activation of B3GALT1 gene with the HDACi SAHA makes it possible for the normal prostatic epithelial cells to generate MUC1 decorated with sLea. On the other hand, PC3 and LNCap C-81 cells which do not express sufficient amounts of MUC1 to allow the production of detectable amount of MUC1 to be decorated with sLea despite the presence of all the enzymes needed for the synthesis of this glycan. Providing MUC1 cDNA to PC3 and LNCaP C-81 cells makes it possible for these cells to produce sufficient amounts of MUC1 to be decorated with sLea. On the other hand, DU145 cells produce high levels of sLea irrespective of SAHA treatment as expected from high basal levels of both MUC1 and B3GALT1 enzyme. These results indicate that each cell type exhibits its own unique expression pattern of mucin and glycosyltransferase genes. Understanding the expression pattern of these genes in a given cell type would afford an opportunity for modulating the synthesis of a specific glycan decorated on a specific mucin in each cell type.
It is of interest to note that without SAHA treatment, RWPE-1 cells could not generate MUC1-associated sLea despite having a very high expression level of the B3GALT5 gene, i.e. 1.04% of GAPDH. This enzyme has been shown to be involved in the synthesis of sLea [43], [44] on CD43 [45] and carcinoembryonic antigen [46], but does not work on MUC1. The results support the idea that each glycosyltransferase has its own set of glycoprotein substrates because each Golgi glycosyltransferase is localized to a specific Golgi compartment according to the glycosylation step in which it participates and also, each glycoprotein has its unique transport path as it is moved from cis to trans-Golgi. Therefore, only the glycoproteins that come in contact with specific glycosyltransferases as they travel through the Golgi apparatus can be decorated with the carbohydrate structures specified by these glycosyltransferases. The important implication of this glycosylation mechanism is that one glycosyltransferase isozyme may not substitute another isozyme even though they show similar substrate specificity when measured in vitro.
A recent report showed that the expression of MUC1 gene in prostatic cancer cells is inversely regulated by the androgen receptor (AR) in a dose-dependent manner [47]. A similar study also demonstrated that high levels of AR inhibited the production of MUC1 and AR-negative prostatic cancer cells expressed high level of MUC1 [48]. This study showed that AR suppressed the expression of MUC1 gene by inhibiting MUC1 promoter and reducing the translation of MUC1 messages by miR-125b induced by AR. However, this study focused only on cancer cells and not non-cancerous prostatic RWPE-1 cells. In our study, we found that RWPE-1 cells, an AR positive cell line, expressed MUC1, a phenomenon contrary to the proposed mode of action of AR in prostatic cancer cells. In addition, the report of Singh et al. [49] also confirms that RWPE-1 cells express MUC1. Our results indicate that the proposed mechanisms for the AR inhibition of MUC1 [48] do not apply to normal prostatic RWPE-1 cells. The mechanism of the failure of AR to inhibit MUC1 production in normal prostatic cells remains to be elucidated.
Histone acetylation and DNA methylation are two well established epigenetic modifications that are targeted by drugs known as HDACi and DNA methyltransferase inhibitors [23], respectively. Numerous HDACi have been developed and many of them have been tested in preclinical and early clinical studies [50]. Several structural classes of HDACi have been identified including short chain fatty acids (butyrate and valproic acid), hydroxamic acids (TSA and SAHA), cyclic tetrapeptides (trapoxin A and apicidin) and benzamides (MS-27-275). Both hydrophobic backbone and carboxyl group of valproic acid play key roles in the inhibition of HDAC [51]. On the other hand, the hydroxamate inhibitors have three common structural characteristics: a zinc binding moiety, an opposite capping group, and a straight chain alkyl, vinyl or aryl linker connecting the two. These functional groups have been shown to interact with the conserved region of the active site of various HDACs to inhibit the activity [52], [53]. SAHA, one of the hydroxamate inhibitors, is non-selective [54] and believed to be a potent inhibitor of class I and class II HDAC enzymes [55]. In our study, we observed upregulation of sLea only in SAHA-treated cells whereas other drugs (valproic acid and TSA) were ineffective. Given that valproic acid is a specific inhibitor of class I HDACs [56], and SAHA and TSA inhibit both class I and II HDACs [55], the SAHA effect probably occurs through inhibition of class II HDACs. Further, TSA induces histone acetylation transiently [57], [58] while SAHA-induced acetylation of histone-3 is long-lasting [58], which could explain the SAHA-induced sLea in RWPE-1 cells is likely the result of sustained inhibition of class II HDACs. In addition, it should be mentioned that acetylation of H3 and H4 and not that of H2A and H2B histones is involved in the activation of B3GALT1 gene in these cells.
SAHA has been shown to inhibit the growth of LNCaP and PC3 cells and also shrink tumors and suppress their growth in mice transplanted with CWR22 human prostate tumor cells [59]. But, its effect on normal prostatic cells has never been reported until now. Our results showing that SAHA can induce the production of a metastasis-promoting selectin ligand sLea in normal prostatic cells is a cause of concern given that this drug has recently been approved by the US Food and Drug Administration for treating cutaneous T-cell lymphoma [60]. Should this observation be reproduced in additional normal prostatic cells, this side effect needs to be carefully monitored when this drug is used for cancer treatment.
Supporting Information
Negative control experiments for proximity ligation assay of MUC1 and sLea in SAHA-treated RWPE-1 cells. RWPE-1 cells treated with 5 µM SAHA for 72 h were exposed to (A) PLA probe only, (B) mouse anti-sLea Ab plus rabbit IgG, or (C) rabbit anti-MUC1 Ab plus mouse IgG and then examined by confocal fluorescence microscopy after PLA assay described in the materials and methods. Bar = 20 µm
(DOCX)
Quantitative real-time PCR analysis of membrane-bound mucin and glycosyltransferase genes. Quantitative real-time PCR analysis was carried out on RWPE-1, PC3, DU1-45 and LNCaP C-81 cells treated with PBS or 5 µM SAHA for 72 h. Relative expression levels of mRNA of different genes were sorted according to ΔCt (see Materials and Methods) method, normalized with GAPDH in same cell preparation and expressed as fold changes ± SEM and then determined by calculating the ratio of the expression level of each gene in SAHA treated vs. that in PBS-treated control cells. Relative amount of each gene versus that of GAPDH (100%) in PBS-treated control cells was given in the parenthesis (n = 3).
(DOCX)
Quantitative real-time PCR analysis of mucin Polypeptide N-acetylgalactosaminyltransferase (GALNT) mRNAs. Fourteen different GALNTs were analyzed by quantitative real-time PCR in RWPE-1 cells treated with PBS or 5 µM SAHA for 72 h. Relative expression level of each GALNT was calculated and plotted as described above.
(DOCX)
Effect of SAHA treatment on levels of acetylated H2A and H2B in RWPE-1 and prostatic cancer cells. Lysates were prepared from RWPE-1, PC3, LNCaP C-81 and DU145 cells treated with PBS or 5 µM SAHA for 72 h. Proteins (100 µg) were separated on 15% SDS-PAGE and blotted onto a PVDF membrane. The acetylated H2A and H2B proteins were detected with respective antibodies. The β-actin from same samples was used as a protein loading control.
(DOCX)
Short interfering RNA (siRNA) sequences.
(DOCX)
Oligonucleotide primers used for quantitative real-time PCR analysis.
(DOCX)
Acknowledgments
We thank Mrs. Helen Cheng for excellent technical assistance. We also thank Dr. Ming-Fong Lin for providing the LNCaP C-81 cells, Janice A. Taylor and James R. Talaska of the Confocal Laser Scanning Microscope Core Facility at the University of Nebraska Medical Center for providing assistance in confocal microscopy and the Nebraska Research Initiative, and the Eppley Cancer Center for their support of the Core Facilities.
Funding Statement
Department of Veteran Affairs 1I1BX000985; the National Institutes of Health 2RO1HL48282; and Nebraska LB506. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Kannagi R, Izawa M, Koike T, Miyazaki K, Kimura N (2004) Carbohydrate-mediated cell adhesion in cancer metastasis and angiogenesis. Cancer Sci 95: 377–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fukuda M (1996) Possible roles of tumor-associated carbohydrate antigens. Cancer Res 56: 2237–2244. [PubMed] [Google Scholar]
- 3. Kim YJ, Borsig L, Han HL, Varki NM, Varki A (1999) Distinct selectin ligands on colon carcinoma mucins can mediate pathological interactions among platelets, leukocytes, and endothelium. Am J Pathol 155: 461–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Paganuzzi M, Bobbio B, Marroni P, Filiberti R, Secco GB, et al. (2003) Prognostic role of serum sialyl Lewisx (CD15s) in colorectal cancer. Oncology 65: 52–59. [DOI] [PubMed] [Google Scholar]
- 5. Sumikura S, Ishigami S, Natsugoe S, Miyazono F, Tokuda K, et al. (2003) Disseminated cancer cells in the blood and expression of sialylated antigen in gastric cancer. Cancer Lett 200: 77–83. [DOI] [PubMed] [Google Scholar]
- 6. Wang QY, Wu SL, Chen JH, Liu F, Chen HL (2003) Expressions of Lewis antigens in human non-small cell pulmonary cancer and primary liver cancer with different pathological conditions. J Exp Clin Cancer Res 22: 431–440. [PubMed] [Google Scholar]
- 7. Kannagi R (2007) Carbohydrate antigen sialyl Lewis a--its pathophysiological significance and induction mechanism in cancer progression. Chang Gung Med J 30: 189–209. [PubMed] [Google Scholar]
- 8. Nakayama T, Watanabe M, Katsumata T, Teramoto T, Kitajima M (1995) Expression of sialyl Lewis(a) as a new prognostic factor for patients with advanced colorectal carcinoma. Cancer 75: 2051–2056. [DOI] [PubMed] [Google Scholar]
- 9. Ugorski M, Laskowska A (2002) Sialyl Lewis(a): a tumor-associated carbohydrate antigen involved in adhesion and metastatic potential of cancer cells. Acta Biochim Pol 49: 303–311. [PubMed] [Google Scholar]
- 10. Kawarada Y, Ishikura H, Kishimoto T, Kato H, Yano T, et al. (2000) The role of sialylated Lewis antigens on hematogenous metastases of human pancreas carcinoma cell lines in vivo. Pathol Res Pract 196: 259–263. [DOI] [PubMed] [Google Scholar]
- 11. Tomlinson J, Wang JL, Barsky SH, Lee MC, Bischoff J, et al. (2000) Human colon cancer cells express multiple glycoprotein ligands for E-selectin. Int J Oncol 16: 347–353. [DOI] [PubMed] [Google Scholar]
- 12. Matsui T, Kojima H, Suzuki H, Hamajima H, Nakazato H, et al. (2004) Sialyl Lewisa expression as a predictor of the prognosis of colon carcinoma patients in a prospective randomized clinical trial. Jpn J Clin Oncol 34: 588–593. [DOI] [PubMed] [Google Scholar]
- 13. Sato M, Narita T, Kimura N, Zenita K, Hashimoto T, et al. (1997) The association of sialyl Lewis(a) antigen with the metastatic potential of human colon cancer cells. Anticancer Res 17: 3505–3511. [PubMed] [Google Scholar]
- 14. Kishimoto T, Ishikura H, Kimura C, Takahashi T, Kato H, et al. (1996) Phenotypes correlating to metastatic properties of pancreas adenocarcinoma in vivo: the importance of surface sialyl Lewis(a) antigen. Int J Cancer 69: 290–294. [DOI] [PubMed] [Google Scholar]
- 15. Fernandez-Rodriguez J, Dwir O, Alon R, Hansson GC (2001) Tumor cell MUC1 and CD43 are glycosylated differently with sialyl-Lewis a and x epitopes and show variable interactions with E-selectin under physiological flow conditions. Glycoconj J 18: 925–930. [DOI] [PubMed] [Google Scholar]
- 16. Hey NA, Aplin JD (1996) Sialyl-Lewis x and Sialyl-Lewis a are associated with MUC1 in human endometrium. Glycoconj J 13: 769–779. [DOI] [PubMed] [Google Scholar]
- 17. Zhang K, Baeckstrom D, Brevinge H, Hansson GC (1997) Comparison of sialyl-Lewis a-carrying CD43 and MUC1 mucins secreted from a colon carcinoma cell line for E-selectin binding and inhibition of leukocyte adhesion. Tumour Biol 18: 175–187. [DOI] [PubMed] [Google Scholar]
- 18. Brockhausen I (2006) Mucin-type O-glycans in human colon and breast cancer: glycodynamics and functions. EMBO Rep 7: 599–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chachadi VB, Cheng H, Klinkebiel D, Christman JK, Cheng PW (2011) 5-Aza-2'-deoxycytidine increases sialyl Lewis X on MUC1 by stimulating beta-galactoside:alpha2,3-sialyltransferase 6 gene. Int J Biochem Cell Biol 43: 586–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ellies LG, Sperandio M, Underhill GH, Yousif J, Smith M, et al. (2002) Sialyltransferase specificity in selectin ligand formation. Blood 100: 3618–3625. [DOI] [PubMed] [Google Scholar]
- 21. Oriol R, Mollicone R, Cailleau A, Balanzino L, Breton C (1999) Divergent evolution of fucosyltransferase genes from vertebrates, invertebrates, and bacteria. Glycobiology 9: 323–334. [DOI] [PubMed] [Google Scholar]
- 22. Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144: 646–674. [DOI] [PubMed] [Google Scholar]
- 23. Feinberg AP (2004) The epigenetics of cancer etiology. Semin Cancer Biol 14: 427–432. [DOI] [PubMed] [Google Scholar]
- 24. Miyamoto K, Asada K, Fukutomi T, Okochi E, Yagi Y, et al. (2003) Methylation-associated silencing of heparan sulfate D-glucosaminyl 3-O-sulfotransferase-2 (3-OST-2) in human breast, colon, lung and pancreatic cancers. Oncogene 22: 274–280. [DOI] [PubMed] [Google Scholar]
- 25. Iwamoto S, Withers DA, Handa K, Hakomori S (1999) Deletion of A-antigen in a human cancer cell line is associated with reduced promoter activity of CBF/NF-Y binding region, and possibly with enhanced DNA methylation of A transferase promoter. Glycoconj J 16: 659–666. [DOI] [PubMed] [Google Scholar]
- 26. Kawamura YI, Toyota M, Kawashima R, Hagiwara T, Suzuki H, et al. (2008) DNA hypermethylation contributes to incomplete synthesis of carbohydrate determinants in gastrointestinal cancer. Gastroenterology 135: 142–151 e143. [DOI] [PubMed] [Google Scholar]
- 27. Yusa A, Miyazaki K, Kimura N, Izawa M, Kannagi R (2010) Epigenetic silencing of the sulfate transporter gene DTDST induces sialyl Lewisx expression and accelerates proliferation of colon cancer cells. Cancer Res 70: 4064–4073. [DOI] [PubMed] [Google Scholar]
- 28. Radhakrishnan P, Beum PV, Tan S, Cheng PW, Richon VM (2007) Butyrate induces sLex synthesis by stimulation of selective glycosyltransferase genes Cancer biology: mechanism of antitumour action of vorinostat (suberoylanilide hydroxamic acid), a novel histone deacetylase inhibitor. Biochem Biophys Res Commun 359: 457–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bolden JE, Peart MJ, Johnstone RW (2006) Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov 5: 769–784. [DOI] [PubMed] [Google Scholar]
- 30. Tassone F, Hagerman RJ, Taylor AK, Gane LW, Godfrey TE, et al. (2000) Elevated levels of FMR1 mRNA in carrier males: a new mechanism of involvement in the fragile-X syndrome. Am J Hum Genet 66: 6–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Beum PV, Singh J, Burdick M, Hollingsworth MA, Cheng PW (1999) Expression of core 2 beta-1,6-N-acetylglucosaminyltransferase in a human pancreatic cancer cell line results in altered expression of MUC1 tumor-associated epitopes. J Biol Chem 274: 24641–24648. [DOI] [PubMed] [Google Scholar]
- 32. Burdick MD, Harris A, Reid CJ, Iwamura T, Hollingsworth MA (1997) Oligosaccharides expressed on MUC1 produced by pancreatic and colon tumor cell lines. J Biol Chem 272: 24198–24202. [DOI] [PubMed] [Google Scholar]
- 33. Dube DH, Bertozzi CR (2005) Glycans in cancer and inflammation--potential for therapeutics and diagnostics. Nat Rev Drug Discov 4: 477–488. [DOI] [PubMed] [Google Scholar]
- 34. Hakomori S (1999) Antigen structure and genetic basis of histo-blood groups A, B and O: their changes associated with human cancer. Biochim Biophys Acta 1473: 247–266. [DOI] [PubMed] [Google Scholar]
- 35. Caretti A, Sirchia SM, Tabano S, Zulueta A, Dall'Olio F, et al. (2012) DNA methylation and histone modifications modulate the beta1,3 galactosyltransferase beta3Gal-T5 native promoter in cancer cells. Int J Biochem Cell Biol 44: 84–90. [DOI] [PubMed] [Google Scholar]
- 36. Chakraborty AK, Sousa Jde F, Chakraborty D, Funasaka Y, Bhattacharya M, et al. (2006) GnT-V expression and metastatic phenotypes in macrophage-melanoma fusion hybrids is down-regulated by 5-Aza-dC: evidence for methylation sensitive, extragenic regulation of GnT-V transcription. Gene 374: 166–173. [DOI] [PubMed] [Google Scholar]
- 37. Kannagi R, Sakuma K, Miyazaki K, Lim KT, Yusa A, et al. (2010) Altered expression of glycan genes in cancers induced by epigenetic silencing and tumor hypoxia: clues in the ongoing search for new tumor markers. Cancer Sci 101: 586–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kim YS, Deng G (2008) Aberrant expression of carbohydrate antigens in cancer: the role of genetic and epigenetic regulation. Gastroenterology 135: 305–309. [DOI] [PubMed] [Google Scholar]
- 39. Serpa J, Mesquita P, Mendes N, Oliveira C, Almeida R, et al. (2006) Expression of Lea in gastric cancer cell lines depends on FUT3 expression regulated by promoter methylation. Cancer Lett 242: 191–197. [DOI] [PubMed] [Google Scholar]
- 40. Giordanengo V, Ollier L, Lanteri M, Lesimple J, March D, et al. (2004) Epigenetic reprogramming of UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase (GNE) in HIV-1-infected CEM T cells. FASEB J 18: 1961–1963. [DOI] [PubMed] [Google Scholar]
- 41. Oetke C, Hinderlich S, Reutter W, Pawlita M (2003) Epigenetically mediated loss of UDP-GlcNAc 2-epimerase/ManNAc kinase expression in hyposialylated cell lines. Biochem Biophys Res Commun 308: 892–898. [DOI] [PubMed] [Google Scholar]
- 42. Demers M, Couillard J, Giglia-Mari G, Magnaldo T, St-Pierre Y (2009) Increased galectin-7 gene expression in lymphoma cells is under the control of DNA methylation. Biochem Biophys Res Commun 387: 425–429. [DOI] [PubMed] [Google Scholar]
- 43. Hayashi N, Nakamori S, Okami J, Nagano H, Dono K, et al. (2004) Association between expression levels of CA 19-9 and N-acetylglucosamine-beta;1,3-galactosyltransferase 5 gene in human pancreatic cancer tissue. Pathobiology 71: 26–34. [DOI] [PubMed] [Google Scholar]
- 44. Mare L, Trinchera M (2004) Suppression of beta 1,3galactosyltransferase beta 3Gal-T5 in cancer cells reduces sialyl-Lewis a and enhances poly N-acetyllactosamines and sialyl-Lewis x on O-glycans. Eur J Biochem 271: 186–194. [DOI] [PubMed] [Google Scholar]
- 45. Holgersson J, Lofling J (2006) Glycosyltransferases involved in type 1 chain and Lewis antigen biosynthesis exhibit glycan and core chain specificity. Glycobiology 16: 584–593. [DOI] [PubMed] [Google Scholar]
- 46. Salvini R, Bardoni A, Valli M, Trinchera M (2001) beta 1,3-Galactosyltransferase beta 3Gal-T5 acts on the GlcNAcbeta 1-->3Galbeta 1-->4GlcNAcbeta 1-->R sugar chains of carcinoembryonic antigen and other N-linked glycoproteins and is down-regulated in colon adenocarcinomas. J Biol Chem 276: 3564–3573. [DOI] [PubMed] [Google Scholar]
- 47. Mitchell S, Abel P, Madaan S, Jeffs J, Chaudhary K, et al. (2002) Androgen-dependent regulation of human MUC1 mucin expression. Neoplasia 4: 9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rajabi H, Joshi MD, Jin C, Ahmad R, Kufe D (2011) Androgen receptor regulates expression of the MUC1-C oncoprotein in human prostate cancer cells. Prostate 71: 1299–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Singh AP, Chauhan SC, Bafna S, Johansson SL, Smith LM, et al. (2006) Aberrant expression of transmembrane mucins, MUC1 and MUC4, in human prostate carcinomas. Prostate 66: 421–429. [DOI] [PubMed] [Google Scholar]
- 50. Lane AA, Chabner BA (2009) Histone deacetylase inhibitors in cancer therapy. J Clin Oncol 27: 5459–5468. [DOI] [PubMed] [Google Scholar]
- 51. Bermudez-Lugo JA, Perez-Gonzalez O, Rosales-Hernandez MC, Ilizaliturri-Flores I, Trujillo-Ferrara J, et al. (2012) Exploration of the valproic acid binding site on histone deacetylase 8 using docking and molecular dynamic simulations. J Mol Model 18: 2301–2310. [DOI] [PubMed] [Google Scholar]
- 52. Finnin MS, Donigian JR, Cohen A, Richon VM, Rifkind RA, et al. (1999) Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature 401: 188–193. [DOI] [PubMed] [Google Scholar]
- 53. Marks PA, Richon VM, Rifkind RA (2000) Histone deacetylase inhibitors: inducers of differentiation or apoptosis of transformed cells. J Natl Cancer Inst 92: 1210–1216. [DOI] [PubMed] [Google Scholar]
- 54. Beckers T, Burkhardt C, Wieland H, Gimmnich P, Ciossek T, et al. (2007) Distinct pharmacological properties of second generation HDAC inhibitors with the benzamide or hydroxamate head group. Int J Cancer 121: 1138–1148. [DOI] [PubMed] [Google Scholar]
- 55. Marks PA, Breslow R (2007) Dimethyl sulfoxide to vorinostat: development of this histone deacetylase inhibitor as an anticancer drug. Nat Biotechnol 25: 84–90. [DOI] [PubMed] [Google Scholar]
- 56. Khan N, Jeffers M, Kumar S, Hackett C, Boldog F, et al. (2008) Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors. Biochem J 409: 581–589. [DOI] [PubMed] [Google Scholar]
- 57. Ishihara K, Hong J, Zee O, Ohuchi K (2004) Possible mechanism of action of the histone deacetylase inhibitors for the induction of differentiation of HL-60 clone 15 cells into eosinophils. Br J Pharmacol 142: 1020–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. De los Santos M, Zambrano A, Aranda A (2007) Combined effects of retinoic acid and histone deacetylase inhibitors on human neuroblastoma SH-SY5Y cells. Mol Cancer Ther 6: 1425–1432. [DOI] [PubMed] [Google Scholar]
- 59. Butler LM, Agus DB, Scher HI, Higgins B, Rose A, et al. (2000) Suberoylanilide hydroxamic acid, an inhibitor of histone deacetylase, suppresses the growth of prostate cancer cells in vitro and in vivo. Cancer Res 60: 5165–5170. [PubMed] [Google Scholar]
- 60. Mann BS, Johnson JR, Cohen MH, Justice R, Pazdur R (2007) FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 12: 1247–1252. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Negative control experiments for proximity ligation assay of MUC1 and sLea in SAHA-treated RWPE-1 cells. RWPE-1 cells treated with 5 µM SAHA for 72 h were exposed to (A) PLA probe only, (B) mouse anti-sLea Ab plus rabbit IgG, or (C) rabbit anti-MUC1 Ab plus mouse IgG and then examined by confocal fluorescence microscopy after PLA assay described in the materials and methods. Bar = 20 µm
(DOCX)
Quantitative real-time PCR analysis of membrane-bound mucin and glycosyltransferase genes. Quantitative real-time PCR analysis was carried out on RWPE-1, PC3, DU1-45 and LNCaP C-81 cells treated with PBS or 5 µM SAHA for 72 h. Relative expression levels of mRNA of different genes were sorted according to ΔCt (see Materials and Methods) method, normalized with GAPDH in same cell preparation and expressed as fold changes ± SEM and then determined by calculating the ratio of the expression level of each gene in SAHA treated vs. that in PBS-treated control cells. Relative amount of each gene versus that of GAPDH (100%) in PBS-treated control cells was given in the parenthesis (n = 3).
(DOCX)
Quantitative real-time PCR analysis of mucin Polypeptide N-acetylgalactosaminyltransferase (GALNT) mRNAs. Fourteen different GALNTs were analyzed by quantitative real-time PCR in RWPE-1 cells treated with PBS or 5 µM SAHA for 72 h. Relative expression level of each GALNT was calculated and plotted as described above.
(DOCX)
Effect of SAHA treatment on levels of acetylated H2A and H2B in RWPE-1 and prostatic cancer cells. Lysates were prepared from RWPE-1, PC3, LNCaP C-81 and DU145 cells treated with PBS or 5 µM SAHA for 72 h. Proteins (100 µg) were separated on 15% SDS-PAGE and blotted onto a PVDF membrane. The acetylated H2A and H2B proteins were detected with respective antibodies. The β-actin from same samples was used as a protein loading control.
(DOCX)
Short interfering RNA (siRNA) sequences.
(DOCX)
Oligonucleotide primers used for quantitative real-time PCR analysis.
(DOCX)