

Abstract
Computational methods for miRNA target prediction vary in the algorithm used; and while one can state opinions about the strengths or weaknesses of each particular algorithm, the fact of the matter is that they fall substantially short of capturing the full detail of physical, temporal and spatial requirements of miRNA::target-mRNA interactions. Here, we introduce a novel miRNA target prediction tool called Targetprofiler that utilizes a probabilistic learning algorithm in the form of a hidden Markov model trained on experimentally verified miRNA targets. Using a large scale protein downregulation data set we validate our method and compare its performance to existing tools. We find that Targetprofiler exhibits greater correlation between computational predictions and protein downregulation and predicts experimentally verified miRNA targets more accurately than three other tools. Concurrently, we use primer extension to identify the mature sequence of a novel miRNA gene recently identified within a cancer associated genomic region and use Targetprofiler to predict its potential targets. Experimental verification of the ability of this small RNA molecule to regulate the expression of CCND2, a gene with documented oncogenic activity, confirms its functional role as a miRNA. These findings highlight the competitive advantage of our tool and its efficacy in extracting biologically significant results.
Keywords: computational prediction, microRNA gene targets, experimental verification, luciferase assays, CCND2, oncogene
Introduction
MicroRNAs (miRNAs) belong to a recently identified group of the large family of noncoding RNAs.1 The mature miRNA is usually 19–27nt long and is derived from a larger precursor that folds into an imperfect stem-loop structure. The mode of action of the mature miRNA in mammalian systems is dependent on complementary base pairing primarily to the 3′-UTR region of the target mRNA, thereafter causing the inhibition of translation and/or the degradation of the mRNA.
Searching through all human genes (~25,000) and/or other species for novel miRNA gene targets is a complicated task for which fast, flexible and reliable identification methods are required. Currently available experimental approaches working toward this goal are complex and sub-optimal.2 Inefficiencies result from various sources, including difficulty in isolating certain miRNAs by cloning due to low expression, stability, tissue specificity and technical difficulties of the cloning and repression assay procedures, while selecting the right 3′UTR to investigate is often a challenging task of its own. Computational prediction of miRNA gene targets from 3′UTR genomic sequences is an alternative technique which offers a much faster, cheaper and effective way of identifying putative miRNA gene targets. Moreover, by predicting the location of a miRNA gene target, these methods enable experimentalists to concentrate their efforts on genomic regions more likely to contain novel genes that undergo miRNA regulation, thus facilitating the discovery process.
Due to the lack of negative data in this specific biological problem, the performance of current miRNA target prediction tools is largely dependent on the overall number of predicted targets. Some tools are very efficient in predicting true target sites (high sensitivity) but at the same time display an extremely large number of overall predictions (low specificity).3-6 In contrast, other tools display an overall high specificity and a relatively low sensitivity.7-9 In order to provide an estimation of a false positive rate, false or mock miRNAs are often generated by randomly shuffling the nucleotide sequence of experimentally supported miRNAs.10 Performing target prediction with these mock miRNAs can provide an estimation of the overall false positive rate of a miRNA target prediction tool.
Accurate prediction of novel miRNA gene targets requires the consideration of certain characteristic properties of the miRNA::target-mRNA interaction. These properties are based on either experimental,11-13 or computational evidence14-18 and can be used to build a classification scheme or predictive model. For example, the foremost nucleotides at the 5′region of a mature miRNA sequence are considered crucial for recognizing and binding to the target mRNA. Research by Kiriakidou et al.19 have shown that almost consecutive complementarity of the first 9 miRNA nucleotides to the 3′UTR of protein coding genes is a prerequisite for translational repression. Moreover, Lewis et al.7 showed that complementary motifs to nucleotides 2–7 of miRNA (commonly referrer to as the miRNA seed region) remain preferentially conserved in several species in a statistically significant manner.20,21
In general, it is believed that binding of at least seven consecutive Watson-Crick (WC) base pairing nucleotides between the foremost 5′region of the miRNA and the mRNA target is required for sufficient repression of protein production.7,19
Based on the above mentioned evidence, miRNA target prediction programs rely heavily on sequence complementarity of the miRNA seed region (nucleotides 2–7) to the 3′UTR sequences of candidate target genes for identifying putative miRNA binding sites.8,22 Furthermore, most prediction tools make use of thermodynamics and evolutionary conservation at the binding site in order to minimize false positives (increase specificity).8,22,23 Some tools utilize additional features such as binding site structural accessibility,4,24,25 nucleotide composition flanking the binding sites26,27 or proximity of one binding site to another within the same 3′ UTR.26,28
In summary, the general features employed for miRNA target prediction are: (1) sequence complementarily at the 5′region of the mature miRNA, better known as the seed region and commonly characterized by nucleotides 2–7, (2) secondary structure of the miRNA::target-mRNA hybrid molecule and the overall thermodynamics of the interaction expressed in free energy (ΔG) and (3) species conservation observed via the use of full genome sequence alignments.
In addition to computational tools, large scale, high throughput transcriptomic and proteomic methods such as microarrays and pSILAC have recently been used, often in conjunction with computational tools, for the identification of novel miRNA gene targets.29,30 These methods are particularly useful as they can provide accurate protein repression data or gene expression data that may be correlated or anti-correlated with miRNA expression. Moreover, if such data are coupled to computational tools, it can facilitate rapid and precise detection of novel miRNA gene targets, while at the same time giving greater credence to computational predictions.
Next Generation Sequencing (NGS) data have also been used for the prediction of miRNA genes and their mature sequences,31 however not all small RNA sequences detected by NGS methods are miRNAs, unless some miRNA regulatory function can be attributed to them. Moreover, multiple small RNA sequences are often missed due to technical difficulties of the sequencing methodology such as library construction. Experimental verification of miRNA targets can be achieved via the use of luciferase assays whereby the miRNA is expressed in vitro while simultaneously expressing and monitoring the target mRNA linked to a luciferase reporter gene.32-34 This assay provides an experimental verification of a direct interaction between the mature miRNA and the target gene and furthermore provides evidence that regulation is mediated via the miRNA silencing pathway. However, the extent to which this interaction takes place in the intact system in vivo cannot be inferred from luciferase assays alone.
In this work, we present an efficient and freely available miRNA target prediction tool (Targetprofiler) where profile HMMs are trained to recognize certain biological features of miRNA::target-mRNA interactions. We validate our computational methodology using protein repression information from a large scale proteomic study (pSILAC)29 as well as experimentally verified miRNA gene targets form Tarbase (v5)35 and compared our results to several existing target prediction tools. We then test Targetprofiler’s ability to identify de novo biologically significant interactions by applying it on a recently identified miRNA candidate (hereafter denoted c-mir-Ch9)36 that is located in a cancer associated genomic region frequently deleted in bladder cancer.37 Finally we predict cyclin D2 (CCND2), a gene with documented oncogenic activity,38 as a key target of c-mir-Ch9 and validate this interaction using luciferase reporter assays.
In addition to our scientific findings, this is the first, to the best of our knowledge, integrative approach in which the prediction of a putative pre-miRNA is followed by the experimental verification of its mature sequence and the computational prediction of a target is experimentally confirmed using reporter assays
Results
Effect of the conservation score on Targetprofiler’s performance
In order to optimize the set of filtering rules applied to Targetprofiler’s output, we investigated the effect of these rules on the tool’s performance. Specifically, we used Targetprofiler to scan all human 3′UTRs for miRNA targets corresponding to 5 benchmark miRNAs and 5 mock miRNAs and assess the tool’s performance in both cases. It is assumed that the predicted miRNA targets for the mock miRNA sequences provide an unbiased estimate of the number of miRNA targets predicted by chance alone. Thus, to obtain an estimate of prediction accuracy (see Materials and Methods) for Targetprofiler we used the results from the mock miRNAs as false positive targets. In particular, using the 5 benchmark miRNAs we generated 5 mock miRNAs as detailed in the Materials and Methods section and use both sets as input to Targetprofiler. Performance was assessed as a function of varying the conservation score threshold for the predicted target site across 8 other organisms (see Materials and Methods). As shown in Figure 1, the number of prediction targets as well as the prediction accuracy was significantly higher for true (grey bars) vs. mock (black bars) miRNAs (p = 0.00103, using a paired, two-tail t-test). Note that, as one would expect, the conservation score is inversely proportional to the number of predicted targets and analogous to the prediction accuracy. Hence, we could infer that selecting candidate predicted target sites with higher conservation scores increases the probability of selecting a true/positive miRNA target site. This can prove to be very useful when selecting predicted target sites for experimental verification.
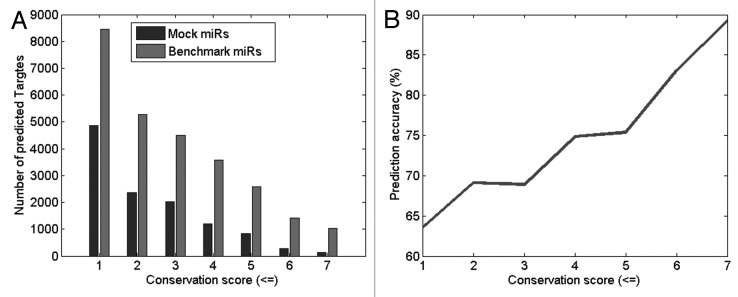
Figure 1. Predicted miRNA targets for all human 3′UTRs when using 5 benchmark miRNAs and 5 Mock miRNAs across different conservation scores for the predicted binding site. (B) Bar chart shows that the number of predictions is significantly higher for the 5 benchmark miRNAs (grey bars) in comparison to the 5 mock miRNAs (black bars). (B) The prediction accuracy (gray line) is shown to increase as the conservation score increases.
Validation of Targetprofiler using a pSILAC protein repression data set
Until recently, a common difficulty in assessing the performance of miRNA target prediction algorithms was the lack of available experimental data that could easily distinguish between true and false targets. However, the recent study of Selbach et al.29 provides both classes of targets (true and false) for five benchmark miRNAs, thus allowing the estimation of both the true positive rate as well as the false positive rate of a prediction algorithm. In the study by Selbach et al.,29 it was observed that there is a correlation between the log2-fold change of protein production with the number of occurrences of the hexamer corresponding to the seed of a miRNA in the 3′UTR. Fold changes are calculated for approximately 5,000 proteins after overexpression of the 5 benchmark miRNAs. Using a log2 fold change cut-off of -0.1 to distinguish between targeted (< -0,1) and non-targeted genes (≥ -0.1), the performance of Targetprofiler as well as three other target prediction tools for different scoring thresholds is assessed and results are presented as a ROC curve (Fig. 2). This analysis shows that Targetprofiler achieves a high true positive rate for values of false positive rates < 0.4, when compared with other tools. This is important as Targetprofiler shows a good balance between sensitivity (true positive rate) and specificity (true negative rate). All tools appear to converge at a false positive rate of ~0.4 after which Pictar and Targetprofiler achieve the highest true positive rates. A closer look at the area under the ROC curves (AUC) for all four tools reveals that Targetprofiler (AUC = 0.5724) performs better than random with p-value 0.021223 using non-directional (two-tailed) test. Similar analyses for the rest of the tools compute the following values: Diana-MiroT–AUC: 0.5297, p-value: 0.293396, TargetScan–AUC: 0.5532, p-value: 0.054999 and PicTar–AUC: 0.5955, p-value: 0.009049. The overall rate of false vs. true positives remains relatively low for all tools, in accordance to previous data,10 however it should be noted that these statistics are irrespective of the use of conservation as a filtering criterion, which is frequently utilized to boost performance of target prediction classifiers (see below).
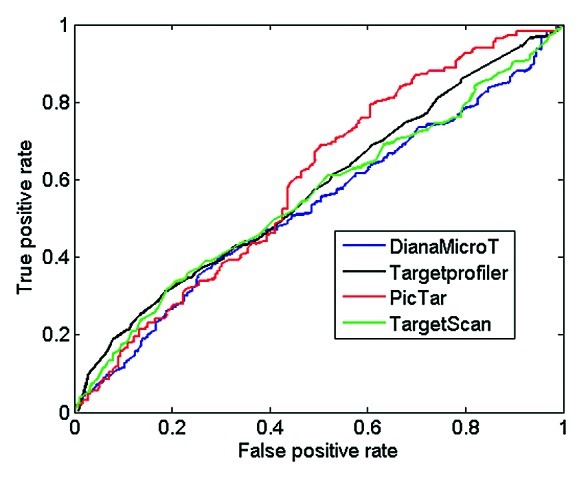
Figure 2. ROC curves using proteomics data from pSILAC. All target sites predicted by Targetprofiler, DianaMicroT, TargetScan and PicTar (conserved and non-conserved) displaying a log2 fold change cut-off of < -0.1 according to the pSILAC mass spectrometry data were used as true positives. The predictions were sorted by classification scores for each individual tool and the sensitivity and specificity were calculated as described in Materials and Methods in order to calculate the true positive (sensitivity) and false positive rate (1-specficity) for different prediction thresholds of all the tools analyzed.
Next, we used the pSILAC data to obtain an indication as to how many of our predicted miRNA targets also show downregulation of the targeted protein when taking into account different conservation thresholds. As shown in Figure 3 there are significantly higher numbers of repressed predicted targets with increasing conservation when compared with non-repressed predicted targets. In Figure 3A there is a clear difference in the distribution of repressed targets with respect to non-repressed targets with increasing conservation (p = 0.07, using a paired, two-tail t-test). Moreover prediction accuracy increases with increasing conservation scores. The overall prediction accuracy is further improved when all filtering criteria are utilized (Fig. 3B). The difference between histograms of repressed vs. non-repressed targets is even more evident (p = 6.75161E-05, using a paired, two-tail t-test). An optimum performance, namely a prediction accuracy of 66.9%, is achieved when the data are filtered, for a conservation score of 6 and an HMM score of 3.
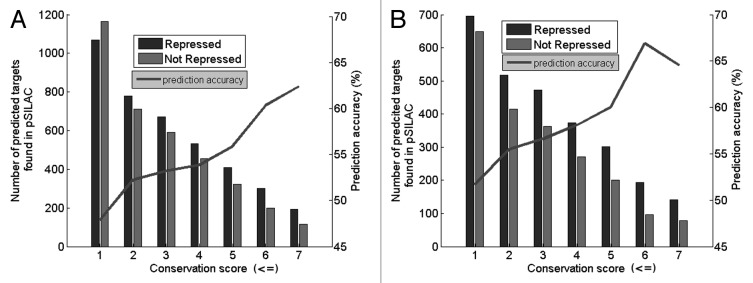
Figure 3. Bar chart of prediction results using the 5 benchmark miRNAs and data form pSILAC. Black bars represent predictions by Targetprofiler found in the Refseq database and shown to be repressed by a log2 fold change cut-off of -0.1. Gray bars are those predictions which exceed the fold change cut-off and hence are considered as non-repressed. Prediction accuracy (gray line) is shown across various conservation scores. (A) Represents data prior to filtering, (B) represents data after filtering. The optimum results (prediction accuracy 66.9%) is obtained when the data are filtered, for a conservation score of 6 and an HMM score of 3.
Comparison to existing Target Prediction Tools
Use of 5 benchmark miRNAs and data form pSILAC
In order to assess Targetprofiler’s performance in comparison to existing state-of-the-art methods, we next compared the optimum results obtained from Targetprofiler using the abovementioned 5 benchmark miRNAs with the optimum results from other prediction tools. As shown in Table 1, Targetprofiler outperforms all listed tools when tested on the pSILAC data set, although the difference from Diana-MicroT3.0 may not be statistically significant. It should be noted that by applying more stringent threshold criteria the number of predictions mapped to pSILAC decreases. It is important to obtain a high performance without utterly diminishing the total number of predictions.
Table 1. Comparison of optimum results from target prediction tools using pSILAC proteomic data.
| Prediction algorithm | Number of predicted targets mapped to Refseq | Number of targets measured by pSILAC | Number of downregulated targets (log2FC < −0.1) |
Fraction of downregulated targets (log2FC < −0.1) |
Reference |
|---|---|---|---|---|---|
| TargetScanS |
2842 |
622 |
381 |
61.25% |
7 |
| PicTar |
3289 |
629 |
386 |
61.37% |
8 |
| rna22 on 3′UTRs |
4112 |
723 |
255 |
35.27% |
3 |
| rna22 on 5′UTRs |
607 |
79 |
20 |
25.32% |
3 |
| PITA top 600 |
3000 |
325 |
139 |
42.77% |
4 |
| PITA top 1000 |
5000 |
572 |
226 |
39.51% |
4 |
| miRbase |
3347 |
658 |
288 |
43.77% |
5 |
| miRanda |
8605 |
1533 |
715 |
46.64% |
6 |
| Diana-MicroT 3.0 |
1678 |
294 |
194 |
65.99% |
9 |
| Targetprofiler | 1879 | 290 | 194 | 66.90% |
Comparing Targetprofiler results with those obtained from different target prediction tools, partly adopted from reference 29. The table shows the number of predicted targets by each tool mapped to Refseq and also the number of predicted targets measured by pSILAC. The overall fraction of downregulated (log ratio < −0.1) or repressed is an indication of the prediction accuracy of each tool. Targetprofiler shows significant improvement in comparison to all of the above tools. The gray row shows the Targetprofiler output when using a relatively stringent threshold (HMM score: 3 and conservation threshold 6.
Use of experimentally verified miRNA targets from Tarbase and common targets analysis
Finally, we used a data set of experimentally supported miRNA targets from Tarbase 535 to assess the performance of our prediction algorithm in comparison to three other state-of-the art prediction tools, namely DIANA-microT 3.0, Pic-Tar and TargetScan 4.2. As evident by the results in Figure 4, all four tools have rather subtle differences in precision levels for experimentally supported targets. However, the precision accuracy (for the same number of predicted targets per miRNA) of Targetprofiler and TargetScan appear to be similar (p = 0.67, using a paired, two-tail t-test) and consistently higher when compared with other tools (Targetprofiler vs. DianaMicroT - p = 0.094, using a paired, two-tail. t-test and Targetprofiler vs.PicTar - p = 0.027, using a paired, two-tail t-test).
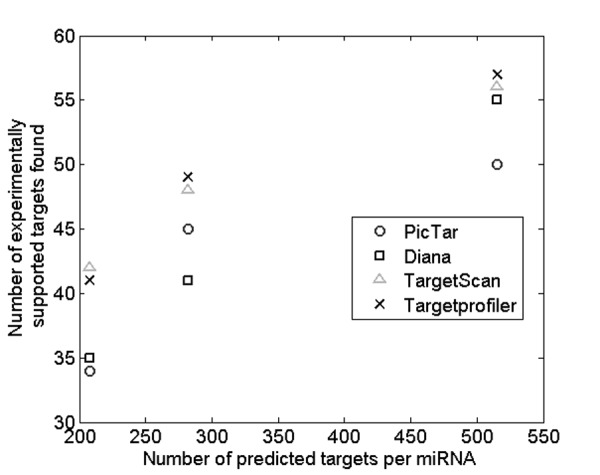
Figure 4. Number of experimentally supported targets found by Targetprofiler across 3 different HMM thresholds using the experimentally supported data set described in Materials and Methods. A comparison is made with 3 other tools (DIANA-microT 3.0, Pic-Tar and TargetScan 4.2) at the same level of predicted targets per miRNA.
As an indication of confidence for any given prediction, it is also important to assess the number of commonly predicted targets. There are considerable variations between the common miRNA targets predicted by Targetprofiler and each of the other programs (Table S1). Only 25.58% of the miRNA targets predicted by Targetprofiler are also predicted by PicTar, while 44.45% and 49.17% of the gene targets predicted by Targetprofiler are also predicted by TargetScan 4.2 and Diana-MicroT 3.0, respectively. In all cases, this leaves a high number of targets (~50%) that are unique to Targetprofiler. It is also interesting to note that of the pairwise comparisons studied, Targetprofiler displays the most common miRNA gene targets (49.17%) to Diana-MicroT relative to the other two prediction tools. Moreover, Diana-MicroT and TargetScan had the highest agreement level (66.32%) than any other tool pair.
In conclusion, our comparison analysis showed that: (1) Conservation threshold has a significant impact on the prediction accuracy of Targetprofiler. (2) The filtering parameters applied successfully boost Targetprofiler performance. (3) Targetprofiler achieved higher prediction accuracy when compared with other publically available tools using both pSILAC as well as experimentally verified miRNAs as benchmark data sets.
Experimental identification of the mature miRNA sequence for a novel miRNA candidate
Following the development and validation of Targetprofiler, our next goal was to test its ability to identify the targets of a novel miRNA gene (c-miR-ch9) recently identified and reported in previous work.36 To achieve this goal, we first needed to extract the mature (functional) miRNA sequence from the potential pre-miRNA of c-miR-ch9 and show that this small RNA molecule is expressed. Unfortunately, according to recently produced deep sequencing data from HeLa cells,31 no small RNA sequence is expressed from the genomic location where the pre-miRNA was detected. As a result, no prior experimental evidence was found regarding the location and/or sequence of the mature miRNA. To address this problem, we used an adjustment of the primer extension methodology for identifying the most probable miRNA mature sequence from a putative precursor. Specifically, instead of using one primer complement to the mature sequence, which in our case was unknown, we designed three different overlapping primers that are complementary to the positive strand of the precursor sequence, namely the strand producing a small RNA. According to the results from primer extension methodology we predicted that the mature sequence for the potential miRNA (c-miR-ch9) is 5′ CUGGCAGGGGGAGAGGUA. In order to verify our prediction we performed a northern blot analysis using an LNA probe complementary to the mature sequence (for details see Fig. S6).
Computational prediction of c-miR-Ch9 targets
Following the experimental verification of the mature miRNA sequence of c-miR-ch9, we used Targetprofiler to scan all human 3′UTRs for potential targets of c-miR-ch9. The scanning procedure was performed as described for the 5 benchmark miRNAs in the Materials and Methods section. A total of 33 predicted targets for c-miR-ch9 achieved an HMM score of 6.2 (maximum score assigned by Targetprofiler = 6.7) or higher (Table S2) and 17 of these where 8mers (as per Guo et al.39). One of these high scoring targets (HMM score: 6.2) was found to be located on a 3′UTR transcribed from chromosome 12. The miRNA::target-mRNA was an 8mer and displayed a low free energy (-23.70ΔG). Moreover, the seed was fully conserved in 7 other organisms, excluding chimp. On selecting a miRNA target site for experimental verification it can be informative to obtain an intersection of predictions from other available target prediction tools. This target site was further confirmed by four other tools (TargetScan, StarMir,24 PITA, DianaMicroT) which were used to perform target prediction using our novel miRNA sequence. The gene corresponding to this 3′UTR was CCND2, a gene with documented oncogenic activity38 that is known to play a role in the G1/S transition of the cell cycle.
Experimental verification of the c-miR-Ch9::CCND2 interaction
Since c-miR-Ch9 was found in a genomic region that is frequently deleted in various cancer types and CCND2 has a documented oncogenic activity,38 the predicted interaction appears, at least in principle, quite plausible. Thus, we next performed experiments using reporter constructs carrying a Firefly luciferase reporter to test whether the predicted interaction is functional. Given that many of the mammalian targets often contain binding sites (b.s.) for multiple miRNAs [23], we used constructs carrying binding sites that were repeated three times (pGL4-10 + wt-Triplet) but also ~1,000 bp of the 3′UTR of CCND2 containing a single copy of the b.s. (pGL4-10 + wt-3′UTR). Moreover, constructs having mutations in the 5′ seed site that disrupt the native pairing within the binding region of the triplet-cassette, as well as within the 3′UTR (designated as pGL4-10 + mut-Triplet and pGL4-10 + mut-3′UTR respectively) were also transfected, in order to provide a negative control. Furthermore, we performed transfection of empty vectors (pGL4-10) as a standardization control. All types of cassettes (constructs) prepared were placed into the pGL4-10 vector, downstream of the luc gene at XbaI site. HeLa cells were subsequently transfected with these reporter vectors carrying potential binding sites for c-miR-Ch9. For every transfection assay, all constructs were tested in parallel: an empty luciferase vector (pGL4-10–Control), a wild-type triplet cassette containing potential binding sites for c-miR-Ch9 (pGL4-10 + wt-Triplet), a wild-type 3′UTR containing a single copy of the potential b.s. for c-miR-Ch9 (pGL4-10 + wt-3′UTR), a mutated triplet cassette containing binding sites with four point mutations (pGL4-10 + mut Triplet) and a mutated 3′UTR containing the same point mutations (pGL4-10 + mut-3′UTR). Since c-miR-Ch9 was previously found to be expressed in HeLa cells at relatively high levels,36 there was no need for miRNA precursor overexpression. Firefly luciferase activity was measured and normalized against Renilla luciferase activity.
The HeLa transfections were repeated three times using triplicate samples and the average relative expression is presented in Figure 5. The reporter construct carrying the wild-type CCND2 potential triplet binding sites (pGL4-10 + wt-Triplet) and the CCND2 wild-type-3′UTR (pGL4-10 + wt-3′UTR) appeared to be efficiently downregulated: the luciferase activity dropped to 49% (2.0-fold reduction–t-test: 1E-07) and 20% (5.0-fold reduction–t-test: 2.22E-12), respectively, compared with 100% in the standardization control (pGL4-10–empty vector). We also assayed the mutated constructs pGL4-10 + mut-Triplet and pGL4-10 + mut-3′UTR, bearing CCND2 binding sites harboring mutations in the “seed” element (at position 3, 4, 6 and 7–see Fig. S5A). The transfection experiments confirmed that the downregulation previously observed was a result of the specific binding sites present in the wt-constructs. Luciferase expression was significantly increased both for the triplet mutated cassette (pGL4-10 + mut-Triplet) as well as the mutated-3′UTR (pGL4-10 + mut-3′UTR). This shows that miRNA-targeted regulation was suppressed due to truncated binding of the miRNA to the targets site(s). Specifically, in the case of pGL4-10 + mut-Triplet there was a ~2 fold increase (t-test: 2.09E-06) in luciferase activity with respect to wt constructs (Fig. 5A). Similarly for pGL4-10 + mut-3′UTR a 2.4 fold increase (t-test: 7.2E-06) was observed with respect to wt conditions (Fig. 5B). It should be noted that, as expected, t-test analysis of pGL4-10 + mut-triplet expression vs. pGL4-10 expression showed that there was no significant difference between the expression of these two constructs (t-test: 0.663864). In the 3′UTR transfection assays, contrary to the triplet cassette assays, the levels of the pGL4-10 + mut-3′UTR expression did not achieve similar expression levels as in the pGL4-10 (empty) vector. One possible explanation for this is that by cloning a large portion of the CCND2 3′UTR we may have included other potential miRNA targets sites hence rendering this construct subject to additional regulation by other miRNAs.

Figure 5. miRNA-sensor assay using luciferase expression as an indicator of miRNA activity after transfection of HeLa cells with various constructs. (A) Relative luciferase expression after transfection of HeLa cells with triplet-cassette constructs: pGL4-10–an empty pGL4-10 vector for standardization control, pGL4-10 + wt-Triplet–vector containing a wild-type triplet cassette containing potential binding sites for c-miR-Ch9, pGL4-10 + mut-Triplet–a vector containing a triplet cassette with mutated binding sites for c-miR-Ch9. (B) Relative luciferase expression after transfection of HeLa cells with 3′UTR constructs. pGL4-10–an empty pGL4-10 vector for standardization control, pGL4-10 + wt-3′UTR–vector containing a wild-type 3′UTR containing a single potential binding site for c-miR-Ch9, pGL4-10 + mut-3′UTR–a vector containing a single mutated potential binding site for c-miR-Ch9. (C) The pGL4-10 + wt-Triplet cassette transfection was repeated with concurrent addition of anti-LNA for our c-miR-Ch9. (D) An average over all transfection experiments performed (total of 5 experiments with 3 triplicates for every condition).
Three additional transfection experiments performed using the pGL4-10 + wt-Triplet constructs together with the anti-c-miR-Ch9 LNA inhibitor (25 nM) in order to block the predicted interaction of c-miR-Ch9 with our reporter constructs, further confirmed the true nature of this regulation. The co-transfection of anti-c-miR-Ch9 LNA resulted in 1.5 fold increase (t-test: 0.004) in luciferase activity in the pGL4-10 + wt-Triplet-plus-LNA transfected constructs with respect to the pGL4-10 + wt-Triplet (Fig. 5C). Co-transfection of anti-c-miR-Ch9 and pGL4-10 + wt-3′UTR was also performed and similar fold increase (1.5) in luciferase activity was observed in the pGL4-10 + wt-3′UTR-plus-LNA transfected constructs with respect to the pGL4-10 + wt-3′UTR (Fig. S7). An average for all the transfection experiments performed (total of five experiments with three triplicates for every condition) is shown in (Fig. 5D). Although standard error bars show greater deviation from the mean in this summary of results, t-test analysis reveals that results remain statistically significant (pGL4-10 vs. pGL4-10 + wt-Triplet–t-test: 5.68E-10, pGL4-10 + wt-Triplet vs. pGL4-10 + mut-Triplet–t-test: 1.68E-05, pGL4-10 + wt-Triplet vs. pGL4-10 + wt-Triplet-plus-LNA–t-test: 0.005805). As previously reported pGL4-10 vs. pGL4-10 + mut-triplet expression is not statistically significant (t-test: 0.221165). While confirmatory of the role of c-miR-Ch9 in targeting and regulating CCND2 targets sites, the lower luciferase expression observed for pGL4-10 + wt-Triplet-plus-LNA with respect to pGL4-10 or pGL4-10 + mut-triplet also suggests the possible regulation of CCND2 by additional miRNAs in the same target site, which is in agreement with computational predictions (see Discussion).
Discussion
This work describes the development of a highly efficient and freely available miRNA target prediction tool (Targetprofiler) which makes use of a HMM algorithm trained on experimentally supported miRNA gene targets. The tool is compared with existing state-of-the-art methodologies uisng two distinct data sets and achieves a very high performance. Specifically, when using the pSILAC mass spectrometry data from,29 Targetprofiler predicts a total of 290 target genes mapped to Refseq and evaluated in the pSILAC experiment, of these 194 (66.90%) show downregulation at the protein level. The downregulation may be attributed to a direct effect, namely the interaction between the mature miRNA and the 3′UTR region of the predicted gene targets, or an indirect effect, for example by the downregulation of transcription factors implicated upstream of the predicted targets. Furthermore, a pairwise ROC curve comparison between Targetprofiler and three other tools (Fig. 2) reveals that the Targetprofiler shows a higher true positive rate at lower levels of false positive rate (high sensitivity and high specificity). When using experimentally verified miRNA targets from Tarbase, the comparison with DIANA-microT 3.0, Pic-Tar and TargetScan 4.2 reveals that Targetprofiler achieves a significant improvement in precision accuracy (Fig. 3). Moreover, we asses the number of common predictions or overlap between the top 4 tools. This comparison is also important because it proves that each tool is unique in its predictions, thus providing a valuable source of computational miRNA target predictions. Overall, our validation experiments show that Targetprofiler is a very efficient tool that competes with other state-of-the-art tools such as TargetScanS, PicTar and Diana-MicroT 3.0.
Targetprofiler is a publicly available tool which includes precompiled predictions for all human miRNAs and gene targets. Furthermore, it provides an optimized prediction algorithm for ad hoc predictions that makes use of several biologically meaningful features of miRNA::target-mRNA interactions, combined with a user friendly interface which allows for user flexibility in filter adjustment and assists in the identification of interactions of interest. The tool, which can be accessed at http://mirna.imbb.forth.gr/Targetprofiler.html, allows for user intervention at various steps of the prediction pipeline and can take into account both conserved and not conserved miRNA targets in accordance to user preferences. Moreover, Targetprofiler provides links to online expression databases for miRNAs and target genes as well as information regarding multiplicity and co-operativity of miRNA binding.
Having shown the capacity of Targetprofiler to predict miRNA targets with high accuracy, competing with other state-of-the-art prediction tools, we take our research one step further and perform experimental verification on computational predictions of biological significance. In previous work36 we showed the prediction and verification (via northern blot analysis) of 4 novel potential miRNA gene candidates. As a follow-up to this work, we utilize our prediction algorithm to identify potential targets for one of these miRNAs. The candidate under investigation (denoted c-miR-Ch9) is located in a cancer associated genomic region commonly deleted in various forms of bladder cancer.37 Importantly, supporting evidence from recent deep sequencing studies do not report an expression for c-miR-Ch9 among the identified microRNA expression signatures of bladder cancer.40 Computational identification of a highly significant and evolutionary conserved target binding site for this potential miRNA in the CCND2 oncogene using Targetprofiler was the initial incentive for performing reporter gene assays. CCND2 is a well known cyclin which functions in the cell cycle and specifically in the G1/S transition. Moreover recent reporter assays have shown that CCND2 is targeted by let-7a and that this interaction inhibits proliferation in human prostate cancer cells both in vitro and in vivo.41 Furthermore, bioinformatics analysis suggested that CCND2 is a putative target for miR-154. Subsequent experiments confirmed that miR-154 directly targets CCND2 in hepatocellular carcinoma (HCC), reduces tumorigenicity and inhibits the G1/S transition in cancer cells.38
In line with these findings, our luciferase reporter assay results show that CCND2 is also targeted by c-miR-Ch9 as depicted by the decreased activity of the reporter gene in wild-type binding site conditions and the increased activity in mutated binding site conditions. Moreover, addition of anti-c-miR-Ch9 LNA to pGL4-10 + wt-Triplet conditions reduced regulation as shown by the observed increase in the reporter gene activity. However, luciferase activity in this case did not achieve the ~2-fold increase observed in the empty pGL4-10 vector or even the pGL4-10 + mut-Triplet constructs. One possible explanation for this is that other miRNA(s) compete for this target site. In fact, the target site under investigation is also a potential target site for 3 other known miRNAs (miR-182, miR-96 and miR-1271) as predicted by Targetprofiler as well as other target prediction tools (TargetScan, Diana-microT). However, using publicly available full genome tiling array42 and next generation sequencing data31 we observed that only miR-182 shows significant expression in HeLa cells. Competition between two miRNAs for the same target site can explain our observed deviations in luciferase activity during LNA silencing of c-miR-Ch9.
At this stage we believe that a comprehensive ranking framework may be valuable for the expert experimental biologist who has a specific interest in miRNA gene target prediction and verification. We provide an intuitive way of ranking miRNA gene targets according to the features provided by Targetprofiler as well as other target prediction tools. Initially, it is necessary to view the score assigned to the prediction (in our case the trained HMM score). This will provide an initial indication as to the likelihood of the prediction. Next the free energy (ΔG) of the interaction may provide additional support for the stability of the hybrid molecule and further strengthen predictions (the lower the ΔG the more stable the interaction). Even manual inspection of the secondary structure, as provided by Targetprofiler may prove to be an asset to the experienced miRNA specialists. As previously documented, conservation is a crucial filtering step in the miRNA target and gene prediction pipelines. The more evolutionary conserved a stretch of DNA the higher the chances that it generates functional, transcribed, RNA sequences. Therefore, conservation is yet another parameter which should be taken in consideration when selecting a potential miRNA target for experimental verification. In this perspective, the cumulative score provided by Targetprofiler, which is a combination of the HMM score and the conservation score, may prove to be helpful. Another additional feature which is provided by Targetprofiler is the number of target sites predicted on the same 3′UTR by the same miRNA. If a miRNA targets a 3′UTR more than once this increases the chances that the given interactions are in fact functional, as in evolutionary terms multiple target sites would not exist in the same 3′UTR unless they were of some functional purpose. Similarly if a given target site is a hotspot for multiple miRNAs then this also increases chances that one of these is in fact regulatory for similar reasoning as described above.
Finally, the results reported here are important for two reasons: first, they confirm that our initial small RNA molecule shown my northern blot in36 is infact a true miRNA gene and second, that this miRNA targets and regulates CCND2. Due to its role in proliferation, it is possible that the recently discovered miRNA may function as a tumor suppressor.43 Bladder cancer patients which exhibit deletion of the region retaining this miRNA may show increased proliferation by their inability to regulate CCND2, causing it to act like an oncogene and leading to failure of cells to arrest in G1/S and hence uncontrollable proliferation. In vivo proliferation assays using bladder cancer cell lines and xenograph implantation using mice models are needed in order to explore these hypotheses.
In conclusion, our study uses an integrative approach in which the prediction of a putative pre-miRNA is followed by the experimental verification of its mature sequence and the computational prediction of a target for this miRNA is experimentally confirmed using reporter assays. Our verified miRNA (c-mir-Ch9) was approved by the miRBase curation team and assigned the official miRNA name—hsa-mir-7150. By capitalizing on the advantages of combining computational with experimental approaches, this work provides novel, validated computational tools along with important experimental findings that are likely to open new avenues for miRNA-related cancer research.
Materials and Methods
miRNA sequences
Five benchmark mirs (let7b, mir155, mir1, mir16 and mir30a) were used for scanning 3′UTRs. Experimentally supported miRNAs and targets from the online database Tarbase version 535 were also used for validation. Only human data were used to train our algoirthm, which, comprised of 100 experimentally verified miRNA target sites and 47 miRNAs.
Human 3′UTRs
The 3UTR genomic sequences were obtained from the UCSC genome browser.44 Alternate transcripts were treated in such a way that overlapping transcripts were eliminated and the longest transcript was used to represent each alternate transcript group.
Mock miRNAs
Mock or artificial miRNAs were generated in order to obtain an indication as to the false positive rate of our prediction methodology. These mock miRNAs were produced as described in reference 10. Briefly, mock miRNA sequences were designed to have approximately the same number of predicted target site sequences as the corresponding real miRNA and were generated by initially scanning all 3′UTR sequences for sites of perfect complementarity to each possible 6 nucleotide (hexamer) long motif (excluding motifs corresponding to positions 1–6, 2–7 and 3–8 of the real miRNAs). The 5 hexamer motifs having the closest number of complementary sites to those of the seed of the real miRNA were selected. These hexamer motifs were then used as the seed for the mock miRNAs. The remaining sequence of the mock miRNAs was consequently produced by randomly shuffling the remaining nucleotides of the real miRNA. The rustling mock miRNA were then used in a similar manner to the true miRNAs in order to predict gene targets.
Multiple alignment files (MAFs)
The multiple genome alignment files have been downloaded from the UCSC Genome Browser.44 The file used for human data (hg17) is the alignment to 8 vertebrate genomes (Human, Chimp, Mouse, Rat, Dog, Cow, Zebrafish and Fugu).
Filtering
Predicted targets are filtered according to a decision rule that is based on a combination of the HMM score, the free energy (as predicted by RNAcofold45) and the conservation of the predicted target site across 8 other organisms. Conservation information was derived form the multiz full genome alignment files provided by UCSC (http://genome.ucsc.edu/). Filtering was assessed for different conservation thresholds, while the score and energy thresholds for these parameters were extracted from experimentally supported miRNA targets (Tarbase version 5). All three filtering parameters were implemented in such a way that they filter data synergistically rather than consecutively (for details see Fig. S1).
A second set of filtering parameters supported by Targetprofiler is associated location of the target site in the 3′UTR. Specifically, the tool filters out all target sites that are located within the nucleotides present in the first 0.3% from the 5′ end or the last 0.2% from the 3′end of the 3′UTR sequences.
In addition to filtering rules, Targetprofiler provides supportive information regarding the multiplicity and cooperativity of binding and expression information for the miRNA and target gene. A single miRNA can target more than one gene (multiplicity), and a single gene can be controlled by more than one miRNA (cooperativity). It is important to note that a given target prediction site may be a hotspot for multiple miRNAs and that the presence of such a hotspot adds further support to putative predictions. Expression information for miRNAs and predicted target genes is available through Targetprofiler by providing access to the Affymetrix transcriptome for long and short RNA fragments available at the UCSC genome browser.46 This expression information is not used as a filtering criterion per se, but the user is provided with a link to the UCSC genome browser which points to the genomic location of all miRNAs and target genes displayed in the results interface of Targetprofiler. This allows for manual inspection of tissue specific expression information for a miRNA or gene target of interest from one of the largest expression databases available to date. As shown recently in reference 39. miRNAs in mammals act predominantly to decrease target mRNA stability so in this respect we would except to observe anti-correlation in the expression levels of miRNA and mRNA targets. However, care must be taken if the user intends to use expression information to pursue experimental verification of putative targets as the main effect of miRNA regulation is thought to be inhibition of translation. The extent to which a mRNA is also destabilized and degraded, upon miRNA binding, heavily depends on the single mRNA and the cellular context (other miRNAs and RNA-binding proteins binding to the mRNA) as well as the overall network of regulation. As detailed for example in reference 47. miRNAs and corresponding targets can exhibit high correlation or high anti-correlation in expression levels, corresponding to the different functional roles attributed to miRNA repression. A high miRNA-target correlation in expression levels can be a consequence of the presence of the miRNA in a feed forward loop, where the miRNA repression opposes the activities of a transcription factor.
Training HMM to recognize features of MiRNA::target-mRNA interactions
Profile Hidden Markov Models48 algorithms were utilized as previously described.36 We used available structure prediction algorithms, namely RNAcofold,45 to capture the secondary structure of 100 experimentally supported miRNA::target-mRNA interactions from Tarbase version 5.49 This information was used to train the HMM and training was validated using a 5-fold cross validation procedure. An outline of the methodology adopted during training as well as the profile HMM utilized is detailed in Figures S2 and S3 respectively. Specifically, the output of RNAcofold is converted into a string representation of Ls (loops) and Ms (matches). A multiple sequence alignment (msa) of the LM string representation is then constructed whereby all miRNA::target-mRNA gene interactions are aligned according to their 5′ region. The aligned structural sequences are used as input to train the profile HMM.
HMM score and validation of predictions using Tarbase 5 miRNAs
To further validate our prediction methodology we utilize the 100 miRNA targets corresponding to 47 miRNAs from Tarbase version 5. This experimentally supported target set allows for an optimum HMM score to be determined which best classifies the true miRNA targets from Tarbase. Using this data set we infer an optimum HMM score threshold of 3. A second validation of predictions is achieved via estimating the classification accuracy on Tarbase miRNA targets, thus allowing the comparison with other tools.
Scanning all human 3′UTRs using 5 benchmark miRNAs from pSILAC
We scanned all human 3′UTR using our trained HMM for miRNA gene targets for 5 benchmark miRNAs (let7b, mir155, mir1, mir16 and mir30a) previously used in a large scale miRNA target verification assay.29 For details see Figure S4. In the abovementioned study, the 5 miRNAs were overexpressed in HeLa cells and the intensity of over 7,000 proteins was measured using mass spectrometry. Measurements were compared with control mock-transfected HeLa cells in order to obtain an indication as to how many of the proteins are downregulated (directly or indirectly) by the 5 miRNA genes. We used this data (referred to as pSILAC) in order to obtain an indication as to how many of our computationally predicted targets, mapped to Refseq and measured by pSILAC, show less than -0.1-fold change in intensity.
Statistical measures
The method’s performance was assessed using sensitivity, specificity and prediction accuracy.
Sensitivity is a measure of how many samples are classified as positive and are really positive (true positives, tp) vs. the number of samples that are positive but classified as negatives (false negatives, fn). While specificity is a measure of the samples that are classified as negative and are really negative (true negatives, tn) vs. the number of samples that are negative but classified as positive (false positives, fp).
Sensitivity = tp/(tp+fn)
Specificity = tn/(tn+fp)
The prediction accuracy is defined as the ratio of correct positive predictions over all positive predictions:
Prediction accuracy = tp /(tp + fp).
In this work, true negatives and false negatives are derived from the pSILAC proteomic data while the average number of target-mRNAs for mock miRNAs provides an estimation of the number of false positive targets predicted.
Mature miRNA prediction by primer extension
We designed three overlapping primers (each 15 nts in length) to bind to the verified positive strand of the c-miR-ch9 precursor sequence. The first primer was designed to bind from the 5th to the 19th nucleotide, the second from the 17th to the 31th and the third from the 29th to the 43nd. The primers were labeled using γ32 ATP and three primer extension reactions were performed under the following conditions: (A) incubation of 4 μg of Hela total RNA with the respective primer at 65°C for 5 min, followed by 1 min on ice; (B) subsequent incubation for 30 min at 16°C; (C) gradual increase in the temperature (0.1°C /sec) to 42°C and incubation for another 30 min at the later temperature. This gradual increase in temperature provides optimum conditions for primer extension and prevents the dehybridization of the primer. The reaction was terminated by incubation for 5 min at 85°C. In order to determine buffer’s, dNTPs’, reverse transcriptase’s and RNase inhibitor’s concentrations, we followed the HT SuperRT-kit manufacturer’s instructions.
Primers:
First, 5–19: ACCAGGGGACACCGT
Second, 17–31: CTGCCAGGTTCCACC
Third, 29–43: TTACCTCTCCCCCTG
RNA extraction and northern blot analysis
Total RNA was extracted from HeLa cells grown in culture using Trizol. Eighty micrograms of total RNA were analyzed with DNA oligonucleotides probes and 30 µg of total RNA were analyzed using LNA oligonucleotides on a 15% denaturing polyacrylamide gel containing 7 M urea and transferred to Nytran N membrane (Schleicher and Schuell). Membranes were probed with standard DNA or LNA oligonucleotides. Two DNA oligonucleotides and one LNA oligonucleotide were used. One DNA probe was complement to the predicted mature sequence and the other was complement to the adjacent sequence, which was used as a negative control. The LNA probe was complement to the predicted mature sequence. Ten picomoles of each DNA oligonucleotide probe and two picomoles of the LNA oligonucleotide probe were end-labeled with [γ–32P] ATP by using T4 polynucleotide kinase. Prehybridization of the ðlters was performed in 7% SDS, 5 × SSC, 1× Denhardt’s solution and 0.02 M Na2 HPO 4 pH 7.2. Hybridizations were performed in the same solution at 50°C after the addition of the radiolabeled DNA oligonucleotide and at 60°C after the addition of the radiolabeled LNA oligonucleotide. Following an overnight hybridization, the membranes were washed at 50°C and 60°C, for DNA and LNA probes respectively, in low stringency buffer [2 x SSC, 0.3% SDS] twice for 30 min. An extra washing step was performed for LNA probes using 1 × SSC, 0.3% SDS, for 15 min, at 60°C. For DNA probes, membranes were stripped by washing in a high stringency buffer (0.1 × SSC and 0.5% SDS) for 30 min at 80°C and reprobed with the negative polarity oligonucleotides.
DNA probes:
Positive: TACCTCTCCCCCTGCCAG
Negative: ACCAGGGGACACCGTGTG
LNA probe:
Positive: TACCTCTCCCCCTGCCAG
Vectors and DNA constructs
To generate reporter vectors bearing miRNA-binding sites, we used two mammalian vectors phRL-TK (Promega, Madison, US) and pGL4-10 carrying the Renilla luciferase gene (hRluc) and firefly luciferase gene (luc), respectively. Specific oligonucleotides having XbaI ends and containing binding sites (b.s.) in triple repeats for the predicted c-miR-Ch9::CCND2 interaction, were generated (Metabion). The phRL-TK vector was used for normalization. The oligos were cloned into the pGL4-10 vector at the XbaI site downstream of the luc gene. For all reporter constructs, two types of cassettes were prepared and studied side by side: wild type (pGL4-10 + wt–Triplet) and carrying mutations (pGL4-10 + mut–Triplet). We further PCR amplified the actual b.s. from the 3′UTR of CCND2 including ~500bp flanking regions on either side of the b.s. Following PCR mutagenesis of this construct (~1000bp), we cloned both wt-3′UTR (pGL4-10 + wt–3′UTR) and mut-3′UTR (pGL4-10 + mut-3′UTR) into the pGL4-10 vector. The empty vector (pGL4-10) was utilized as a control to observe the effect of our miRNA on the construct per se. All constructs were verified by sequencing. Additionally, anti-c-miR-Ch9 LNA (Exiqon, Berlin, Germany) was used to inhibit the expression of c-miR-Ch9. The sequences used in our studies are listed in Supplementary Material (Fig. S5). Positions of mutations in the mutated constructs are indicated in bold.
Transfection assay
Human HeLa 229 cell lines (LGC Promochem, ATCC Number: CCL-2.1) were grown in Dulbecco's Modified Eagle's Medium (DMEM) at 37°C, in a humidified atmosphere of 5% CO2. The cells were transfected in the 24-well plates in serum-free DMEM by using Lipofectamine 2000 (Invitrogen) according to manufacturers' instructions. For each transfection experiment, 350 ng of appropriate reporter construct, 50 ng of normalization vector and 400 ng of pBSK(+) as a carrier plasmid, were used in order to obtain optimal results. HeLa cells were also transfected with the empty pGL4-10 vector which was used as a reference point. Cells were harvested 48h after transfection and assayed both for firefly and Renilla luciferase activity using Dual Luciferase Assay System (Promega). The luciferase activity was measured using Dual Luciferase Assay System (Promega) with a FB 12 Luminometer (Berthold Detection Systems). For the inhibition of endogenous c-miR-Ch9 miRNA in HeLa cells the transfection of anti-c-miR-Ch9 LNA (Dharmacon) at varying concentrations ranging from 25–50 nM was performed, using Lipofectamine 2000 according to manufacturer’s instructions. Final expression values form transfection assays reported here were calculated by averaging all repeats for the particular construct. Values for error bars were calculated using the following formula for estimating the standard error of the mean: σM = σ/√N, where σ is the standard deviation of the original distribution and N is the sample size (the repetition number).
Supplementary Material
Acknowledgments
We would like to thank all additional members from the three collaborating laboratories for the successful completion of this manuscript. Furthermore, we thank the laboratories of Dr. J. Papamatheakis (especially George Vrentzos) and Dr. D. Kardassis for assistance with cell culture and luciferase readings respectively. This work was supported in part by the European Commission under EU FP7 project High-Performance Computing Infrastructure for South East Europe’s Research Communities (HP-SEE) (under contract number 261499). This research was also co-financed by the European Union (European Social Fund–ESF) and Greek national funds through the Operational Program “Education and Lifelong Learning” of the National Strategic Reference Framework (NSRF)–Research Funding Program: Heracleitus II. Investing in knowledge society through the European Social Fund.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Author’s Contributions
A.O. performed the development of the miRNA target prediction pipeline (Targetprofiler), performed the statistical analysis and also performed the putative target predictions for the novel miRNAs under investigation. He participated in the design and execution of the reporter assays; he also drafted the major part of this manuscript. N.K. contributed in the development of Targetprofiler and in the luciferase reporter assays and drafted the manuscript. A.L. performed the luciferase reporter assays for the 3′UTR. Y.P., K.K. and I.I. participated in the design and coordination of the study and also conceived of the study and helped to draft the manuscript. All authors read and approved the final manuscript.
Footnotes
Previously published online: www.landesbioscience.com/journals/rnabiology/article/21725
References
- 1.Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75:843–54. doi: 10.1016/0092-8674(93)90529-Y. [DOI] [PubMed] [Google Scholar]
- 2.Hüttenhofer A, Vogel J. Experimental approaches to identify non-coding RNAs. Nucleic Acids Res. 2006;34:635–46. doi: 10.1093/nar/gkj469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miranda KC, Huynh T, Tay Y, Ang YS, Tam WL, Thomson AM, et al. A pattern-based method for the identification of MicroRNA binding sites and their corresponding heteroduplexes. Cell. 2006;126:1203–17. doi: 10.1016/j.cell.2006.07.031. [DOI] [PubMed] [Google Scholar]
- 4.Kertesz M, Iovino N, Unnerstall U, Gaul U, Segal E. The role of site accessibility in microRNA target recognition. Nat Genet. 2007;39:1278–84. doi: 10.1038/ng2135. [DOI] [PubMed] [Google Scholar]
- 5.Griffiths-Jones S, Grocock RJ, van Dongen S, Bateman A, Enright AJ. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006;34(Database issue):D140–4. doi: 10.1093/nar/gkj112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Betel D, Wilson M, Gabow A, Marks DS, Sander C. The microRNA.org resource: targets and expression. Nucleic Acids Res. 2008;36(Database issue):D149–53. doi: 10.1093/nar/gkm995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 8.Krek A, Grün D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, et al. Combinatorial microRNA target predictions. Nat Genet. 2005;37:495–500. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- 9.Maragkakis M, Reczko M, Simossis VA, Alexiou P, Papadopoulos GL, Dalamagas T, et al. DIANA-microT web server: elucidating microRNA functions through target prediction. Nucleic Acids Res. 2009;37(Web Server issue):W273-6. doi: 10.1093/nar/gkp292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maragkakis M, Alexiou P, Papadopoulos GL, Reczko M, Dalamagas T, Giannopoulos G, et al. Accurate microRNA target prediction correlates with protein repression levels. BMC Bioinformatics. 2009;10:295. doi: 10.1186/1471-2105-10-295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Khvorova A, Reynolds A, Jayasena SD. Functional siRNAs and miRNAs exhibit strand bias. Cell. 2003;115:209–16. doi: 10.1016/S0092-8674(03)00801-8. [DOI] [PubMed] [Google Scholar]
- 12.Lee Y, Jeon K, Lee JT, Kim S, Kim VN. MicroRNA maturation: stepwise processing and subcellular localization. EMBO J. 2002;21:4663–70. doi: 10.1093/emboj/cdf476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lim LP, Glasner ME, Yekta S, Burge CB, Bartel DP. Vertebrate microRNA genes. Science. 2003;299:1540. doi: 10.1126/science.1080372. [DOI] [PubMed] [Google Scholar]
- 14.Helvik SA, Snøve O, Jr., Saetrom P. Reliable prediction of Drosha processing sites improves microRNA gene prediction. Bioinformatics. 2007;23:142–9. doi: 10.1093/bioinformatics/btl570. [DOI] [PubMed] [Google Scholar]
- 15.Hertel J, Stadler PF. Hairpins in a Haystack: recognizing microRNA precursors in comparative genomics data. Bioinformatics. 2006;22:e197–202. doi: 10.1093/bioinformatics/btl257. [DOI] [PubMed] [Google Scholar]
- 16.Lim LP, Lau NC, Weinstein EG, Abdelhakim A, Yekta S, Rhoades MW, et al. The microRNAs of Caenorhabditis elegans. Genes Dev. 2003;17:991–1008. doi: 10.1101/gad.1074403. b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sewer A, Paul N, Landgraf P, Aravin A, Pfeffer S, Brownstein MJ, et al. Identification of clustered microRNAs using an ab initio prediction method. BMC Bioinformatics. 2005;6:267–81. doi: 10.1186/1471-2105-6-267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yousef M, Nebozhyn M, Shatkay H, Kanterakis S, Showe LC, Showe MK. Combining multi-species genomic data for microRNA identification using a Naive Bayes classifier. Bioinformatics. 2006;22:1325–34. doi: 10.1093/bioinformatics/btl094. [DOI] [PubMed] [Google Scholar]
- 19.Kiriakidou M, Nelson PT, Kouranov A, Fitziev P, Bouyioukos C, Mourelatos Z, et al. A combined computational-experimental approach predicts human microRNA targets. Genes Dev. 2004;18:1165–78. doi: 10.1101/gad.1184704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Friedman RC, Farh KK, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19:92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Witkos TM, Koscianska E, Krzyzosiak WJ. Practical Aspects of microRNA Target Prediction. Curr Mol Med. 2011;11:93–109. doi: 10.2174/156652411794859250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microRNA targets. Cell. 2003;115:787–98. doi: 10.1016/S0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
- 23.Enright AJ, John B, Gaul U, Tuschl T, Sander C, Marks DS. MicroRNA targets in Drosophila. Genome Biol. 2003;5:R1. doi: 10.1186/gb-2003-5-1-r1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Long D, Lee R, Williams P, Chan CY, Ambros V, Ding Y. Potent effect of target structure on microRNA function. Nat Struct Mol Biol. 2007;14:287–94. doi: 10.1038/nsmb1226. [DOI] [PubMed] [Google Scholar]
- 25.Marín RM, Vanícek J. Efficient use of accessibility in microRNA target prediction. Nucleic Acids Res. 2011;39:19–29. doi: 10.1093/nar/gkq768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grimson A, Farh KK, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell. 2007;27:91–105. doi: 10.1016/j.molcel.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schmidt T, Mewes HW, Stümpflen V. A novel putative miRNA target enhancer signal. PLoS One. 2009;4:e6473. doi: 10.1371/journal.pone.0006473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baek D, Villén J, Shin C, Camargo FD, Gygi SP, Bartel DP. The impact of microRNAs on protein output. Nature. 2008;455:64–71. doi: 10.1038/nature07242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Selbach M, Schwanhäusser B, Thierfelder N, Fang Z, Khanin R, Rajewsky N. Widespread changes in protein synthesis induced by microRNAs. Nature. 2008;455:58–63. doi: 10.1038/nature07228. [DOI] [PubMed] [Google Scholar]
- 30.Betel D, Koppal A, Agius P, Sander C, Leslie C. Comprehensive modeling of microRNA targets predicts functional non-conserved and non-canonical sites. Genome Biol. 2010;11:R90. doi: 10.1186/gb-2010-11-8-r90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Friedländer MR, Chen W, Adamidi C, Maaskola J, Einspanier R, Knespel S, et al. Discovering microRNAs from deep sequencing data using miRDeep. Nat Biotechnol. 2008;26:407–15. doi: 10.1038/nbt1394. [DOI] [PubMed] [Google Scholar]
- 32.Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Shimizu M, et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci U S A. 2005;102:13944–9. doi: 10.1073/pnas.0506654102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mayr C, Hemann MT, Bartel DP. Disrupting the pairing between let-7 and Hmga2 enhances oncogenic transformation. Science. 2007;315:1576–9. doi: 10.1126/science.1137999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sylvestre Y, De Guire V, Querido E, Mukhopadhyay UK, Bourdeau V, Major F, et al. An E2F/miR-20a autoregulatory feedback loop. J Biol Chem. 2007;282:2135–43. doi: 10.1074/jbc.M608939200. [DOI] [PubMed] [Google Scholar]
- 35.Papadopoulos GL, Reczko M, Simossis VA, Sethupathy P, Hatzigeorgiou AG. The database of experimentally supported targets: a functional update of TarBase. Nucleic Acids Res. 2009;37(Database issue):D155–8. doi: 10.1093/nar/gkn809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oulas A, Boutla A, Gkirtzou K, Reczko M, Kalantidis K, Poirazi P. Prediction of novel microRNA genes in cancer-associated genomic regions--a combined computational and experimental approach. Nucleic Acids Res. 2009;37:3276–87. doi: 10.1093/nar/gkp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Simoneau M, Aboulkassim TO, LaRue H, Rousseau F, Fradet Y. Four tumor suppressor loci on chromosome 9q in bladder cancer: evidence for two novel candidate regions at 9q22.3 and 9q31. Oncogene. 1999;18:157–63. doi: 10.1038/sj.onc.1202277. [DOI] [PubMed] [Google Scholar]
- 38.Wang W, Peng B, Wang D, Ma X, Jiang D, Zhao J, et al. Human tumor microRNA signatures derived from large-scale oligonucleotide microarray datasets. Int J Cancer. 2011;129:1624–34. doi: 10.1002/ijc.25818. [DOI] [PubMed] [Google Scholar]
- 39.Guo H, Ingolia NT, Weissman JS, Bartel DP. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature. 2010;466:835–40. doi: 10.1038/nature09267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Han Y, Chen J, Zhao X, Liang C, Wang Y, Sun L, et al. MicroRNA expression signatures of bladder cancer revealed by deep sequencing. PLoS One. 2011;6:e18286. doi: 10.1371/journal.pone.0018286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dong Q, Meng P, Wang T, Qin W, Qin W, Wang F, et al. MicroRNA let-7a inhibits proliferation of human prostate cancer cells in vitro and in vivo by targeting E2F2 and CCND2. PLoS One. 2010;5:e10147. doi: 10.1371/journal.pone.0010147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kapranov P, Cheng J, Dike S, Nix DA, Duttagupta R, Willingham AT, et al. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science. 2007;316:1484–8. doi: 10.1126/science.1138341. [DOI] [PubMed] [Google Scholar]
- 43.Wang D, Qiu C, Zhang H, Wang J, Cui Q, Yin Y. Human microRNA oncogenes and tumor suppressors show significantly different biological patterns: from functions to targets. PLoS One. 2010;5:5. doi: 10.1371/journal.pone.0013067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Karolchik D, Hinrichs AS, Kent WJ. The UCSC Genome Browser. Curr Protoc Bioinformatics 2007; Chapter 1:Unit 1 4. [DOI] [PubMed] [Google Scholar]
- 45.Hofacker IL. Vienna RNA secondary structure server. Nucleic Acids Res. 2003;31:3429–31. doi: 10.1093/nar/gkg599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fujita PA, Rhead B, Zweig AS, Hinrichs AS, Karolchik D, Cline MS, et al. The UCSC Genome Browser database: update 2011. Nucleic Acids Res. 2011;39(Database issue):D876–82. doi: 10.1093/nar/gkq963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Osella M, Bosia C, Corá D, Caselle M. The role of incoherent microRNA-mediated feedforward loops in noise buffering. PLoS Comput Biol. 2011;7:e1001101. doi: 10.1371/journal.pcbi.1001101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eddy SR. Profile hidden Markov models. Bioinformatics. 1998;14:755–63. doi: 10.1093/bioinformatics/14.9.755. [DOI] [PubMed] [Google Scholar]
- 49.Sethupathy P, Corda B, Hatzigeorgiou AG. TarBase: A comprehensive database of experimentally supported animal microRNA targets. RNA. 2006;12:192–7. doi: 10.1261/rna.2239606. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.