

Abstract
Copper transfer to cuproproteins located in vesicular compartments of the secretory pathway depends on activity of the copper translocating ATPase (ATP7A or ATP7B) but the mechanism of transfer is largely unexplored. Copper-ATPase ATP7A is unique in having a sequence rich in histidine and methionine residues located on the lumenal side of the membrane. The corresponding fragment binds Cu(I) when expressed as a chimera with a scaffold protein, and mutations or deletions of His and/or Met residues in its sequence inhibit dephosphorylation of the ATPase, a catalytic step associated with copper release. Here we present evidence for a potential role of this lumenal region of ATP7A in copper transfer to cuproenzymes. Both Cu(II) and Cu(I) forms were investigated since the form in which copper is transferred to acceptor proteins is currently unknown. Analysis of Cu(II) using EPR demonstrated that at Cu:P ratios below 1:1, 15N-substituted protein had Cu(II) bound by 4 His residues, but this coordination changed as the Cu(II) to protein ratio increased towards 2:1. XAS confirmed this coordination via analysis of the intensity of outer-shell scattering from imidazole residues. The Cu(II) complexes could be reduced to their Cu(I) counterparts by ascorbate, but here again, as shown by EXAFS and XANES spectroscopy, the coordination was dependent on copper loading. At low copper Cu(I) was bound by a mixed ligand set of His + Met while at higher ratios His coordination predominated. The copper-loaded loop was able to transfer either Cu(II) or Cu(I) to peptidylglycine monooxygenase in the presence of chelating resin, generating catalytically active enzyme in a process that appeared to involve direct interaction between the two partners. The variation of coordination with copper loading suggests copper-dependent conformational change which in turn could act as a signal for regulating copper release by the ATPase pump.
Introduction
Mammalian cuproenzymes such as peptidylglycine α-amidating monooxygenase (PAM) 1, dopamine β-monooxygenase (DBM) 2, tyrosinase 3 and extracellular superoxide dismutase (SOD3) 4 mature within vesicles of the secretory pathway. These enzymes contain copper centers, which cycle between the Cu(I) and Cu(II) states during catalysis. PAM catalyzes the C-terminal amidation of glycine-extended neuropeptides, while DBM catalyzes the benzylic hydroxylation of dopamine to nor-epinephrine. Their catalytic cores contain two mononuclear copper centers (CuH and CuM), with CuH coordinated by three histidine residues and CuM coordinated by two histidines and a methionine 5-8. Tyrosinase contains a coupled binuclear copper center with each Cu coordinated to three histidines 9 and catalyzes the hydroxylation of catechols to quinones ultimately forming the pigment melanin. SOD3 contains an active site similar to the Cu-Zn containing SOD1 family, with a mononuclear Cu center coordinated by four histidines one of which bridges to the Zn atom, and is found in significant amounts in endothelial cells where it performs a protective role against the effects of excess superoxide 10.
While the mechanism of copper insertion into these proteins is largely unexplored, an increasing amount of data points to a key role for copper transporting ATPases 10-13. These are members of the P1B family of heavy metal transporters and are found in all forms of life from bacteria to mammals where they function in copper export across membranes. The proteins have a multidomain structure with an N-terminal regulatory domain, a cytosolic ATP binding domain, and a transmembrane domain which is usually comprised of eight transmembrane helices 14. The bacterial CopA from Legionella pneumophila 15 is currently the only copper transporting ATPase for which a crystal structure is available, and provides a template for understanding the transport mechanism. However, the mammalian homologues ATP7A and 7B differ from CopA in the presence of a more complex N-terminal regulatory domain comprised of six metal binding subdomains, each of which binds Cu(I), and a small domain between TMs 1and 2 which extends outwards from the lumenal side of the membrane (Scheme 1). Cuproprotein loading is a catalytic process beginning with a high affinity conformer of the ATPase, E1, with copper bound at a transmembrane site. This activates ATP-dependent phosphorylation of the protein which in turn drives a conformational change leading to the occlusion of copper from the cytosolic side. Copper release on the lumenal side is accompanied by dephosphorylation, and the generation of the low affinity E2 form, ready to rebind copper and restart the catalytic cycle 16,17.
Scheme 1.
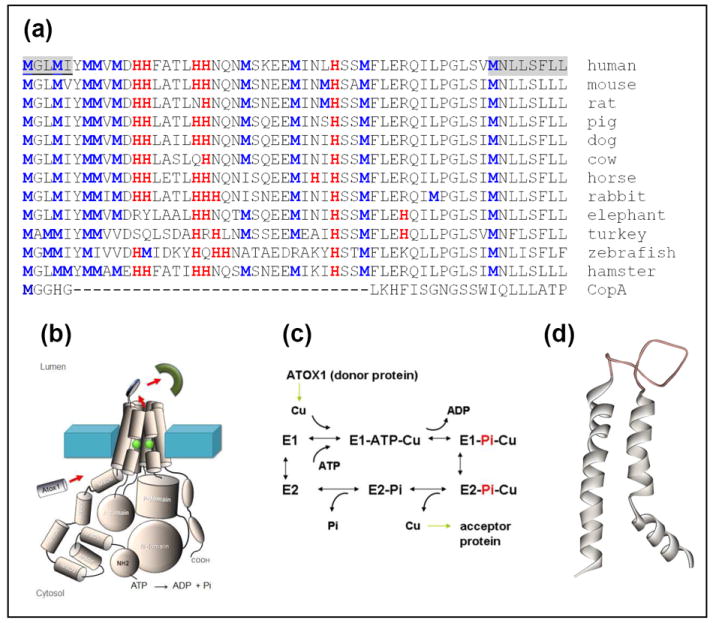
(a) Sequence comparisons of the lumenal loop region of ATP7A from different organisms with the end of TM1 and start of TM2 shaded in the first sequence. (b) Cartoon of the domain structure of P1B type ATPases. (c) Catalytic mechanism for PIB type ATPases; (d) Modeled structure of TM helices 1 and 2 of ATP7B showing the location of the lumenal loop. (Note that this modeled structure does not contain the more extended loop found in ATP7A). Model based on that developed in Schushan M, Bhattarjee A, Ben-Tal N, Lutsenko S (2012) A structural model of the copper ATPase ATP7B: interpretation of Wilson diseasecausing mutations and details of the transport mechanism, Metallomics (submitted).
Whereas cytosolic cuproproteins are metallated via copper chaperones 18-20, existing evidence suggests that copper delivery within the lumen of secretory granules may not rely on specific chaperones 21, rather that ATP7A is able to transfer copper directly to vesicular cuproenzymes with different structures and copper coordination. Additionally, since the cuproenzyme targets are redox active and cycle between Cu(I) and Cu(II), the oxidation state required for copper loading is unknown. While it is possible that a specific chaperone for vesicular copper transport may yet be discovered, we have put forward an alternative hypothesis that the lumenal loop between transmembrane segments 1 and 2 acts as an important sensor for acceptor protein loading, and may be involved in direct ATPase-acceptor interactions 16. As shown in Scheme 1(a) this loop is rich in Met and His residues. Met-rich domains are ubiquitous in copper transport proteins, and often signal areas of the protein involved in Cu(I) binding or selectivity 22-25 whereas His-rich domains often bind Cu(II). We have therefore proposed that the lumenal loop in ATP7A binds copper as it exits the membrane, and selectively hands it off to apo-cuproproteins either as Cu(I), or as Cu(II) formed by redox chemistry at the lumenal copper binding sites.
In support of our hypothesis we recently showed that mutations of His and/or Met residues within the lumenal loop of ATP7A had a major effect on the rate of dephosphorylation and thus directly affect the catalytic step in involving copper release into the lumenal space 26. Investigation of copper binding to the lumenal loop of full-length ATP7A is complicated by Cu(I) binding to the N-terminal and intramembrane domains, making it difficult to separate binding events in the lumenal loop from those elsewhere in the protein. Therefore we prepared a “model system” in which the lumenal peptide was inserted as a loop of sequence within a small soluble protein scaffold, which itself contained no metal binding Cys, Met or His residues and demonstrated the ability of the insert to bind copper 26. In the present paper, we more accurately define the Cu(I) and Cu(II) binding sites using XAS and EPR spectroscopy, and show that the metal loaded chimera transfers copper to the apo form of the catalytic core of the monooxygenase domain of PAM, (hereafter termed apo-PHM) in a facile manner.
Materials and Methods
All buffers used were reagent grade, purchased from Sigma-Aldrich at a minimum purity of 99%. Sodium ascorbate, copper (II) sulfate pentahydrate and tetrakis(acetonitrile)copper(I) hexafluorophosphate were also purchased from Sigma-Aldrich. Beef liver catalase was from Roche. The enzyme substrate, dansyl-Tyr-Val-Gly (dansyl-YVG), was obtained from American Peptide Co. The copper chelating agent, Chelex-100 resin (100-200 mesh, sodium form), was obtained from Bio-Rad Laboratories. The resin was cleaned by soaking overnight in 5M HCl, equilibrated with 50 mM sodium phosphate buffer at pH 8.0, and air dried. Distilled-deionized water used throughout the experiments was purified to a resistivity of 17-18 MΩ with a Barnstead Nanopure II system.
Construction of ScoHM Chimera
The chimeric protein, ScoHM, was constructed by replacing the C45XXXC49 copper binding motif of a Met52/Met56Ile-His55/His135A quadruple mutant of Bacillus subtilis Sco (BSco) 27 protein with the lumenal loop Histidine-Methionine (HM) rich peptide (MDHHFATLHHNQNMSKEEMINLHSSM) of the Menkes protein (ATP7A) as previously described.
Preparation of Strep-tag II /ScoHM/pTXB-3 fusion protein
Strep-tag II /ScoHM fusion protein was constructed by polymerase chain reaction amplification of ScoHM plasmid to include a Strep-tag II affinity tag (pASK-IBA5, Genosys Biotechnologies Inc) at the N-terminus of ScoHM by using the primers 5’-TATTACCATGGCTAGCGCTTGGAGCCACCCGCAGTTCGAAAAAGGACACCACATTAAAGGACAGCAGATTAAAG ATCCG-3’ (strep-tag underlined), and the MXE intein reverse primer. The forward primer introduced a new NcoI site containing an ATG start codon followed by the strep-tag II, thus eliminating the original NcoI site in ScoHM. The MXE intein reverse primer located in pTXB3 expression vector (New England BioLabs) includes a unique SpeI site. The polymerase chain reaction product containing the Strep-tag II affinity tag, the complete ScoHM, and a small portion of pTXB-3 with a unique SpeI site was digested with NcoI SpeI and cloned into NcoI SpeI sites in pTXB-3. The pTXB-3 expression vector allows for a translational fusion of an MxeGyrA intein tag to the C-terminus of the Strep-tag II/ScoHM fusion protein. The validity of construct was checked by DNA sequencing. The ligation mixture of the Strep-tag II /ScoHM/pTXB-3 fusion protein was termed 5’-Stag ScoHM.
Expression and Purification of ScoHM and its 5’-Stag ScoHM variant
ScoHM plasmid and 5’Stag ScoHM were transformed, separately, into the Escherichia coli strain ER2566 (New England BioLabs). Protein expression and purification were carried out using protocols described previously 27. Briefly, cells expressing the soluble proteins were grown in a 1 liter LB-glucose medium containing 100μg/ml of ampicillin at 37 °C to a final A600 between 0.6 and 0.9. Protein expression was induced by adding 500μM isopropyl-β-thiogalactopyranoside (IPTG) at 17 °C with shaking for 20h. The cells were harvested by centrifugation in a Sorvall GS-3 rotor at 8000 × g for 30 min, and frozen at -80 °C (as needed). Apoproteins were purified from the soluble lysate by resuspending the cells in 50 mM phosphate buffer, 500 mM NaCl at pH 7.3 (Buffer A) containing EDTA-free protease inhibitor (Roche), lysed in a French Press at 1000 psi, and centrifuged at 8500 × g for 30 min. The supernatants were loaded onto an affinity column containing chitin beads (New England Biolabs). The apoproteins were cleaved from the intein by incubating overnight at 4 °C with 50 mM 2-mercaptoethanesulfonate (MESNA) in Buffer A. The eluted protein fractions were assayed for purity by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) analysis on an Amersham Biosciences PHAST system using gradient10-15 gel (GE Healthcare). Fractions containing the proteins were collected and then concentrated to about 10 ml in an Amicon Ultra-15 centrifugal filter with a molecular mass cutoff of 5 kDa. The protein concentrations were determined by Bradford analysis. The concentration as measured by Bradford assay was comparable to that measured at OD280 using the calculated extinction coefficient of 19940 M-1 cm-1 28.
15N -Enriched ScoHM
15N -Enriched ScoHM was grown using in-house made M9 minimal medium containing 15NH4Cl as the source of nitrogen-15. The sterilized M9 minimal medium contained 0.5 g/L 15NH4Cl, 10 ml of 40% glucose, 2 ml of 1M MgSO4, 100μl of IM CaCl2, 1 ml of 5% thiamine, 10 ml of 0.1% biotin, 1 ml of 10% ampicillin, 200 ml 5x M9 salts, and 778 ml sterile deionized water. Apo- 15N -Enriched ScoHM was expressed and purified with modification to the method described previously 29. In a typical expression experiment, 15N-enriched ScoHM was grown by inoculating a 10ml LB-glucose medium with a single colony of cells expressing ScoHM at 37 °C. After 8 hr, a 100 μl aliquot of LB culture was diluted into 10 ml of N15-enhanced M9 minimal medium and grown overnight at 37 °C. The overnight culture was then transferred into 1L of N15-enhanced M9 minimal medium, and let grow at 37 °C until OD600 was between 0.5 and 0.9. Expression of 15N-enriched ScoHM was induced by the addition of 0.5 mM IPTG at 17 °C shaking for 20h. The cells were harvested by centrifugation in a Sorvall GS-3 rotor at 8000 × g for 30 min, and frozen at -80 °C. The 15N-enriched ScoHM was purified using the method described above.
Reconstitution with Cu(II) and pure 65Cu(II) isotope
Apo-ScoHM and its 5’-Stag ScoHM variant were dialyzed into three buffer changes of 50mM phosphate buffer at pH 8.0 (buffer B) to remove excess MESNA present in the protein solution. Cu(II) reconstitution was performed by adding at least 3 molar equivalents of CuSO4(aq) to the apoproteins at room temperature. Excess Cu(II) was removed by dialysis against three changes of metal-free buffer B. Fully reconstituted 65Cu(II)- ScoHM and 15N -enriched ScoHM proteins were prepared by adding stoichiometric equivalents of 65CuCl2 to the apoproteins. 65CuCl2 was prepared from 65CuO (Oak Ridge) as described previously 30,31. Cu(II) titration experiments with either natural-abundance Cu or 65Cu were achieved by adding, incrementally, the required fractional amounts of copper to the protein solutions.
Reconstitution with Cu(I)
Cu(I) reconstitution of ScoHM protein was performed in an anaerobic chamber to prevent the oxidation and/or disproportionation reaction of Cu(I) under aerobic conditions. To further avoid the presence of oxygen in the protein solution, all buffer solutions used were pre-degassed by purging with argon in an airtight container for at least an hour. Degassed solutions were then transferred into the anaerobic chamber. Apo-protein samples were also degassed by dialyzing overnight into degassed 50mM phosphate buffer at pH 8.0. The sample was reconstituted with Cu(I) by slow infusion of the required molar equivalents of [Cu(I)CH3CN]PF6 dissolved in 100% acetonitrile. After the addition of Cu(I), the reconstituted protein was quickly spun through 3 sets of pre-equilibrated desalting spin columns (Pierce) to remove excess, unbound Cu(I). The immediate use of the desalting spin column prevented ScoHM from precipitation in the presence of excess Cu(I) 32. The concentration of copper bound to the proteins was measured on a Perkin Elmer Optima 2000 DV inductively coupled plasma optical emission spectrometer (ICPOES). The same reconstitution steps were repeated for the Cu(I) reconstitution of 5’-Stag ScoHM variant. Cu(I)-ScoHM samples were also prepared by treating the Cu(II)-bound forms with a five-fold excess of ascorbate buffered at the same pH.
Analysis of ATP7A membrane fraction on Blue Native gels
HEK293Trex cells were treated with 100uM BCS or 100uM CuCl2 for 3 hours. Cells were then resuspended in 2 ml of the lysis buffer (25 mM imidazole pH 7.4, 250 mM sucrose, 5 mM DTT, 2 mM AEBSF, and 1 tablet of Roche Complete protease inhibitor cocktail (EDTA-free) per 50 ml buffer). Cells were homogenized in Dounce homogenizer, and centrifuged at 500 × g for 10 minutes. Supernatant was collected and centrifuged at 20000g for 45 min to sediment microsomal membranes. Pelleted membranes were solubilized in 50mM Bis-Tris pH 7.0, 50mM NaCl, 10% Glycerol, 0.5% DDM (v/v) on ice for 1 hour, then centrifuged at 20000 × g for 10 min. Supernatant (10ug of protein) was analysed by Blue-Native gel (4-16% Invitrogen NativePage Novex Bis-Tris Gel, 15 wells) after adding 1 volume of 10 × loading buffer (0.5M Aminocaproic Acid and 5% Brilliant Blue G250).
______The proteins separated on Blue Native gels were transferred to PVDF membrane with transfer buffer of 25mM Tris and 192mM Glycine at 200mA for 4 hours. Membranes were blocked with 5% milk in PBS at room temperature for 3 hours, briefly washed and incubated with primary antibodies at 1:5000 dilution in 1% milk/PBS in the cold room overnight, or at room temperature for 1 hour. Secondary antibodies were used at 1:10000 dilution in PBS/Tween. Bands were detected using SuperSignal West PICO Chemiluminscent substrate from Thermo Scientific.
EPR Spectroscopic Measurements
Qualitative EPR spectra on ScoHM samples were recorded on a Bruker Elexsys E500 spectrometer equipped with a Bruker ER049X SuperX microwave bridge, and an E27H lock-in detector. Spectra were recorded at X-band frequency of 9.4 GHz, and at temperature of 100 - 120 K, which was maintained by continuous cooling of the cryostat and sample with liquid nitrogen. Data were collected under non saturating microwave power conditions, set at 60 db receiver gain, 100 KHz modulation frequency, and modulation amplitude of 4G. A total of 3 scans, consisting of 2048 points at a sweep time of 167 s were averaged for each data set. The concentrations of paramagnetic copper in the samples were determined by reference of the double integral of the protein samples to Cu(II)-EDTA standard curve of known concentration (200-1000 μM). High-resolution isotope-dependent spectra for simulation (15N-labeled Sco-HM reconstituted with 65Cu(II)) were recorded at the Illinois EPR Center on a Varian E-122 spectrometer. The samples were run as frozen glasses at ~ 110 K using a continuous nitrogen flow cryostat system. Magnetic fields were calibrated with an NMR gaussmeter. Simulations were carried out using the SIMPIPM program developed at the University of Illinois 33.
X-ray Absorption Spectroscopy (XAS), Data Collection, and Analysis
Cu K-edge data (8.980 keV) data were collected in February 2009, June 2009 and May 2010 at the Stanford Synchrotron Radiation Lightsource. The extended X-ray absorption fine structure (EXAFS) and the X-ray absorption near edge structure (XANES) data were measured on beam line 9-3, operating at 3 GeV with beam currents either between 200 and 160 mA (2010) or between 100mA and 80 mA (2009). The beamline was configured with a Si[220] monochromator (crystal orientation φ = 90°), and a Rhodium (Rh)-coated mirror upstream of the monochromator with a 13 keV (Cu) energy cutoff to reject harmonics. A second Rh mirror downstream of the monochromator was used to focus the beam. Data were collected in fluorescence mode using a liquid nitrogen-cooled, high-count-rate Canberra 100-element (2010) or 30-element (2009) Ge array detector with maximum count rates below 120 Khz. Soller slits with a Z-1 metal oxide (Ni) filter were placed in front of the detector to selectively attenuate the elastic scatter peak. Under these conditions, no dead time corrections were necessary. Energy calibration was achieved by reference to the first inflection point of a copper foil (8980.3 eV) placed between the second and third ionization chamber. Four to six scans of a sample containing only sample buffer (50 mM NaPO4, pH 8.0) were collected, averaged, and subtracted from the averaged data of the protein samples to remove Z-1 (Ni) Kβ fluorescence and produce a flat pre-edge baseline. Protein samples (80μl) were measured as aqueous glasses (containing ≥ 20% ethylene glycol) at 10-15 K in a liquid helium cryostat. The number of scans collected for each sample varied from 6 - 10, depending on the concentration of copper in the samples. The scans were collected to k = 12.8Å-1 at the copper K-edge to avoid possible interference by traces of zinc in the samples.
Data reduction and background subtraction were performed with the program modules of EXAFSPAK34. Data from each detector were inspected for glitches, drop-outs before inclusion in the final average. Spectral simulation was carried out by least-squares curve fitting using the program EXCURVE 9.2 as previously described7,35,36. The quality of the fits was evaluated by the goodness-of-fit parameter, F, obtained the end of the simulation. F, as defined, is also referred to as the fit index.
Copper Transfer from ScoHM to PHM
The PHM catalytic core was isolated from Chinese hamster ovary cell line and purified as described previously 37,38. To assess copper transfer from ScoHM to PHM, pre-degassed apo-PHM was added to a reaction vial containing one mole-equivalent (on a per copper basis) of 5’-Stag ScoHM fully loaded with either Cu(I) or Cu(II). The mixture was then incubated at room temperature under strictly anaerobic conditions. After 1 hr, 1 ml of the ScoHM-PHM mixture was transferred onto a 1 ml strep-tactin resin column (IBA BioTAGnology) equilibrated with buffer (50 mM NaPO4, 150 mM NaCl, pH 8.0) and incubated for another hour. Proteins were separated by washing 5 times with 1 column volume of buffer, followed by elution of bound 5’-Stag ScoHM with 2.5 mM desthiobiotin in the same buffer. All wash and elution fractions were collected and analyzed for copper and protein content. The protein content in each of the elution fractions was analyzed by SDS-PAGE. To demonstrate that copper transfer to PHM resulted from the direct interaction of PHM with copper loaded ScoHM, a control experiment was performed in which chelex resin was added to the reaction mixture. In the control experiment, the appropriate amount of chelex resin (≈ 0.002 g) was added to chelate an amount of free Cu(II) equivalent to the total amount of protein-bound copper. After an hour of gentle agitation of the reaction mixture with a magnetic stirrer, the protein mixture was spun down for 1 min at 3000 rpm. The supernatant solution containing the protein mixture was transferred onto the strep-tactin column and subjected to the procedure described above.
Determination of PHM activity after copper transfer from ScoHM
PHM activity was measured by HPLC using dansyl-Tyr-Val-Gly as fluorescent substrate, or by oxygen consumption in an O2- sensitive electrode, as described in detail previously 39.
Results
Binding of Cu(II) to the ScoHM lumenal loop
When ScoHM was incubated with an excess of cupric ion, followed by exhaustive dialysis or desalting, it bound 1.9 ± 0.2 mole equivalents of Cu(II) per protein. This suggests the presence of two binding sites in the fully loaded complex. EPR and XAS studies were undertaken to determine the coordination environment of each of these sites.
EPR of Cu(II)-binding to ScoHM
Fig. 1 shows an EPR titration of ScoHM with increasing amounts of cupric ion. At copper to protein ratios below 1:1, a type 2 EPR spectrum is obtained with well-resolved superhyperfine splittings in the g⊥ region. As the copper to protein ratio is increased above 1, new features appear, accompanied by the loss of superhyperfine structure. The final spectra are at least two-component and confirm that two separate Cu(II) species are present. At the highest copper to protein (2.5:1) the superhyperfine is lost, although the spectrum integrates to 100 percent Cu(II). This suggests that addition of a second Cu(II) to the loop causes line broadening, perhaps due to dipolar relaxation effects, but that the two copper centers cannot be close enough to cause exchange coupling of their respective spins.
Fig 1. EPR titration and simulation of Cu(I)) ScoHM complexes.
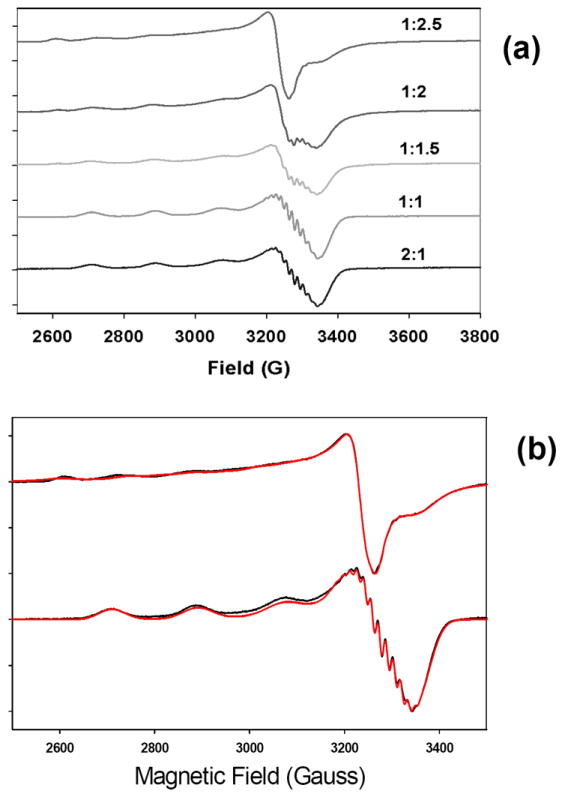
(a) EPR titration of apo-ScoHM with Cu(II). Apo-ScoHM was titrated with 0.5 mole equivalents of Cu(II) to generate spectra with protein to Cu(II) ratios as shown. (b) Simulation of ScoHM+Cu(II) EPR spectra. Bottom: 1:1 Protein to Cu(II) as a single component with four equivalent nitrogens gx = 2.075, gy = 2.040, gz = 2.254, Ax = -59, AY= - 27, AZ= -567 MHz, Ax(N) = -28, AY(N) = -42, AZ(N) = -24 MHz, microwave frequency 9.40 GHz, receiver gain 60db, microwave power 2 mW, modulation amplitude 4G, T =100-120 K.
Simulations were carried out on spectra collected from 65Cu labeled protein at 1:1 and 2.5:1 copper to protein. At 1:1, good fits (Fig. 1b) could be obtained by assuming a single species with gz = 2.254, gx=2.075, gy=2.040. The superhyperfine splittings in the g⊥ region of the spectrum could be simulated equally well by 2-4 equivalent N ligands. To distinguish between these possibilities higher resolution EPR spectra were collected at 0.45: 1 and at 0.9:1 Cu to protein from a sample obtained from cells globally labeled with 15N and these are compared with 14N spectra in Figure 2(a). Close inspection reveals differences in the superhyperfine structure on both the low-field g∥ line and in the g⊥ region for 15N and 14N spectra respectively. The 15N spectra (Fig. 2b) have 5 superhyperfine low-field lines while the 14N spectra (Fig. 2c) are less well-resolved, but appear to have 9 lines. From these empirical data we predict that the Cu(II) is coordinated to four equivalent N ligands where the number of lines n is equal to 2nI + 1. Simulation of 14N (Fig. S1(b)) and 15N (Fig S1(c and d)) gives good fits with 4 equivalent N ligands and spin Hamiltonian parameters listed in Table S1.
Fig 2. Isotope dependence of EPR spectra for 1:1 65Cu(II)-HM complex and the formation of dimeric species.
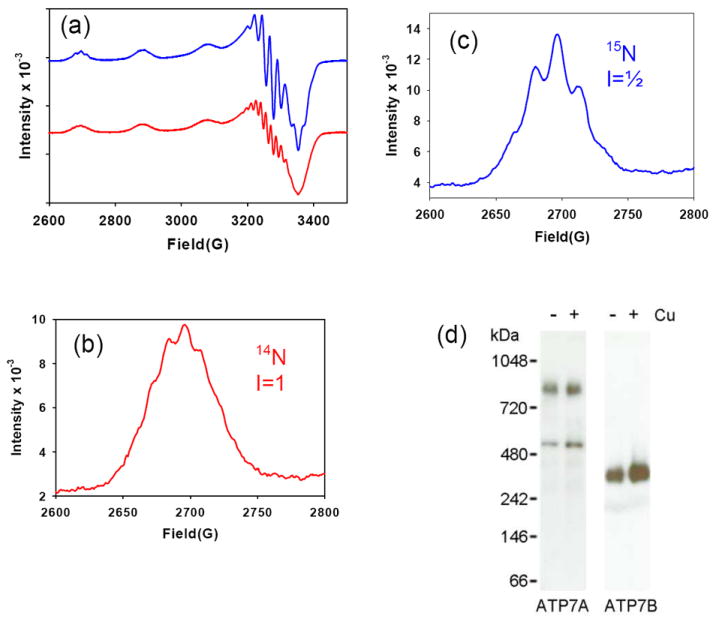
(a) Comparison of 14N- (red) and 15N- (blue) substituted ScoHM at 1:1 Cu:P. (b) and (c) Low-field hyperfine line expanded to reveal superhyperfine structure for 14N- and 15N-substituted proteins respectively. EPR instrumental settings were as listed in the legend to Figure 1. (d) Western blots of ATP7A membranes separated on blue native gels showing the presence of oligomeric forms of ATP7A. ATP7B treated in the same fashion is shown as a control. The following soluble proteins were used as molecular weight markers: 1048 kDa, IgM Pentamer; 720 kDa, Apoferritin Band 1; 480 kDa, Apoferritin Band 2; 242 kDa, B-phycoerythrin; 146 kDa, Lactate Dehydrogenase; 66 kDa, Bovine Serum Albumin.
At ratios above 1:1 copper to protein, the spectra broaden eventually losing all superhyperfine splittings at or above 2:1. The 65Cu/14N EPR spectrum of this final spectrum was collected under lower resolution and simulated as shown in Fig 1(b). The simulations required the presence of two distinct species present at approximately 1:1 ratio both of which differed from that observed at or below 1:1, Spin Hamiltonian parameters are listed in the legend to Fig 1.
Dimerization of the lumenal loop and ATP7A
These data are consistent with the formation of multiple distinct coordination environments for Cu(II) as the copper to protein ratios increase. At low ratios, a species with 4 equivalent N ligands (most likely imidazole side chains from His residues) predominates. During the titration, this species appears to be almost fully formed at a ratio of 0.5 coppers per protein, and its spectral intensity changes little between 0.5 and 1:1 ratios. Comparison of 15N spectra at 0.43 and 0.90 equivalents Cu(II) per protein are shown in Fig. S1(a). The 0.45 and and 0.9 Cu:P spectral simulations differ only in the presence of an additional broad structureless background in the 0.9:1 data, which accounts for the increase in the total cupric content. This suggests a dimeric form, in which Cu(II) is coordinated by two His residues from each of two protein molecules. Attempts to confirm the presence of a dimer using high performance gel filtration were unsuccessful, showing instead only monomeric species (data not shown). However, the dimer is unlikely to survive the gel filtration process if it is held together solely by copper binding, and could dissociate under conditions where free cupric ions are no longer in equilibrium with the CuL2 species. As additional Cu(II) loads into the protein, two new sites are formed with distinct g values and unresolved hyperfine and superhyperfine splittings. This behavior suggests that additional Cu loading breaks apart the dimer, and causes Cu(II) binding at two distinct sites in each monomer. The loss of hyperfine is consistent with dipolar broadening due to the presence of two spins at an intermediate distance (>5Å).
The question arises whether the formation of a dimeric species has physiological relevance with respect to copper release into the lumen. This would require ATP7A to be able to of form dimeric or oliogomeric structures within the membrane. To address this issue we used HEK293 cells that express ATP7A endogenously. These cells were treated with either BCS to deplete copper or with extra copper. We then prepared the ATP7A-containing membranes, solubilized protein with a mild detergent (0.5% dodecyldecamaltoside) and examined the ATP7A oligomeric state on the blue native gels under non-denaturing conditions. The data are shown in Figure 2(d). It is apparent that ATP7A migrates as two bands: a minor low molecular weight band and the major higher molecular weight band with a size double to that of the minor band. Consequently, we conclude that the predominant form of ATP7A even in copper-depleted cells is an oligomer. It should be noted that in the blue native gels the molecular weight markers do not accurately reflect masses of membrane proteins. ATP7B (165 kDa) which is slightly smaller than ATP7A (180 kDa) is shown as a control. Although formally we cannot exclude the presence of higher order oligomers, our current interpretation is that the low ATP7A band represents a monomer, and the higher band - a dimer.
Characterization of the Copper Sites by XAS
Further characterization of 1:1 and 2:1 complexes was carried out by X-ray absorption spectroscopy. Fig 3 compares Fourier transforms and EXAFS spectra for the 1:1 and 2:1 complexes respectively. The spectra are dominated by an intense peak at ~2 Å, but have additional satellite peaks at 3 and 4 Å respectively which are fingerprints for imidazole ligation. These spectra therefore establish that Cu(II) is bound by His residues from the loop region. Establishing the ratio of His to non-His O/N ligands depends on accurate simulation of the multiple scattering (MS) interactions which lead to the outer-shell satellite peaks, and generally is only semi-quantitative due to correlations between scattering amplitudes and Debye-Waller terms, and the sensitivity of the MS to small differences in orientation of each Cu-His interaction. Notwithstanding these uncertainties, inspection of the relative amplitudes of the outer-shell transform peaks indicates more His residues coordinated (higher intensity) at 1:1 than at 2:1 ratio (Fig. 4). This is confirmed by simulation where the 1:1 ratio sample simulates best with 4 ± 1 His residues, while the 2:1 ratio sample simulates best with 2 His + 2 non-His residues, again with ~25% uncertainty in coordination numbers of each shell. These data therefore support the model developed above in which initial copper loading forms a dimeric structure involving two His residues from each monomer which on further copper addition, breaks apart to form two distinct 4-coordinate monomeric sites with 2 His ligands, and additional ligands from either solvent or non-his protein side-chain/main-chain donors. Best-fit simulations of the EXAFS data and the metrical parameters used in the fits are shown in Fig 3 and Table 1 respectively.
Fig 3. EXAFS of Cu(II) complexes: top 1:1, bottom, 2:1.
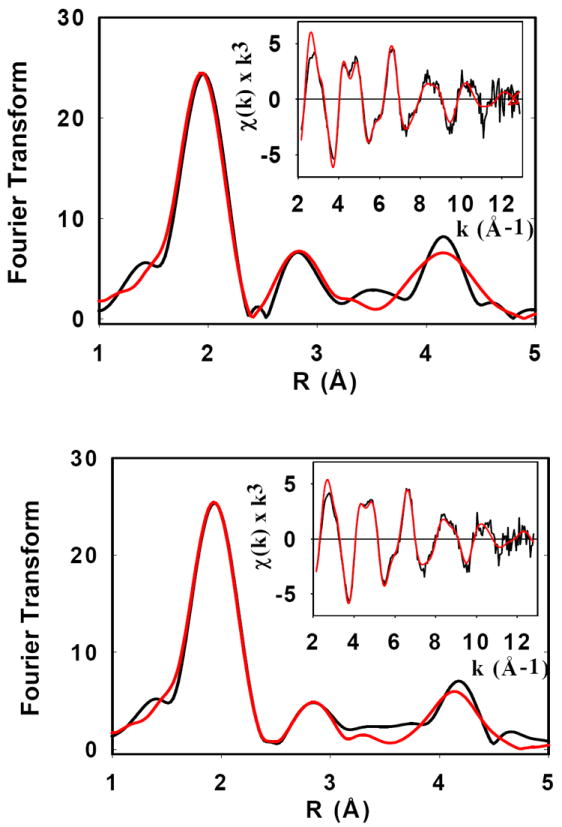
Fourier transforms and EXAFS (inserts) of Cu(II) complexes of ScoHM: top 1:1, bottom, 2:1. Black lines represent experimental spectra, red lines represent simulated spectra. Parameters used in the fit are listed in Table 1.
Fig 4. Comparison of transform intensity for 1:1 and 2:1 complexes.
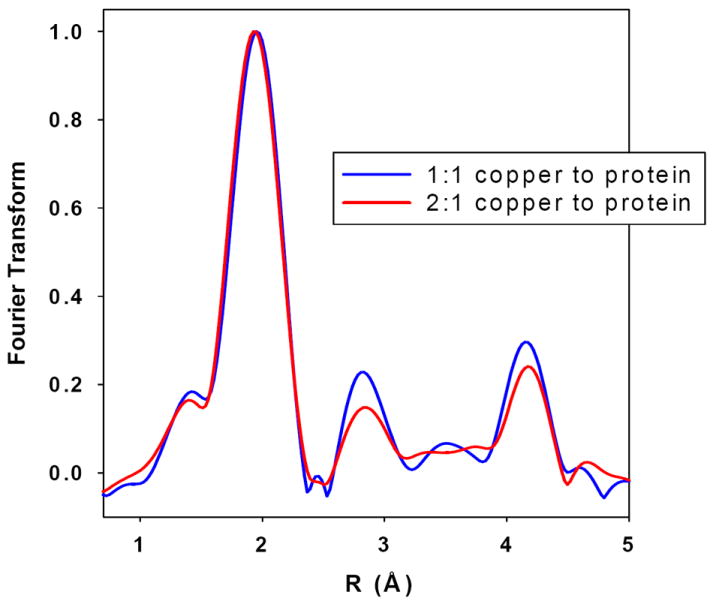
Comparison of the Fourier transform intensity for Cu(II) to protein ratios of 1:1 (blue) and 2:1 (red) complexes of ScoHM complexes.
Table 1.
Fits obtained to the EXAFS of oxidized and ascorbate-reduced forms of ScoHM
| Fa | Noc | R (Å)d | DW (Å2) | Noc | R (Å)d | DW (Å2) | Noc | R (Å)d | DW (Å2) | -E0 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Cu(II) ScoHM | |||||||||||
|
|
|||||||||||
| Cu-N(His)b | Cu-O/N | Cu-S | |||||||||
|
|
|||||||||||
| 1:1 | 0.637 | 4 | 1.99 | 0.015 | 4.80 | ||||||
| 1:2 | 0.357 | 2 | 1.93 | 0.009 | 2 | 2.03 | 0.009 | 5.04 | |||
| Cu(I) ScoHM prepared by ascorbate reduction | |||||||||||
|
|
|||||||||||
| Cu-N(His1)b | Cu-O/N | Cu-S | |||||||||
|
|
|||||||||||
| 1:1 | 0.569 | 2 | 1.87 | 0.016 | 1 | 2.19 | 0.018 | -4.3 | |||
| 2:1 | 0.879 | 2 | 1.87 | 0.011 | -1.20 | ||||||
F is a least-squares fitting parameter defined as
Fits modeled histidine coordination by an imidazole ring, which included single and multiple scattering contributions from the second shell (C2/C5) and third shell (C3/N4) atoms respectively. The Cu-N-Cx angles were as follows: Cu-N-C2 126°, Cu-N-C3 -126°, Cu-N-N4 163°, Cu-N-C5 -163°.
Coordination numbers are generally considered accurate to ± 25%
In any one fit, the statistical error in bond-lengths is ±0.005 Å. However, when errors due to imperfect background subtraction, phase-shift calculations, and noise in the data are compounded, the actual error is probably closer to ±0.02 Å.
In previous work 26 we reported initial data on Cu(I) binding to the ScoHM loop, where different coordination environments were observed at low (1:1) and high (2.5:1) Cu(I) to protein ratios. In the present work, the availability of stable Cu(II) adducts allowed us to use reduction of these Cu(II) species by ascorbate as an alternative route for preparation of the Cu(I)-bound derivatives. Fig. 5 shows Fourier transforms and EXAFS spectra for ascorbate-reduced samples at 2:1 (bottom) and 1:1 (top) copper:protein ratios, respectively. The 2:1 samples can be analyzed as a homogeneous 2-coordinate environment with Cu(I) coordinated to two His residues at a characteristically short (1.88 Å) distance. The 1:1 sample on the other hand shows a split first-shell peak due to a mixture of Cu-N and Cu-S coordination, with the best fit simulation predicting two His and one S(Met) ligand at 1.88 and 2.19 Å. Comparison of the absorption edges confirms these assignments, showing an intense 8983 eV feature from the 2-coordinate site in the 2:1 sample, and a decreased intensity feature attributable to greater 3-coordinate character in the 1:1 sample (Fig. 6). The data suggest that at low copper, the dimeric species dissociates on reduction, and the Cu(I) redistributes between His and Met residues, while at higher Cu:P ratios the 4-coordinate Cu(II)-(His)2(O/N)2 centers reduce cleanly to a pair of 2-coordinate Cu(I)-(His)2 sites. A likely possibility is that the non-His O/N ligands are solvent molecules which would be expected to dissociate on reduction of the Cu(II) to Cu(I).
Fig. 5. Ascorbate reduction, top 1:1, bottom 2:1.
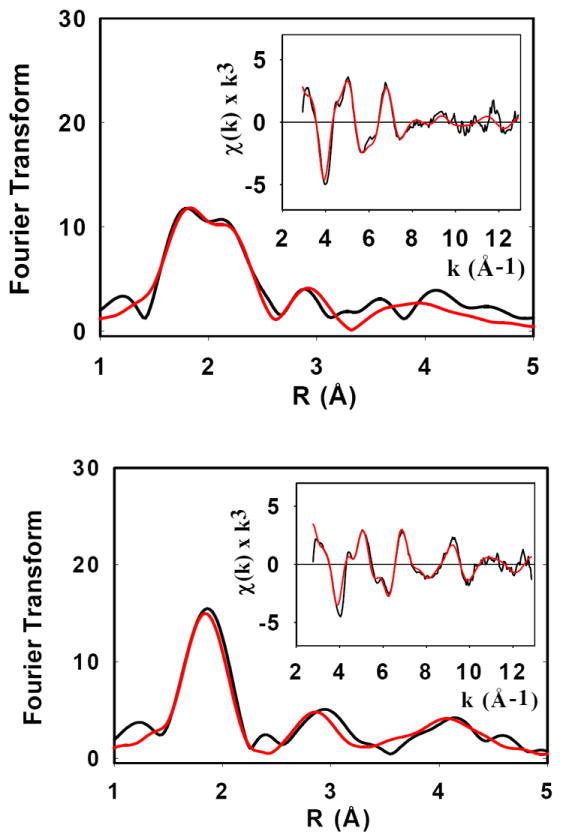
Fourier transforms and EXAFS (inserts) of Cu(I) complexes of ScoHM prepared by ascorbate reduction of their Cu(II) homologues. Top panel Cu(I) to protein 1:1, bottom panel Cu(I) to protein 2:1. Black lines represent experimental spectra, red lines represent simulated spectra. Parameters used in the fit are listed in Table 1.
Fig 6. Comparison of absorption edges for oxidized and reduced forms.
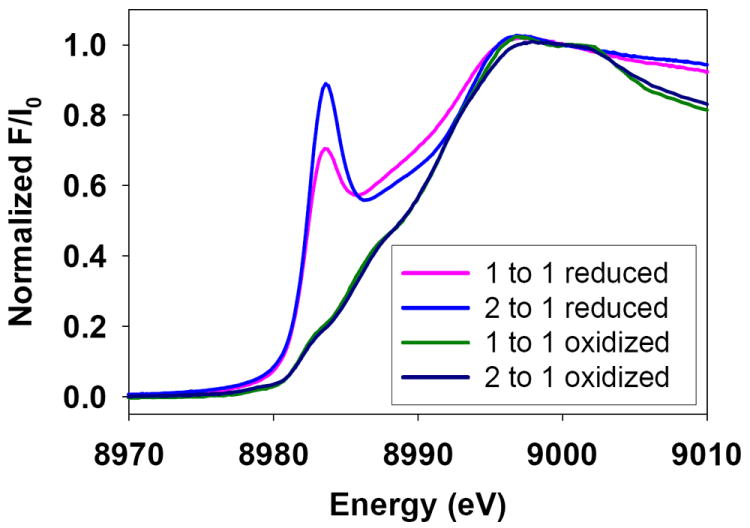
Comparison of XANES for oxidized and reduced forms. Spectra from the bottom are as follows: black trace 2:1 Cu(II) to protein, green trace 1:1 Cu(II) to protein, pink trace 1:1 Cu(I) to protein, blue trace 2:1 Cu(I) to protein.
Transfer of copper to peptidylglycine monooxygenase
We tested the ability of the lumenal loop to transfer copper to apo-PHMcc via quantitative measurement of the recovery of enzyme activity using an oxygen sensitive electrode. We first constructed a ScoHM chimera with a strep-tag fused to the N-terminus. The strep-tag-ScoHM chimera was loaded with Cu(II), mixed with a molar equivalent (on a per Cu basis) of apo PHMcc, and allowed to react for 1 hr. Aliquots of the resulting mixture were added to the assay reagents and catalytic activity was measured by determining the rate of oxygen consumption, at saturating concentrations of N-Ac-YVG and ascorbate. Determining transfer efficiency is complicated by the fact that apo-PHM can be fully reconstituted by aqueous Cu(II) ions in vitro. Therefore it is possible that copper could dissociate from ScoHM as aqueous Cu(II) and subsequently react with apo PHM to form fully metallated enzyme. To guard against this possibility we determined conditions under which added chelex resin would bind free copper quantitatively, yet be unable to remove Cu(II) from either PHMcc or ScoHM. It was found that incubation of 300 μM PHM or ScoHM with 1 mg of chelex for 4 hr resulted in no loss of PHM-bound Cu(II) and less than 10 percent of ScoHM-bound Cu(II), but removed aqueous Cu(II) quantitatively from solution (Fig. S2). Rates of oxygen consumption were then determined for PHM reconstituted with ScoHM in the presence and absence of 1 mg chelex, together with negative controls consisting of apo-PHM, and Cu(II)-loaded ScoHM with no PHM, and a positive control consisting of PHM fully reconstituted with aqeous Cu(II), using established procedures. The results (Table 2 and Fig. S3) show that Cu(II)-loaded ScoHM reconstitutes the enzyme to full activity, yet neither apo-PHM or ScoHM on its own generate activity levels above background. Addition of chelex results in a small decrease (~20 percent) in activity suggesting that 80% of copper transfer occurs by direct transfer between the lumenal loop and apo PHM.
Table 2.
PHM activity in samples reconstituted with Cu(II)-loaded ScoHM.
| Protein sample | mg of Protein | Activity (μmoles of O2·mg-1·min-1) |
|---|---|---|
| Cu(II)-ScoHM | 0.0124 | 0.043 |
| Apo PHM | 0.0124 | 0.234 |
| Cu(II)-PHM1 | 0.0124 | 3.216 |
| Cu(II)-ScoHM-PHM Mix2 | 0.0124 | 3.096 |
| Cu(II)-PHM fraction3 | 0.0186 | 1.830 |
Positive control composed of a sample of native PHM reconstituted with aqueous Cu(II) to a ratio of 2.0 copper per protein.
Mixture of equimolar amounts of apo PHM and Cu(II)-loaded ScoHM.
Reconstituted PHM fraction after separation from strep-tagged ScoHM on a streptavidin column.
We also investigated whether copper transfer from the ScoHM generated native forms of PHM as judged by their spectroscopic signatures, and by the catalytic activity of the PHM product after separation from the ScoHM copper donor. The proteins were again allowed to react with either Cu(II) or Cu(I) for 1 hr and then separated on a strep-tactin affinity column. The untagged PHM did not bind, and was collected in the flow-through fraction and buffer washes. The ScoHM fraction carrying the tag bound to the column and was eluted with desthiobiotin. The results of a typical experiment (Fig. 7) show that this protocol resulted in a reconstituted PHM protein containing 1.2 Cus per protein, while the copper content of the ScoHM had decreased from 2 to 0.5 Cus per ScoHM, indicative of 60-75% transfer. The reconstituted PHM was active as indicated by the HPLC chromatogram in panel (c) where substrate dansyl-YVG (peak on right) is converted into product dansylYVG-OH (peak in center) as a function of reaction time. For Cu(I) transfer (data not shown), the reconstituted PHM was also analyzed by XAS to ensure correct assembly of the active site. Table S2, (Supporting Information) compares fits to the Cu(I) transfer samples with fully reduced WT PHM.
Fig 7. Transfer to PHM.
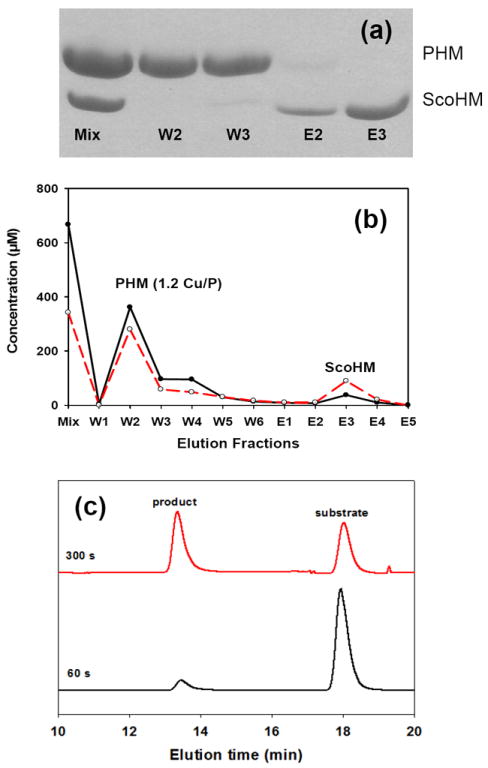
Transfer of Cu(II) from Cu(II)-loaded ScoHM to apo-PHM (a) Separation of proteins on PAGE after mixing and transfer: initial mixture (lane 1), buffer washes (lane 2-3) and the desthiobiotin elution fractions (lane 4-5). (b) HPLC gel permeation chromatography of separated fractions in (a): black traces represent total copper concentration and red traces represent protein concentrations. (c) Conversion of substrate (dansyl-YVG) into product (dansyl-YVG-OH) catalyzed by PHM fraction W2.
Discussion
Maturation of cuproproteins such as tyrosinase, PAM, DBM and SOD3 involves trafficking of the immature proteins through the TGN into storage vesicles or to the plasma membrane where they react with substrates and accumulate product 40,41. The maturation process requires metallation with Cu ions, and involves interaction with the ATPase ATP7A which co-locates with the enzyme in the intracellular compartments 10,11,21. Little is known about the process by which ATP7A hands off copper into the lumen of the secretory pathway (where the soluble domains of the enzymes reside), but available data suggests that specificity is achieved entirely through spatial colocation, and does not require the intermediacy of a metallochaperone 21. Recently we reported on the biochemical and spectroscopic properties of a characteristic His and Met loop of ATP7A which is located between TMs1 and 2 and extends into the lumenal space 26. We showed that this loop bound both Cu(I) and Ag(I), and that mutations in key His or Met residues inhibited the dephosphorylation step of ATP7A catalysis, suggesting a role in copper release. Because the full-length ATP7A binds copper to each of its six N-terminal subdomains as well as to sites within the membrane, and the loop sequence was unstable as an isolated peptide, we created a chimera with a scaffold protein by substituting the loop sequence for the CETIC copper-binding motif of B. subtilis Sco, in which all other His and Met residues had been mutated to Ala or Ile. In the present paper we have used XAS and EPR to characterize (a) the species formed when the loop binds Cu(II), (b) the species formed when the Cu(II)-bound forms are reduced by ascorbate and (c) the ability of both Cu(II) and Cu(I) forms to transfer copper to the PHM catalytic core.
EPR and EXAFS data of Cu(II)-ScoHM both support a model in which at least two different Cu(II)-binding species form as a function of copper loading. At low Cu, 15N superhyperfine splittings confirm the presence of four equivalent N ligands, whereas at higher Cu to protein ratios species with fewer His residues per Cu predominate. Since the Cu-His4 species appears to be fully formed at Cu/P ratios below 1, a possible explanation is a dimer formed by one HH doublet from each of two ScoHM molecules. This situation could be an artifact of the small size of the scaffold which holds the loop, which would have little relevance to physiological transport if the full length ATP7A existed solely as a monomer in the membrane. However, we have shown using blue-native gels that ATP7A certainly forms higher order oligomers which are in all probability dimers. Therefore the idea that copper release might involve a transient binding site at the interface of two lumenal loop structures from each of two ATP7A molecules is not excluded by our data. Alternatively, it is possible that all four His residues are derived from the two HH pairs in a single molecule. Copper binding to peptides and peptide fragments has been studied in other systems, and offers informative comparisons with the ATP7A lumenal loop peptide. The HH motif has been shown to bind Cu(II) in a variety of systems which include a fragment of the Alzheimers amyloidogenic peptide Aβ 42, but also in enzymes such as PHM H-site 6, and the CuB center of cytochrome c oxidase 43. However to our knowledge no case of two pairs of HH motifs binding to Cu(II) has been reported previously.
As the Cu:P ratio increases above 1:1, the mode of binding changes, involving two binding sites each of which appears to have fewer histidine residues. This is reminiscent of Cu(II) binding to the Prion protein, where different Cu(II)-bound species are observed at different ratios 44-46. At low copper, the octarepeat region, a domain comprised of four or more tandem repeats of the motif PHGGGWGQ, binds a single Cu(II) in a square planar environment involving four histidine residues, (one from each repeat). At higher Cu(II) to protein ratios, the domain binds four Cu(II) ions with one His, two deprotonated amide nitrogens and a main-chain carbonyl oxygen as ligands. The amylidogenic unstructured region of the Prion protein contains an additional two His residues, and both of these have been proposed to bind Cu(II) via the His and three amide N ligands. Binding of Cu(II) by a single His residue and additional amide N donors is also found in the ATCUN (amino terminal Cu and Ni binder) 47 where a His residue occurs at position 3 in the sequence.
The present study has shown that the Cu(II) bound lumenal loop is readily reducible by ascorbate to a Cu(I)-bound form, the coordination of which is also dependent on the degree of copper loading. Reduction of 1:1 Cu(II)/P samples generates a Cu(I)-binding site with both His and Met coordination, whereas reduction of the 2:1 Cu(II)/P sample generates a 2-coordinate Cu(I)His2 species. These findings are similar to our published data on Cu(I) binding induced by reaction of apo-protein with copper(I) tetrakis acetonitrile [Cu(I)(MeCN)4+], and confirm that multiple conformational states exist for Cu(I) binding as a function of copper loading involving both homogeneous Cu(I)-His2 and mixed Cu(I)-(His)-(Met) environments. Both of these Cu(I)-binding environments are common for Cu(I). Mixed His/met coordination is found in the transporters/metallochaperones CusF 48,49, PCuAC 50, PcoC51,52, and CopC53,54, as well as enzyme active sites such as the catalytic site of PHM 6, DBM 8, and TBM 55. Cu(I)His2 coordination is also well-documented, and appears to be particularly stable. Two independent studies have shown that an HH doublet present in a synthetic fragment of the Aβ Alzheimer’s peptide forms a stable linear 2-coordinate complex with Cu(I) which is highly resistant to oxidation by O242,56.
The Cu-bound forms of the lumenal loop are able to transfer copper in either oxidation state to the cuproprotein acceptor PHM to form fully active enzyme when added to an assay mixture as a 1:1 mixture of fully loaded ScoHM and apo-PHM. Addition of chelex resin in quantities sufficient to bind all of the copper (if present as aqueous Cu(II)) has a minimal effect on the resulting catalytic activity of the reconstituted PHM, suggesting that the transfer occurs via a direct protein-protein interaction, or at least by an “inner-sphere” pathway, and does not involve dissociation of Cu(II) from the donor into the aqueous phase followed by capture by the acceptor protein. Transfer also occurs when the reaction is carried out at higher protein concentrations, and the products separated by affinity chromatography. However, under the latter conditions, the transfer is incomplete, proceeding to approximately 70%. This not unusual for transfer between chaperone-target pairs where the shallow thermodynamic gradient often leads to an equilibrium distribution of copper over both proteins 36,57. Analysis of the XAS spectra of the Cu(I) form of the reconstituted PHM product revealed essentially no differences from enzyme fully loaded with 2 Cu, suggesting that the H and M centers of the reconstituted enzyme are both equally populated, thereby generating active enzyme. As yet we have not been successful in identifying the ligand environment of the copper remaining in the ScoHM. Since both PHM and ScoHM each contain copper centers which are chemically/structurally distinct, the question remains whether there is specificity in the metalation of the H and M centers in PHM by the different copper centers in ScoHM. Additionally, we have not as yet identified any protein-protein complexes, nor have we been able to monitor the kinetics of the transfer although experiments to achieve each of these goals are ongoing. However, despite the fact PHM is easily reconstituted by Cu(II)-aquo ions, it does not accept copper from simple chaperones such as Cu(I)-ATOX1, even though the Cu(I) chelator BCA is able to compete effectively with both proteins in the same concentration range (unpublished data). This emphasizes that interprotein specificity is important and further suggests that kinetic rather than thermodynamic effects are controlling the transfer process.
Our findings have important physiological implications. High levels of ATP7A are present in tissues such as pituitary and cerebral cortex which also express high levels of PAM 11. Likewise, tissues of mice with inactivated atp7a show reduced levels of amidated peptides but contain normal levels of PAM which is fully active when assayed in the presence of exogenous copper. The inference is that ATP7A is the source of the catalytic copper in vivo 11. However, copper delivery within the lumen of secretory granules does not appear to rely on specific chaperones 21. When expressed in yeast, PHM colocates with the ATP7A homologue CCC2 in the TGN, and is fully loaded with copper in an ATX1-dependent fashion. Since yeast does not contain a PHM or DBM homologue and therefore would not be expected to express a PHM-specific transporter, these results mitigate against a requirement for a lumenal copper chaperone. Tyrosinase requires ATP7A for activity 12 and is processed via the secretory pathway of melanocytes where it likewise colocates with ATP7A13.
These and other data 10 underscore the apparent ability of ATP7A to transfer copper directly to vesicular cuproenzymes with different structures and copper coordination. The observation that (when expressed in a scaffold protein) the ATP7A lumenal loop is able to bind both Cu(II) and Cu(I) in a variety of different conformations raises the possibility that the loop acts as a dynamic copper donor which can select a copper conformation appropriate for the particular acceptor protein. The apparent ability of ATP7A to oligomerize may be an additional factor imparting selectivity to the process, allowing copper to bind at the interface of two protomeric loop structures, as suggested for the ScoHM chimera at low Cu to protein ratios. In this way selectivity could be achieved entirely through colocation, without the need for unique chaperone-mediated transfer. The ability of the loop to bind copper in both oxidation states may allow it to present copper to the lumenal space in an oxidation state appropriate for the redox conditions within the vesicle. PHM and DBM which are packaged in dense-core synaptic vesicles with an internal pH of 5 require both ascorbate and oxygen for catalytic activity. Whereas the high levels of ascorbate may favor transfer of Cu as Cu(I), the more oxidizing environment of the vesicle, and the stability of the cupric forms of these enzymes do not preclude transfer as Cu(II). Tyrosinase, located in melanosomes at pH 7, does not require an external reductant, instead using the catechol product as a reductant. This makes it more likely that the preferred state for transport of copper into the oxidizing environment of the melanosome could be the Cu(II) form. These conclusions are further supported by studies on model peptides which suggest that sequences containing both Met and His residues are best able to stabilize copper in both oxidation states. 47,58,59. Further work is underway to clarify the mechanism of selectivity in transfer from ATP7A to cuproenzymes.
Supplementary Material
Acknowledgments
This work was funded by grants from the National Institute of Health P01GM01067166 (SL) and R01 NS27583 (NJB). We gratefully acknowledge the use of facilities at the Stanford Synchrotron Radiation Lightsource which is supported by the National Institutes of Health Biomedical Research and Technology Program Division of Research Resources, and by the US Department of Energy Office of Biological and Environmental Research.
Footnotes
Supporting Information
Three figures describing the simulation of the 15N EPR spectrum of ScoHM at Cu:P of 1:1, chelex treatment of ScoHM and PHM, and rates of oxygen uptake by ScoHM reconstituted samples; one Table of EXAFS parameters for Cu(I)-PHM prepared by ScoHM transfer. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Prigge ST, Mains RE, Eipper BA, Amzel LM. Cell Mol Life Sci. 2000;57:1236–1259. doi: 10.1007/PL00000763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Klinman JP. J Biol Chem. 2006;281:3013–3016. doi: 10.1074/jbc.R500011200. [DOI] [PubMed] [Google Scholar]
- 3.Rosenzweig AC, Sazinsky MH. Curr Opin Struct Biol. 2006;16:729–735. doi: 10.1016/j.sbi.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 4.Antonyuk SV, Strange RW, Marklund SL, Hasnain SS. J Mol Biol. 2009;388:310–326. doi: 10.1016/j.jmb.2009.03.026. [DOI] [PubMed] [Google Scholar]
- 5.Prigge ST, Kolhekar AS, Eipper BA, Mains RE, Amzel LM. Nature Struct Biol. 1999;6:976–983. doi: 10.1038/13351. [DOI] [PubMed] [Google Scholar]
- 6.Prigge ST, Kolhekar AS, Eipper BA, Mains RE, Amzel LM. Science. 1997;278:1300–1305. doi: 10.1126/science.278.5341.1300. [DOI] [PubMed] [Google Scholar]
- 7.Blackburn NJ, Rhames FC, Ralle M, Jaron S. J Biol Inorg Chem. 2000;5:341–353. doi: 10.1007/pl00010663. [DOI] [PubMed] [Google Scholar]
- 8.Blackburn NJ, Hasnain SS, Pettingill TM, Strange RW. J Biol Chem. 1991;266:23120–23127. [PubMed] [Google Scholar]
- 9.Matoba Y, Kumagai T, Yamamoto A, Yoshitsu H, Sugiyama M. J Biol Chem. 2006;281:8981–8990. doi: 10.1074/jbc.M509785200. [DOI] [PubMed] [Google Scholar]
- 10.Qin Z, Itoh S, Jeney V, Ushio-Fukai M, Fukai T. FASEB J. 2006;20:334–336. doi: 10.1096/fj.05-4564fje. [DOI] [PubMed] [Google Scholar]
- 11.Steveson TC, Ciccotosto GD, Ma X-M, Mueller GP, Mains RE, Eipper BA. Endocrinology. 2003;144:188–200. doi: 10.1210/en.2002-220716. [DOI] [PubMed] [Google Scholar]
- 12.Petris MJ, Strausak D, Mercer JF. Hum Mol Genet. 2000;9:2845–2851. doi: 10.1093/hmg/9.19.2845. [DOI] [PubMed] [Google Scholar]
- 13.Setty SR, Tenza D, Sviderskaya EV, Bennett DC, Raposo G, Marks MS. Nature. 2008;454:1142–1146. doi: 10.1038/nature07163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lutsenko S, Barnes NL, Bartee MY, Dmitriev OY. Physiol Rev. 2007;87:1011–1046. doi: 10.1152/physrev.00004.2006. [DOI] [PubMed] [Google Scholar]
- 15.Gourdon P, Liu XY, Skjorringe T, Morth JP, Moller LB, Pedersen BP, Nissen P. Nature. 2011;475:59–64. doi: 10.1038/nature10191. [DOI] [PubMed] [Google Scholar]
- 16.Lutsenko S, LeShane ES, Shinde U. Arch Biochem Biophys. 2007;463:134–148. doi: 10.1016/j.abb.2007.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bartee MY, Lutsenko S. Biometals. 2007;20:627–637. doi: 10.1007/s10534-006-9074-3. [DOI] [PubMed] [Google Scholar]
- 18.Lutsenko S. Curr Opin Chem Biol. 2010;14:211–217. doi: 10.1016/j.cbpa.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boal AK, Rosenzweig AC. Chem Rev. 2009;109:4760–4779. doi: 10.1021/cr900104z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim BE, Nevitt T, Thiele DJ. Nat Chem Biol. 2008;4:176–185. doi: 10.1038/nchembio.72. [DOI] [PubMed] [Google Scholar]
- 21.El Meskini R, Culotta VC, Mains RE, Eipper BA. J Biol Chem. 2003;278:12278–12284. doi: 10.1074/jbc.M211413200. [DOI] [PubMed] [Google Scholar]
- 22.Kim EH, Rensing C, McEvoy MM. Nat Prod Rep. 2010;27:711–719. doi: 10.1039/b906681k. [DOI] [PubMed] [Google Scholar]
- 23.Davis AV, O’Halloran TV. Nat Chem Biol. 2008;4:148–151. doi: 10.1038/nchembio0308-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Long F, Su CC, Zimmermann MT, Boyken SE, Rajashankar KR, Jernigan RL, Yu EW. Nature. 2010;467:484–488. doi: 10.1038/nature09395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.De Feo CJ, Aller SG, Siluvai GS, Blackburn NJ, Unger VM. Proc Natl Acad Sci USA. 2009;106:4237–4242. doi: 10.1073/pnas.0810286106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barry AN, Otoikhian A, Bhatt S, Shinde U, Tsivkovskii R, Blackburn NJ, Lutsenko S. J Biol Chem. 2011;286:26585–26594. doi: 10.1074/jbc.M111.229039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Andruzzi L, Nakano M, Nilges MJ, Blackburn NJ. J Am Chem Soc. 2005;127:16548–16558. doi: 10.1021/ja0529539. [DOI] [PubMed] [Google Scholar]
- 28.Gill SC, von Hippel PH. Anal Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 29.Yan Z, Caldwell GW, McDonell PA. Biochem Biophys Res Commun. 1999;262:793–800. doi: 10.1006/bbrc.1999.1304. [DOI] [PubMed] [Google Scholar]
- 30.Siluvai GS, Mayfield M, Nilges MJ, DeBeer George S, Blackburn NJ. J Am Chem Soc. 2010;132:5215–5226. doi: 10.1021/ja910759v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Siluvai GS, Nakano M, Mayfield M, Nilges MJ, Blackburn NJ. Biochemistry. 2009;48:12133–12144. doi: 10.1021/bi901480g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yatsunyk LA, Rosenzweig AC. J Biol Chem. 2007;282:8622–8631. doi: 10.1074/jbc.M609533200. [DOI] [PubMed] [Google Scholar]
- 33.Nilges MJ. Illinois EPR Research Center (IERC) University of Illinois; Urbana-Champaign: 1979. [Google Scholar]
- 34.George GN Stanford Synchrotron Radiation Laboratory. Menlo Park, CA: 1995. [Google Scholar]
- 35.Ralle M, Lutsenko S, Blackburn NJ. J Biol Chem. 2003;278:23163–23170. doi: 10.1074/jbc.M303474200. [DOI] [PubMed] [Google Scholar]
- 36.Bagai I, Rensing C, Blackburn NJ, McEvoy MM. Biochemistry. 2008;47:11408–11414. doi: 10.1021/bi801638m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bauman AT, Jaron S, Yukl ET, Burchfiel JR, Blackburn N. Biochemistry. 2006;45:11140–11150. doi: 10.1021/bi060905a. [DOI] [PubMed] [Google Scholar]
- 38.Bauman AT, Ralle M, Blackburn N. Protein Expression and Purification. 2007;51:34–38. doi: 10.1016/j.pep.2006.06.016. [DOI] [PubMed] [Google Scholar]
- 39.Bauman AT, Broers BA, Kline CD, Blackburn NJ. Biochemistry. 2011;50:10819–10828. doi: 10.1021/bi201193j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Milgram SL, Johnson RC, Mains RE. J Cell Biol. 1992;117:717–728. doi: 10.1083/jcb.117.4.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sobota JA, Ferraro F, Back N, Eipper BA, Mains RE. Mol Biol Cell. 2006;17:5038–5052. doi: 10.1091/mbc.E06-07-0626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shearer J, Szalai VA. J Am Chem Soc. 2008;130:17826–17835. doi: 10.1021/ja805940m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tiefenbrunn T, Liu W, Chen Y, Katritch V, Stout CD, Fee JA, Cherezov V. PLoS One. 2011;6:e22348. doi: 10.1371/journal.pone.0022348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Viles JH, Klewpatinond M, Nadal RC. Biochem Soc Trans. 2008;36:1288–1292. doi: 10.1042/BST0361288. [DOI] [PubMed] [Google Scholar]
- 45.Klewpatinond M, Davies P, Bowen S, Brown DR, Viles JH. J Biol Chem. 2008;283:1870–1881. doi: 10.1074/jbc.M708472200. [DOI] [PubMed] [Google Scholar]
- 46.Chattopadhyay M, Walter ED, Newell DJ, Jackson PJ, Aronoff-Spencer E, Peisach J, Gerfen GJ, Bennett B, Antholine WE, Millhauser GL. J Am Chem Soc. 2005;127:12647–12656. doi: 10.1021/ja053254z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Haas KL, Putterman AB, White DR, Thiele DJ, Franz KJ. J Am Chem Soc. 2011;133:4427–4437. doi: 10.1021/ja108890c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xue Y, Davis AV, Balakrishnan G, Stasser JP, Staehlin BM, Focia P, Spiro TG, Penner-Hahn JE, O’Halloran TV. Nat Chem Biol. 2008;4:107–109. doi: 10.1038/nchembio.2007.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Loftin IR, Franke S, Blackburn NJ, McEvoy MM. Protein Sci. 2007;16:2287–2293. doi: 10.1110/ps.073021307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Abriata LA, Banci L, Bertini I, Ciofi-Baffoni S, Gkazonis P, Spyroulias GA, Vila AJ, Wang S. Nat Chem Biol. 2008;4:599–601. doi: 10.1038/nchembio.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wernimont AK, Huffman DL, Finney LA, Demeler B, O’Halloran TV, Rosenzweig AC. J Biol Inorg Chem. 2003;8:185–194. doi: 10.1007/s00775-002-0404-9. [DOI] [PubMed] [Google Scholar]
- 52.Peariso K, Huffman DL, Penner-Hahn JE, O’Halloran TV. J Am Chem Soc. 2003;125:342–343. doi: 10.1021/ja028935y. [DOI] [PubMed] [Google Scholar]
- 53.Zhang L, Koay M, Maher MJ, Xiao Z, Wedd AG. J Am Chem Soc. 2006;128:5834–5850. doi: 10.1021/ja058528x. [DOI] [PubMed] [Google Scholar]
- 54.Arnesano F, Banci L, Bertini I, Thompsett AR. Structure. 2002;10:1337–1347. doi: 10.1016/s0969-2126(02)00858-4. [DOI] [PubMed] [Google Scholar]
- 55.Hess CR, Klinman JP, Blackburn NJ. J Biol Inorg Chem. 2010;15:1195–1207. doi: 10.1007/s00775-010-0677-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Himes RA, Park GY, Siluvai GS, Blackburn NJ, Karlin KD. Angew Chem Int Ed Engl. 2008;47:9084–9087. doi: 10.1002/anie.200803908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huffman DL, O’Halloran TV. J Biol Chem. 2000;275:18611–18614. doi: 10.1074/jbc.C000172200. [DOI] [PubMed] [Google Scholar]
- 58.Rubino JT, Riggs-Gelasco P, Franz KJ. J Biol Inorg Chem. 2010;15:1033–1049. doi: 10.1007/s00775-010-0663-9. [DOI] [PubMed] [Google Scholar]
- 59.Jiang J, Nadas IA, Kim MA, Franz KJ. Inorg Chem. 2005;44:9787–9794. doi: 10.1021/ic051180m. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.