

Abstract
Diabetic nephropathy is characterized by enhanced glomerular and tubulointerstitial deposition of extracellular matrix proteins, which are bound together by tissue transglutaminase (TG2). Huang et al. demonstrate that infusion of a novel TG2 inhibitor in diabetic rats prevented renal scarring and albuminuria and preserved glomerular filtration rate. These studies confirm the role of TG2 in the pathogenesis of diabetic nephropathy and add to an emerging literature that demonstrates that TG2 is an attractive therapeutic target for sclerosing kidney diseases.
Diabetes is recognized as the most common cause of chronic kidney disease worldwide, with the majority of incident cases of end-stage renal disease in the United States attributed to diabetic nephropathy. The pathogenesis of diabetic nephropathy is complex but is largely characterized by glomerular fibrosis. The predominant histologic lesion is glomerular enlargement due to deposition of interstitial collagens and a variety of other ‘scar matrix’ proteins, which accumulate within the thickened glomerular basement membranes, in a mesangial pattern, and/or as Kimmelstiel-Wilson nodules in the glomerular periphery. Interstitial fibrosis, which is composed of similar extracellular matrix proteins between tubules, is also an important predictor of diabetic nephropathy outcomes. Mainstay therapies such as glucose and blood pressure control can slow diabetic nephropathy progression, and some of these regimens may be particularly effective because the pharmacologic agents have the additive effect of interrupting scar matrix protein deposition. However, specific antiflbrotic agents have not been convincingly shown to be beneficial for diabetic nephropathy, in part because the mechanism by which multiple extracellular matrix proteins become intertwined within glomerular and interstitial scars is not well understood.
Transglutaminases (TGs) are a family of eight enzymes (factor XIII and TG1–TG7) that catalyze formation of covalent bonds between free ε-amino groups (usually from a lysine residue) and a γ-car-boxyamine group from a glutamine on adjacent peptides or proteins (Figure 1). Tissue transglutaminase (tTG, or TG2) is an 80-kDa protein that resides mainly in the cytosol and has several enzymatic functions, including transamidase (cross-linking), deaminase, isopeptidase, GTPase, protein kinase, and protein disulfide isomerase activities.1 Despite lacking a signal peptide sequence, TG2 is unique among family members because a small fraction of total cellular TG2 is secreted. The abundance of extracellular TG2 is enhanced in the context of cellular injury. In the environment of relatively high extracellular Ca2+ concentration, TG2 acts as a biological glue by binding and covalently cross-linking extracellular matrix proteins, such as collagens and fibronectin, to form large heteromeric complexes (Figure 1), which are resistant to proteolysis.2 This cross-linking function of TGs has been exploited in the food industry to provide texture to pasta and milk products, and as ‘meat glue’ to bind pieces of meat to form sausages, hot dogs, and steaks.
Figure 1. Cross-linking of extracellular matrix proteins by tissue transglutaminase-regulated transamidation.
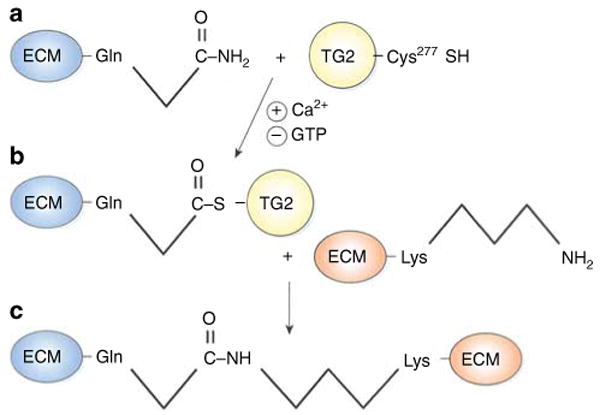
(a) Secreted tissue transglutaminase (TG2), which is stabilized by extracellular calcium, forms a thioester bond between a cysteine (Cys277) within its active site and an extracellular matrix (ECM) glutamine (Gln) residue on its substrate <2>. Both substrate–TG2 binding and TG2 activity are enhanced by calcium and inhibited by guanosine triphosphate (GTP <3>). (b) The TG2–substrate intermediate then interacts with another ECM molecule, and TG2 catalyzes an acyl transfer reaction to form an isopeptide linkage between Gln and lysine (Lys) residues on adjacent ECM proteins (c). The net effect of enhanced TG2 activity is that multiple ECM proteins become glued together to form sclerotic scars <4> <5>.
TG2 has been associated with multiple physiologic functions, many of which are relevant to glomerular diseases, including cell adhesion and migration, apoptosis, wound healing, insulin secretion, and transforming growth factor-β (TGF-β) activation.1,2 TGM2−/− mice have no overt phenotype, perhaps because of compensation by other TG isoforms, whereas mice with TGM2 transgene overexpression under the control of a cardiac-specific promoter developed cardiac fibrosis (reviewed by Griffin et al.2). In humans, TG2 has been most notably implicated in the pathogenesis of celiac disease, wherein IgA and IgG autoantibodies are generated against TG2 cross-linked to the gliadin fraction of gluten.3 TG2 has also been associated with neurologic disorders, such as Huntington's, Parkinson's, and Alzheimer's diseases, as well as in carcinogenesis and fibrosis of the liver, lung, heart, and kidney.
Now, the University of Sheffield group (Huang et al.,4 this issue) extends its ongoing investigation of the role of TG2 in the pathogenesis of diabetic glomerulosclerosis and interstitial fibrosis by demonstrating that pharmacologic inhibition of TG2 ameliorated pathologic lesions and renal dysfunction in a rat model of diabetic nephropathy. The model used was the streptozotocin-induced diabetic rat, which underwent uninephrectomy to accelerate disease progression. Osmotic minipumps containing the irreversible TG2 inhibitor N-benzyloxycarbonyl-L-phenylalanyl-6-dimethylsulfonium-5-oxo-L-norleucine (NTU281) were inserted into the renal parenchyma; this was followed by experiments demonstrating uniform distribution of NTU281 in a control kidney. Experimental and control rats were examined for renal TG activity pathology and function after 1, 4, and 8 months of diabetes. Curiously kidney TG activity was increased three- to fourfold in the untreated diabetic, uninephrectomized rats at 1 and 8 months, but not at 4 months. Nevertheless, this does not diminish the impressive main findings, that NTU281 infusion nearly normalized serum creatinine and albuminuria, as well as glomerular and tubulointerstitial scarring at 8 months.
Although these results are very encouraging, they should be viewed with caution before NTU281 could be realistically considered as a therapeutic agent for diabetic nephropathy. First, there is the generic concern that NTU281-loaded osmotic minipumps were inserted directly into the renal parenchyma and before the onset of diabetes. This is an initial and obligatory proof-of-principle approach, though it is obviously not applicable to human diabetic nephropathy, which is routinely not recognized until many years after diabetes is diagnosed. With the advent of biomarker and susceptibility-gene discovery, one could envision that prophylactic therapy for at-risk diabetic patients might one day be realistic. Until such tools become available, a rational next step would be to determine whether NTU281 might have an effect in animals with established diabetes or diabetic nephropathy. Second, scant information is provided about the specificity of NTU281 for TG2, other than that the compound binds the active site of TG2 and is exclusively effective against extracellular TGs, which are factor XIII and TG2. Even if NTU281 is specific for TG2, the possibility of off-target effects with systemic administration is significant, since TG2 is ubiquitously expressed in other tissues. Third, the TG2-regulated pathway that was altered by NTU281 was not investigated. The implication is that NTU281 administration improved diabetic nephropathy by inhibiting TG2 cross-linking of extracellular scar matrix proteins, consistent with previous studies describing increased TG2 expression and transamidase activity in human renal biopsies, as well as diabetic and nondiabetic rat models of chronic renal disease.5–7 Interestingly, NTU281 treatment also inhibited whole-kidney type III and IV collagen mRNA expression, suggesting that the drug effects may be more complex than simply post-translational modification of TG2 substrates.
Because TG2 interacts with over 100 substrates,1 definitive identification of the specific target or targets affected by NTU281 could be a bewildering task In addition to extracellular matrix proteins, another pertinent substrate is latent TGF-β-binding protein-1 (LTBP-1), since TG2-catalyzed anchoring of LTBP-1 to extracellular matrix facilitates subsequent TGF-β activation,8 and TGF-β has been implicated in the pathogenesis of diabetic glomerulosclerosis.9 In studies with TGM2−/− mice, Schweke et al. recently showed that TGF-β activity was the major mechanism of TG2-dependent interstitial fibrosis in a ureteral obstruction model.10 Improved clarification of the relative roles of TG2 in cross-linking of extracellular matrix proteins versus LTBP-1 anchoring in diabetic nephropathy, as well as the impact of NTU281 on these two pathways, would be welcome.
So although it would be premature at this point to consider NTU281 as a therapeutic agent for diabetic nephropathy, the use of this novel drug as a tool to interrogate the role of TG2 in diabetic nephropathy in rats was appropriate. This approach is akin to using targeted gene deletion strategies in mice to decipher functions of specific proteins, but it has the advantage that streptozotocin administration mimics diabetic nephropathy more faithfully in rats than in mice. Especially when combined with multiple other publications by this group about TGs in kidney disease, the paper by Huang et al.4 clearly extends the body of data implicating TG2 in diabetic nephropathy pathogenesis. This is a particularly exciting avenue to pursue since current therapies target pathways that are unrelated to post-translational extracellular matrix protein modification. Therefore, if effective treatments can be devised to safely target TG2 in the kidney, one could envision that there could be additive benefits to existing regimens for diabetic nephropathy, as well as other scarring renal diseases.
Footnotes
Disclosure: The author declared no competing interests.
References
- 1.Facchiano F, Facchiano A, Facchiano AM. The role of transglutaminase-2 and its substrates in human diseases. Front Biosci. 2006;11:1758–1773. doi: 10.2741/1921. [DOI] [PubMed] [Google Scholar]
- 2.Griffin M, Casadio R, Bergamini CM. Transglutaminases: nature's biological glues. Biochem J. 2002;368:377–396. doi: 10.1042/BJ20021234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dieterich W, Ehnis T, Bauer M, et al. Identification of tissue transglutaminase as the autoantigen of celiac disease. Nat Med. 1997;3:797–801. doi: 10.1038/nm0797-797. [DOI] [PubMed] [Google Scholar]
- 4.Huang L, Haylor JL, Hau Z, et al. Transglutaminase inhibition ameliorates experimental diabetic nephropathy. Kidney Int. 2009;76 doi: 10.1038/ki.2009.230. in this issue. [DOI] [PubMed] [Google Scholar]
- 5.Johnson TS, El-Koraie AF, Skill NJ, et al. Tissue transglutaminase and the progression of human renal scarring. J Am Soc Nephrol. 2003;14:2052–2062. doi: 10.1097/01.asn.0000079614.63463.dd. [DOI] [PubMed] [Google Scholar]
- 6.Skill NJ, Griffin M, El Nahas AM, et al. Increases in renal ε-(γ-glutamyl)-lysine crosslinks result from compartment-specific changes in tissue transglutaminase in early experimental diabetic nephropathy: pathologic implications. Lab Invest. 2001;81:705–716. doi: 10.1038/labinvest.3780279. [DOI] [PubMed] [Google Scholar]
- 7.Johnson TS, Fisher M, Haylor JL, et al. Transglutaminase inhibition reduces fibrosis and preserves function in experimental chronic kidney disease. J Am Soc Nephrol. 2007;18:3078–3088. doi: 10.1681/ASN.2006070690. [DOI] [PubMed] [Google Scholar]
- 8.Kojima S, Nara K, Rifkin DB. Requirement for transglutaminase in the activation of latent transforming growth factor-β in bovine endothelial cells. J Cell Biol. 1993;121:439–448. doi: 10.1083/jcb.121.2.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yamamoto T, Noble NA, Cohen AH, et al. Expression of transforming growth factor-β isoforms in human glomerular diseases. Kidney Int. 1996;49:461–469. doi: 10.1038/ki.1996.65. [DOI] [PubMed] [Google Scholar]
- 10.Shweke N, Boulos N, Jouanneau C, et al. Tissue transglutaminase contributes to interstitial renal fibrosis by favoring accumulation of fibrillar collagen through TGF-β activation and cell infiltration. Am J Pathol. 2008;173:631–642. doi: 10.2353/ajpath.2008.080025. [DOI] [PMC free article] [PubMed] [Google Scholar]