

Abstract
Interleukin-4 (IL-4) is an important immune regulatory protein that possesses potent anti-osteoclastogenic properties, and does so via the transcription factor STAT6. Previous studies have shown that IL-4 selectively blocks RANKL-induced activation of NF-κB and mitogen-activated protein kinase (MAPK) pathway molecules, suggesting that the cytokine arrests osteoclastogenesis by blockade of these signaling cascades. However, the fact that the inhibitory effect on these pathways requires prolonged IL-4 pretreatment, and that the cytokine fails to exert an anti-osteoclastogenic effect after short-term pre-exposure of RANKL to osteoclast precursors, suggests that an additional, more immediate mechanism may also be involved. In this study, we found that simultaneous exposure of IL-4 did not alter RANKL-dependent activation of NF-κB or MAPKs, whereas the cytokine did block RANKL-induced nuclear factor activated T cells c1 (NFATc1), a master osteoclastogenic transcription factor. This inhibitory effect of IL-4 required STAT6, consistent with its functional role in osteoclastogenesis. In addition, the cytokine also partially impaired RANKL-stimulated bone resorption. Furthermore, IL-4 suppressed expression of RANKL-induced osteoclast specific genes in a STAT6-dependent manner, but failed to do so when osteoclast precursors were pre-exposed to RANKL. Thus, we provide the first evidence that IL-4 inhibits osteoclast formation by inhibiting RANKL induction of NFATc1 via STAT6 as an early event, in addition to its suppression of other signaling pathways. The inhibitory effect is ultimately regulated at the gene expression transcriptional level.
Keywords: RANKL, IL-4, STAT6, NFATc1, BONE MARROW MACROPHAGE, OSTEOCLAST
Physiological bone resorption is always coupled with bone formation, permitting the essential bone remodeling cycle. This equilibrium is disrupted in inflammatory osteolysis, in which limited bone formation cannot overcome accelerated resorptive activity prompted by proinflammatory cytokines [Teitelbaum, 2005; Wei and Siegal, 2008; Walsh and Gravallese, 2010]. The rapid degradation of periarticular bone is achieved by osteoclasts, which are abundant in affected joints in conditions such as rheumatoid or psoriatic arthritis, periodontal disease and orthopedic implant loosening [Scott et al., 2000; Ritchlin et al., 2003; Teitelbaum, 2005]. Furthermore, bone destruction represents the most difficult target in the treatment of rheumatoid arthritis. The stimulated osteoclastogenesis in inflammatory conditions is largely due to the increased expression of RANKL produced by bone marrow stromal cells, activated T cells and synovial fibroblasts [Teitelbaum, 2005; Wei and Siegal, 2008; Walsh and Gravallese, 2010]. TNF and IL-1 play a key role in inflammatory osteolysis. Their importance is underscored by the therapeutic success achieved by inhibiting either cytokine [Zwerina et al., 2004].
In addition to the action of destructive mediators, the deleterious process seems to also be under the control of a number of regulatory mediators. Among them, interleukin-4 (IL-4), a Th2 cytokine, has been proved to be a potent anti-osteoclastogenic agent in numerous previous studies. Over-expression of IL-4 in vivo prevents bone erosion in animal models of inflammatory arthritis [Lubberts et al., 2000; Woods et al., 2001; Saidenberg-Kermanac’h et al., 2004]. These findings may have significant implications for the prevention of bone loss in arthritis. IL-4 abrogates osteoclast formation in vitro directly by affecting the commitment of its precursors to osteoclast differentiation, an event mediated by the transcription factor STAT6 [Abu-Amer, 2001; Wei et al., 2002; Moreno et al., 2003], and indirectly by blunting the proinflammatory cytokine TNF- and IL-1-induced RANKL expression under the aegis of p38 mitogen-activated protein kinase (MAPK) in bone marrow stromal cells [Wei et al., 2005].
The molecular mechanism by which IL-4 inhibits RANKL-induced osteoclastogenesis in primary osteoclast precursors, namely bone marrow marcrophages (BMMs), has remained controversial. A previous study found that the cytokine suppressed RANK mRNA expression in the developing precursor cells [Moreno et al., 2003]. In contrast, IL-4-mediated down-regulation of protein levels of RANK or TRAF6, an adaptor molecule that plays a key role in osteoclastogenesis, was not observed in our early study [Wei et al., 2002]. Moreover, IL-4 showed no effect on RANK mRNA expression in primary mature osteoclasts [Mangashetti et al., 2005]. Dissecting the down-stream RANK signaling pathways in BMMs reveals that IL-4 selectively dampens RANKL-induced activation of NF-κB and all three MAPK members, pathway molecules that are essential to osteoclastogenesis, suggesting that the cytokine arrests osteoclastogenesis by blockade of these signaling cascades [Wei et al., 2002]. However, these events take place with a lapse of time and do not occur without prolonged IL-4 pretreatment (i.e., ≥ 24 h). These observations, along with the fact that the cytokine fails to exert an anti-osteoclastogenic effect after short-term exposure of RANKL to BMMs, suggest that an additional, more immediate mechanism may also be involved.
A major breakthrough in osteoclast biology was the identification of nuclear factor activated T cells c1 (NFATc1) as a master osteoclastogenic transcription factor. This molecule, in turn, highlights the essential role of Ca2+ signaling activation in osteoclast differentiation [Negishi-Koga and Takayanagi, 2009]. Thus, the present study was designed to investigate if NFATc1 plays a role in the suppressive effect of IL-4 in osteoclastogenesis in primary osteoclast precursors. We have found that the cytokine blocks RANKL-induced NFATc1, an event that also requires STAT6. Further, the cytokine does so by inhibiting the expression of RANKL-induced, osteoclast-specific genes. In addition, exposure of IL-4 substantially suppresses in vitro RANKL-induced osteoclastic bone resorption. Thus, the previous in vivo finding that IL-4-prevented bone destruction in inflammatory osteolysis likely involves two distinct mechanisms: by targeting early osteoclast precursors to impact osteoclast differentiation and by acting directly on mature osteoclasts to hinder their function.
MATERIALS AND METHODS
ANIMALS
Ethical approval for animal experiments was approved by Institutional Animal Care and Use Committee (IACUC) of University of Alabama at Birmingham. Wild type (WT) C57BL/6 mice were obtained from Harlan Industries (Indianapolis, IN). The STAT6 deficient mice with a background of C57BL/6 were purchased from the Jackson Laboratory (Bar Harbor, ME).
REAGENTS
Recombinant GST-RANKL was purified as previously described [McHugh et al., 2000]. Mouse M-CSF was prepared from the M-CSF-producing cell line, CMG14-12, constructed and provided by Dr. Sunao Takeshita [Takeshita et al., 2000]. Recombinant murine IL-4 was purchased from R&D Systems, Inc. (Minneapolis, MN). Polyclonal anti-IκBα, anti-phospho-IκBα, anti-p44/42ERK, anti-phospho-p44/42ERK, anti-JNK, anti-phospho-JNK, anti-p38, and anti-phospho-p38 were all purchased from Cell Signaling Technology, Inc. (Beverly, MA). Monoclonal anti-NFATc1 was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). All other chemicals were obtained from the Sigma Chemical Co. (St. Louis, MO).
CELL CULTURE
BMMs were isolated from the long bones of 4–6 week old mice and were maintained in α-minimal essential medium (α-MEM) in the absence of heat-inactivated fetal bovine serum, at 37°C, in 5% CO2, as previously described [Liu et al., 2005]. In Brief, after 24 h in culture, M-CSF–dependent macrophages were derived by culturing the nonadherent marrow cells in α-MEM supplemented with 10% heat-inactivated fetal bovine serum, in the presence of M-CSF (220 ng/ml), in petri dishes for 3 days, in order to obtain sufficient cells for subsequent experimental use.
CHARACTERIZATION OF OSTEOCLASTS
To generate osteoclasts from BMMs, 5 × 104 cells per well were plated in 24-well tissue culture plates and cultured in the presence of M-CSF (44 ng/ml) and RANKL (100 ng/ml) as well as IL-4, as appropriate. The osteoclastogenic cultures were stained for tartrate resistant acid phosphatase (TRAP) activity with Leukocyte Acid Phosphatase Kit from Sigma on day 4. All assays were performed in triplicate and repeated at least two times.
IN VITRO BONE RESORPTION ASSAYS
BMMs (5 × 104) were plated on bovine cortical bone slices in 24-well plates, and cultured with M-CSF (44 ng/ml) and RANKL (100 ng/ml) as well as IL-4, as appropriate, for 4 days to stimulate osteoclast formation. The cultures were then continued for 3 more days to allow osteoclasts to resorb bone. The cells were then removed from the bone slices with 0.25 M ammonium hydroxide and mechanical agitation. Bone resorption pits were imaged using a Leica DMIRBE inverted UV SP1 Confocal Microscope System with Leica Confocal software at the Imaging Facility of the University of Alabama at Birmingham. Four areas in bone slices from each bone resorption assay were chosen randomly, and the percentage of resorbed area was determined using ImageJ analysis software obtained from NIH (http://rsb.info.nih.gov/ij/). The assays were performed in triplicate.
IMMUNOBLOTTING
BMMs were cultured in serum-free α-MEM in the presence of M-CSF for 16 h before treatment with RANKL and/or IL-4 for various times as indicated. Cells were washed twice with ice-cold PBS and then lysed in the lysis buffer (Cell Signaling Technology) containing protease inhibitor mixtures (Sigma). Cell lysates was boiled in the presence of SDS sample buffer (0.5 M Tris–HCl, pH 6.8, 10% (w/v) SDS, 10% glycerol, 0.05% (w/v) bromphenol blue) for 5 min and subjected to electrophoresis on 10% SDS–PAGE. Proteins were transferred to nitrocellulose membranes using a semidry blotter (Bio-Rad, Hercules, CA), and incubated in blocking solution (5% nonfat dry milk in TBS containing 0.1% Tween 20) for 1 h to prevent nonspecific binding. Membranes were then exposed to primary antibodies overnight at 4°C, washed three times and incubated with secondary antibody for 1 h. Membranes were washed extensively, and an enhanced chemiluminescence detection assay was performed using the SuperSignal West Dura kit from Pierce (Rockford, IL).
RNA EXTRACTION AND SEMI-QUANTITATIVE RT-PCR
Total RNA was isolated from BMMs using Trizol reagent (Invitrogen Co., Carlsbad, CA). One microgram total RNA was reversed transcribed to cDNA with oligo (dT) using the ThermoScript™ RT-PCR System (Invitrogen). The reverse transcription (RT) was carried out in a 20 μl volume at 55°C, for 55 min, followed by enzyme inactivation and RNA H digestion. Five microliters of the RT reaction mixture was subjected to PCR using Go-Taq DNA polymerase (Promega, Madison, WI) in a 50 μl reaction volume, set for various cycles to monitor the linearity of the amplification, with an annealing temperature at 58°C. The oligonucleotide primers used were as follows: 5′-CTTCTTCTCTGGACGTCAAATG-3′ and 5′-CATTTTGGAAACTCACACGCC-3′ for matrix metallopeptidase 9 (MMP9), 5′-AGAGAACTGGCACAAGGACTT-3′ and 5′-CCTCCTTTC-AGCACTGCATTGT-3′ for carbonic anhydrase II (Car2), 5′-GATGCT-TACCCATATGTGGGC-3′ and 5′-CATATCCTTTGTTTCCCCAGC-3′ for cathepsin K (Ctsk), 5′-GCCAAGATGGATTCATGGGTGG-3′ and 5′-CAGAGACATGATGAAGTCAGCG-3′ for TRAP, 5′-ACATCAT-CCCTGCATCCACTG-3′ and 5′-TCATTGAGAGCAATGCCAGC-3′ for GAPDH. Twenty microliters of the PCR reaction mixture was loaded on a 2% agarose gel for electrophoretic analysis.
STATISTICAL ANALYSIS
Statistical analysis was performed using the Student t-test. A P-value of 0.05 was considered statistically significant.
RESULTS
IL-4 INHIBITS RANKL-INDUCED OSTEOCLASTOGENESIS BY TARGETING AN EARLY STAGE OF OSTEOCLASTOGENESIS
IL-4 is a known potent anti-osteoclastogenic cytokine. To delineate the precise role of IL-4 in osteoclastogenesis, we first carried out assays to examine at which stage IL-4 inhibits osteoclastogenesis. To that end, IL-4 was absent or added to osteoclastgenerating cultures at the dose indicated. TRAP staining was performed on day 4. As seen in Figure 1, optimal concentrations of M-CSF and RANKL induced confluent TRAP-positive multinucleated osteoclasts, while IL-4 exerted a maximum inhibitory effect at a very low dose (4 ng/ ml), thus indicating its potency and verifying our previous findings (top panel) [Wei et al., 2002]. Attesting to function, substantial resorption pits were seen when cells were plated on bovine bone slices in a parallel study, whereas only minimal bone resorption was present in the presence of IL-4 (lower panel). Conversely, the cytokine failed to block osteoclastogenesis after a 36-h exposure of BMMs to RANKL, even at a much higher dose (Fig. 2A, top panel), recapitulating the previous findings [Wei et al., 2002]. Thus, IL-4 inhibits RANKL-induced osteoclastogenesis by targeting an early stage of osteoclastogenesis.
Fig. 1.
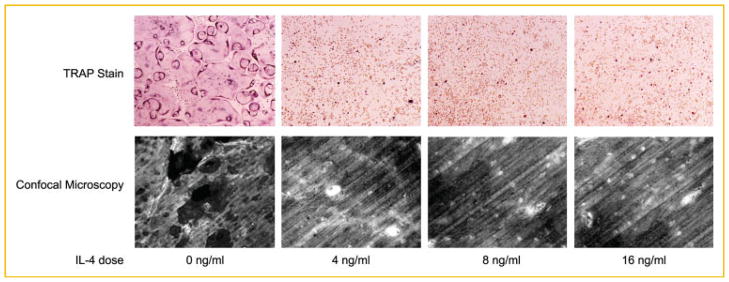
IL-4 inhibits RANKL-induced osteoclastogenesis by targeting an early stage of osteoclastogenesis. Osteoclasts were generated in the presence of M-CSF (44 ng/ml) and RANKL (100 ng/ml) in the absence and presence of IL-4 at the dose indicated. TRAP staining was performed on day 4 (top panel). In parallel, the same cultures were carried out on the bovine bone slices, followed by removal of the cells from the slices and image analysis as described in the Materials and Methods Section (lower panel). [Color figure can be seen in the online version of this article, available at http://wileyonlinelibrary.com/journal/jcb]
Fig. 2.

IL-4 partially impairs RANKL-induced bone resorption. A: BMMs were cultured in the presence of M-CSF and RANKL in tissue culture dishes (top panels) and on bovine bone slices (lower panels) for 36 h. The cultures were then continued in the absence and presence of IL-4 at the dose indicated. TRAP staining was performed on day 4 (top panels). Bone slices were subjected to image analysis for resorption pits as described in the Materials and Methods Section (lower panels). B: Quantification of the bone resorption assays. Four areas in the bone slices from each bone resorption assay shown in (A) were randomly chosen, and the percentage of resorbed area was determined. Data are presented as means ± standard deviation (*P <0.001 vs. absence of IL-4). No statistical significance was found between the conditions with presence of IL-4. [Color figure can be seen in the online version of this article, available at http://wileyonlinelibrary.com/journal/jcb]
IL-4 INHIBITS RANKL-INDUCED OSTEOCLAST ACTIVATION
We next asked if IL-4 inhibits RANKL-induced osteoclast activation. Osteoclasts were generated in the presence of M-CSF and RANKL on bone slices to allow osteoclastic bone resorption, and IL-4 was added to the cultures after 36 h at the dose indicated. The magnitude of bone resorption, as expressed by a percent of the resorption area/ bone surface, was significantly reduced in the presence of IL-4 (Fig. 2A, lower panel, and 2B). Similarly, IL-4 exerted its maximum inhibitory effect on bone resorption at a very low dose (4 ng/ml). However, different from the potent inhibitory effect on osteoclast differentiation, IL-4 suppression of RANKL-induced osteoclast activation was incomplete, as the extent of inhibition remained even when IL-4 dose was increased substantially.
RANKL-INDUCED ACTIVATION OF NF-κB AND MAPKs REMAINS INTACT WHEN BMMs ARE SIMULTANEOUSLY EXPOSED TO IL-4
We next explored the molecular mechanisms by which IL-4 inhibits osteoclastogenesis by dissecting the RANKL-activated signaling cascades. RANKL binds to its receptor RANK on osteoclast precursors to activate a number of signaling pathway molecules involved in osteoclast differentiation, including NF-κB and all three MAPK members [Teitelbaum, 2007]. While our previous study showed the inhibitory effects of IL-4 on RANKL-induced activation of NF-κB and MAPK, these events are not immediate and do not occur without prolonged IL-4 pretreatment [Wei et al., 2002]. In this study, simultaneous exposure of cells to IL-4 failed to impact these aspects of RANKL-induced signaling (Fig. 3). Thus, the fact that IL-4 targets only early osteoclast precursors to block osteoclastogenesis but does not blunt immediate RANKL-induced activation of NF-κB and MAPK suggests that there might be another signaling pathway involved in this process.
Fig. 3.

RANKL-induced activation of NF-κB and MAPKs remains intact when BMMs are simultaneously exposed to IL-4 and RANKL. BMMs were treated with IL-4 (4 ng/ml) and RANKL (100 ng/ml) as indicated. Cells were lysed and analyzed by Western blotting using antibodies against phospho-IκB (p-IκB), phospho-ERK (p-ERK), phospho-JNK (p-JNK), and phospho-p38 (p-p38). The same volume of lysates were run and then probed with antibodies against IκB, ERK, JNK, and p38 as loading controls. [Color figure can be seen in the online version of this article, available at http://wileyonlinelibrary.com/journal/jcb]
IL-4 INHIBITS RANKL-INDUCED NFATc1 ACTIVATION IN A STAT6-DEPENDENT MANNER
We next turned to determine if IL-4 alters RANKL-activated NFATc1 signaling. Given that IL-4 inhibits osteoclastogenesis via STAT6, we also asked whether the effect of IL-4 on RANKL-induced NFATc1 signaling, if any, is STAT6-dependent. To this end, osteoclast precursors were stimulated by RANKL for 24 h in the absence or presence of IL-4, and cell lysates were subjected to Western blot analysis. As seen in Figure 4, while simultaneous exposure of IL-4 substantially dampens RANKL-induced NFATc1 activation, the cytokine fails to affect NFATc1 signaling in osteoclast precursors derived from STAT6-deficient mice. Thus, the findings suggest that IL-4 inhibition of osteoclastogenesis involves a novel mechanism—blocking of RANKL-induced activation of NFATc1.
Fig. 4.
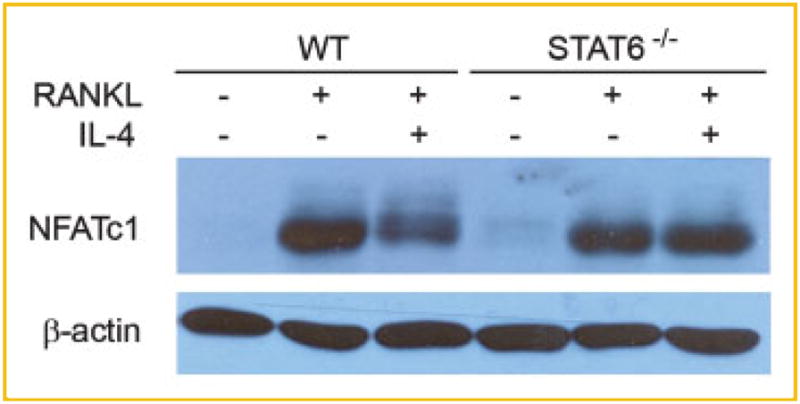
IL-4 inhibits RANKL-induced NFATc1 activation via STAT6. BMMs from WT and STAT6 deficient mice (STAT6−/−) were treated with RANKL for 24 h in the absence or presence of IL-4 (4 ng/ml). Cells were lysed and subjected to Western blotting using antibodies against NFATc1. Blots were stripped and then reprobed with β-actin antibody as the loading control. [Color figure can be seen in the online version of this article, available at http://wileyonlinelibrary.com/journal/jcb]
IL-4 INHIBITS RANKL-INDUCED GENE EXPRESSION OF MMP9, Car2, Ctsk, AND TRAP VIA STAT6
Gene expression profiling studies have demonstrated that RANKL directs osteoclastogenesis by inducing expression of a subset of genes [Cappellen et al., 2002; Ishida et al., 2002; Rho et al., 2002]. Notably, RANKL greatly activates the genes encoding MMP9, Car2, Ctsk, and TRAP [Cappellen et al., 2002; Rho et al., 2002]. Thus, the expression of these genes is widely used as markers for osteoclasts [Teitelbaum and Ross, 2003]. To further explore the molecular basis of the roles of IL-4 in RANKL-induced osteoclastogenesis, we examined the effect of IL-4 on the expression of these genes. Whereas 1-day RANKL treatment markedly increased mRNA levels of these genes (2 days in case of Ctsk), the presence of IL-4 substantially diminished transcription of these genes induced by RANKL (Fig. 5). Moreover, the inability of IL-4 to arrest transactivation of these molecules in cells lacking STAT6 further supports the previous observation that the transcription factor mediates IL-4 inhibition of osteoclastogenesis. In addition, IL-4 failed to exert this inhibitory effect when exposed to the cells at a later time, thus reflecting the fact that the cytokine targets only early stage of osteoclastogenesis (Fig. 6). These findings indicate that IL-4 inhibits osteoclastogenesis by preventing the expression of RANKL-induced osteoclast-specific genes, and once activated, these genes are refractory to the inhibitory effect of IL-4.
Fig. 5.
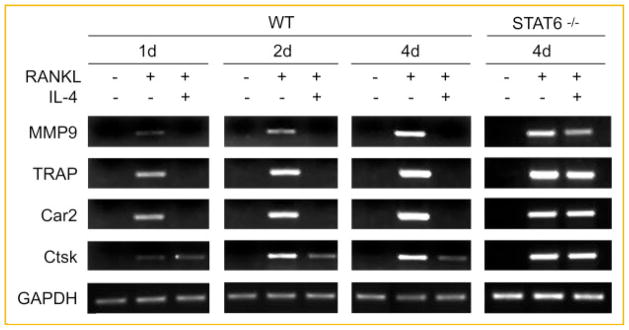
IL-4 inhibits RANKL-induced gene expression of MMP9, Car2, Ctsk, and TRAP via STAT6. BMMs from WT and STAT6−/− mice were maintained in the absence or presence of RANKL (100 ng/ml) and/or IL-4 (4 ng/ml) for various periods of time as indicated. Total RNA was extracted from the cells and applied for semi-quantitative RT-PCR of the several osteoclast-specific genes as indicated.
Fig. 6.

RANKL-induced osteoclast-specific genes are not inhibited by subsequent treatment of IL-4. BMMs were maintained in the absence or presence of RANKL (100 ng/ml) for 2 days without IL-4. The cell cultures were then continued in the absence (left and middle lanes) or presence of IL-4 (4 ng/ml; right lanes) for 2 more days. Gene expression was determined by semi-quantitative RT-PCR.
DISCUSSION
RANKL and IL-4 are unique cytokines in leukocyte biology in that they both induce fusion of mononuclear precursors of the monocyte/macrophage lineage, which leads to multinucleation [Nelms et al., 1999]. Yet, the polykaryons induced by the latter are those characteristic of foreign body giant cells [McNally and Anderson, 1995]. We have previously shown that the IL-4 directly inhibits in vitro osteoclastogenesis by selectively arresting RANKL-induced activation of NF-κB and MAPK pathways in primary osteoclast precursors [Wei et al., 2002]. In this study, we provided an additional mechanism for its potent anti-osteoclastogenic effect.
NFATc1 is a recently identified master regulator of RANKL-induced osteoclast differentiation. The expression of NFATc1 is up-regulated robustly during osteoclastogenesis [Asagiri et al., 2005]. NFATc1-deficient embryonic stem cells fail to differentiate into osteoclasts in response to RANKL [Takayanagi et al., 2002]. We found that IL-4 diminished RANKL-induced NFATc1 activation in primary BMMs at a concentration sufficient to inhibit osteoclastogenesis (4 ng/ml). This finding is in keeping with a previous study using a substantially higher dose of IL-4 (100 ng/ml) [Kamel Mohamed et al., 2005]. More importantly, we demonstrated, for the first time, that IL-4 blockade of RANKL-induced NFATc1 activation is STAT6-dependent, thus reflecting the mechanism of IL-4 inhibition of osteoclastogenesis.
The finding that IL-4 fails to prevent osteoclastogenesis in RANKL-pretreated BMMs is also of mechanistic importance. In the current study, we have shown that IL-4 blocks RANKL-induced expression of four osteoclast-specific genes, MMP9, Car2, Ctsk, and TRAP, via STAT6. The lack of this inhibitory effect in RANKL-pretreated BMMs indicates that the early commitment of osteoclast precursors initiated by RANKL renders these cells refractory to IL-4, and may do so by altering the osteoclast-specific genes into a state in which IL-4 is incapable to suppress their activation. Interestingly, a number of these genes are regulated by NFATc1, further supporting our hypothesis that IL-4 inhibits osteoclastogenesis by targeting, at least in part, NFATc1. Specifically, NFATc1 binds directly to the promoter region of a number of osteoclast-specific genes, including MMP9 and Ctsk [Sundaram et al., 2007; Song et al., 2009]. Over-expression of NFATc1 also markedly transactivates the TRAP gene promoter [Ikeda et al., 2004]. Moreover, knockdown of NFATc1 by short interfering RNA caused TRAP expression to be down-regulated, and ectopic expression of NFATc1 abrogated the IL-4-induced down-regulation of TRAP [Yu et al., 2009]. Taken together, NFATc1 plays a key role in mediating the inhibitory effect of IL-4 in osteoclast differentiation. Thus, IL-4 inhibits RANKL-induced osteoclastogenesis at multiple levels, involving NFATc1, NF-κB, and all three MAP kinase members, at different stages of differentiation.
The effect of IL-4 in osteoclast activation has not been fully understood. We found that IL-4 partially inhibited RANKL-induced bone resorption, in keeping with a previous observation [Mangashetti et al., 2005]. However, different from another study [Moreno et al., 2003], a near complete inhibition of bone resorption was not found in our study. Interestingly, IL-4 inhibition of bone resorption occurs, at least in part, through prevention of RANKL-induced intracellular ionized calcium changes [Mangashetti et al., 2005], suggesting that IL-4 may exert its inhibitory effect by inactivation of NFATc1 in mature osteoclasts. In opposition to this hypothesis, the ablation of NFATc1 in mature osteoclasts did not prevent bone resorption activity [Kim et al., 2006]. Thus, the exact molecular mechanisms by which IL-4 inhibits bone resorption remains to be elucidated.
In summary, we provide the first evidence that IL-4 inhibits osteoclast formation by blocking RANKL induction of NFATc1 via the transcription factor STAT6. The inhibitory effect is ultimately regulated at the gene expression level. Further studies aimed at the interaction of STAT6 and NFATc1 may provide new insights into novel therapeutic targets in preventing and treating bone loss in various inflammatory conditions.
Acknowledgments
Grant sponsor: Center for Metabolic Bone Disease at the University of Alabama at Birmingham; Grant sponsor: National Scholarship for Building a High Level University from the Chinese Ministry of Education; Grant sponsor: NIAMS (AR47830).
The work was supported in part by a pilot grant from the Center for Metabolic Bone Disease at the University of Alabama at Birmingham (to SW), the National Scholarship for Building a High Level University from the Chinese Ministry of Education (to JC), and a NIAMS grant AR47830 (to XF).
Abbreviations
- RANKL
receptor activator of NF-κB ligand
- M-CSF
macrophage colony-stimulating factor
- IL-4
interleukin-4
- STAT6
signal transducer and activator of transcription 6
- BMM
bone marrow macrophage
- TNF
tumor necrosis factor
- IL-1
interleukin-1
- MAPK
mitogen-activated protein (MAP) kinase
- NFATc1
nuclear factor activated T cells c1
- MMP9
matrix metallopeptidase 9
- Car2
carbonic anhydrase II
- Ctsk
cathepsin K
- TRAP
tartrate resistant acid phosphatase
References
- Abu-Amer Y. IL-4 abrogates osteoclastogenesis through STAT6-dependent inhibition of NF-kappaB. J Clin Invest. 2001;107:1375–1385. doi: 10.1172/JCI10530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asagiri M, Sato K, Usami T, Ochi S, Nishina H, Yoshida H, Morita I, Wagner EF, Mak TW, Serfling E, Takayanagi H. Autoamplification of NFATc1 expression determines its essential role in bone homeostasis. J Exp Med. 2005;202:1261–1269. doi: 10.1084/jem.20051150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappellen D, Luong-Nguyen NH, Bongiovanni S, Grenet O, Wanke C, Susa M. Transcriptional program of mouse osteoclast differentiation governed by the macrophage colony-stimulating factor and the ligand for the receptor activator of NFkappa B. J Biol Chem. 2002;277:21971–21982. doi: 10.1074/jbc.M200434200. [DOI] [PubMed] [Google Scholar]
- Ikeda F, Nishimura R, Matsubara T, Tanaka S, Inoue J, Reddy SV, Hata K, Yamashita K, Hiraga T, Watanabe T, Kukita T, Yoshioka K, Rao A, Yoneda T. Critical roles of c-Jun signaling in regulation of NFAT family and RANKL-regulated osteoclast differentiation. J Clin Invest. 2004;114:475–484. doi: 10.1172/JCI19657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida N, Hayashi K, Hoshijima M, Ogawa T, Koga S, Miyatake Y, Kumegawa M, Kimura T, Takeya T. Large scale gene expression analysis of osteoclastogenesis in vitro and elucidation of NFAT2 as a key regulator. J Biol Chem. 2002;277:41147–41156. doi: 10.1074/jbc.M205063200. [DOI] [PubMed] [Google Scholar]
- Kamel Mohamed SG, Sugiyama E, Shinoda K, Hounoki H, Taki H, Maruyama M, Miyahara T, Kobayashi M. Interleukin-4 inhibits RANKL-induced expression of NFATc1 and c-Fos: A possible mechanism for downregulation of osteoclastogenesis. Biochem Biophys Res Commun. 2005;329:839–845. doi: 10.1016/j.bbrc.2005.02.049. [DOI] [PubMed] [Google Scholar]
- Kim MS, Day CJ, Selinger CI, Magno CL, Stephens SR, Morrison NA. MCP-1-induced human osteoclast-like cells are tartrate-resistant acid phosphatase, NFATc1, and calcitonin receptor-positive but require receptor activator of NFkappaB ligand for bone resorption. J Biol Chem. 2006;281:1274–1285. doi: 10.1074/jbc.M510156200. [DOI] [PubMed] [Google Scholar]
- Liu W, Wang S, Wei S, Sun L, Feng X. Receptor activator of NF-kappaB (RANK) cytoplasmic motif, 369PFQEP373, plays a predominant role in osteoclast survival in part by activating Akt/PKB and its downstream effector AFX/FOXO4. J Biol Chem. 2005;280:43064–43072. doi: 10.1074/jbc.M509006200. [DOI] [PubMed] [Google Scholar]
- Lubberts E, Joosten LA, Chabaud M, van Den Bersselaar L, Oppers B, Coenen-De Roo CJ, Richards CD, Miossec P, van Den Berg WB. IL-4 gene therapy for collagen arthritis suppresses synovial IL-17 and osteoprotegerin ligand and prevents bone erosion. J Clin Invest. 2000;105:1697–1710. doi: 10.1172/JCI7739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangashetti LS, Khapli SM, Wani MR. IL-4 inhibits bone-resorbing activity of mature osteoclasts by affecting NF-kappa B and Ca2+ signaling. J Immunol. 2005;175:917–925. doi: 10.4049/jimmunol.175.2.917. [DOI] [PubMed] [Google Scholar]
- McHugh KP, Hodivala-Dilke K, Zheng MH, Namba N, Lam J, Novack D, Feng X, Ross FP, Hynes RO, Teitelbaum SL. Mice lacking beta3 integrins are osteosclerotic because of dysfunctional osteoclasts. J Clin Invest. 2000;105:433–440. doi: 10.1172/JCI8905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNally AK, Anderson JM. Interleukin-4 induces foreign body giant cells from human monocytes/macrophages. Differential lymphokine regulation of macrophage fusion leads to morphological variants of multinucleated giant cells. Am J Pathol. 1995;147:1487–1499. [PMC free article] [PubMed] [Google Scholar]
- Moreno JL, Kaczmarek M, Keegan AD, Tondravi M. IL-4 suppresses osteoclast development and mature osteoclast function by a STAT6-dependent mechanism: Irreversible inhibition of the differentiation program activated by RANKL. Blood. 2003;102:1078–1086. doi: 10.1182/blood-2002-11-3437. [DOI] [PubMed] [Google Scholar]
- Negishi-Koga T, Takayanagi H. Ca2+-NFATc1 signaling is an essential axis of osteoclast differentiation. Immunol Rev. 2009;231:241–256. doi: 10.1111/j.1600-065X.2009.00821.x. [DOI] [PubMed] [Google Scholar]
- Nelms K, Keegan AD, Zamorano J, Ryan JJ, Paul WE. The IL-4 receptor: Signaling mechanisms and biologic functions. Annu Rev Immunol. 1999;17:701–738. doi: 10.1146/annurev.immunol.17.1.701. [DOI] [PubMed] [Google Scholar]
- Rho J, Altmann CR, Socci ND, Merkov L, Kim N, So H, Lee O, Takami M, Brivanlou AH, Choi Y. Gene expression profiling of osteoclast differentiation by combined suppression subtractive hybridization (SSH) and cDNA microarray analysis. DNA Cell Biol. 2002;21:541–549. doi: 10.1089/104454902320308915. [DOI] [PubMed] [Google Scholar]
- Ritchlin CT, Haas-Smith SA, Li P, Hicks DG, Schwarz EM. Mechanisms of TNF-alpha- and RANKL-mediated osteoclastogenesis and bone resorption in psoriatic arthritis. J Clin Invest. 2003;111:821–831. doi: 10.1172/JCI16069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saidenberg-Kermanac’h N, Bessis N, Lemeiter D, de Vernejoul MC, Boissier MC, Cohen-Solal M. Interleukin-4 cellular gene therapy and osteoprotegerin decrease inflammation-associated bone resorption in collagen-induced arthritis. J Clin Immunol. 2004;24:370–378. doi: 10.1023/B:JOCI.0000029116.12371.bf. [DOI] [PubMed] [Google Scholar]
- Scott DL, Pugner K, Kaarela K, Doyle DV, Woolf A, Holmes J, Hieke K. The links between joint damage and disability in rheumatoid arthritis. Rheumatology (Oxford) 2000;39:122–132. doi: 10.1093/rheumatology/39.2.122. [DOI] [PubMed] [Google Scholar]
- Song I, Kim JH, Kim K, Jin HM, Youn BU, Kim N. Regulatory mechanism of NFATc1 in RANKL-induced osteoclast activation. FEBS Lett. 2009;583:2435–2440. doi: 10.1016/j.febslet.2009.06.047. [DOI] [PubMed] [Google Scholar]
- Sundaram K, Nishimura R, Senn J, Youssef RF, London SD, Reddy SV. RANK ligand signaling modulates the matrix metalloproteinase-9 gene expression during osteoclast differentiation. Exp Cell Res. 2007;313:168–178. doi: 10.1016/j.yexcr.2006.10.001. [DOI] [PubMed] [Google Scholar]
- Takayanagi H, Kim S, Koga T, Nishina H, Isshiki M, Yoshida H, Saiura A, Isobe M, Yokochi T, Inoue J, Wagner EF, Mak TW, Kodama T, Taniguchi T. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev Cell. 2002;3:889–901. doi: 10.1016/s1534-5807(02)00369-6. [DOI] [PubMed] [Google Scholar]
- Takeshita S, Kaji K, Kudo A. Identification and characterization of the new osteoclast progenitor with macrophage phenotypes being able to differentiate into mature osteoclasts. J Bone Miner Res. 2000;15:1477–1488. doi: 10.1359/jbmr.2000.15.8.1477. [DOI] [PubMed] [Google Scholar]
- Teitelbaum S. Osteoclasts; culprits in inflammatory osteolysis. Arthritis Res Ther. 2005;29:201. doi: 10.1186/ar1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teitelbaum SL. Osteoclasts: What do they do and how do they do it? Am J Pathol. 2007;170:427–435. doi: 10.2353/ajpath.2007.060834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teitelbaum SL, Ross FP. Genetic regulation of osteoclast development and function. Nat Rev Genet. 2003;4:638–649. doi: 10.1038/nrg1122. [DOI] [PubMed] [Google Scholar]
- Walsh NC, Gravallese EM. Bone remodeling in rheumatic disease: A question of balance. Immunol Rev. 2010;233:301–312. doi: 10.1111/j.0105-2896.2009.00857.x. [DOI] [PubMed] [Google Scholar]
- Wei S, Siegal GP. Mechanisms modulating inflammatory osteolysis: A review with insights into therapeutic targets. Pathol Res Pract. 2008;204:695–706. doi: 10.1016/j.prp.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei S, Wang MW, Teitelbaum SL, Ross FP. Interleukin-4 reversibly inhibits osteoclastogenesis via inhibition of NF-kappa B and mitogen-activated protein kinase signaling. J Biol Chem. 2002;277:6622–6630. doi: 10.1074/jbc.M104957200. [DOI] [PubMed] [Google Scholar]
- Wei S, Kitaura H, Zhou P, Ross FP, Teitelbaum SL. IL-1 mediates TNF-induced osteoclastogenesis. J Clin Invest. 2005;115:282–290. doi: 10.1172/JCI23394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods JM, Katschke KJ, Volin MV, Ruth JH, Woodruff DC, Amin MA, Connors MA, Kurata H, Arai K, Haines GK, Kumar P, Koch AE. IL-4 adenoviral gene therapy reduces inflammation, proinflammatory cytokines, vascularization, and bony destruction in rat adjuvant-induced arthritis. J Immunol. 2001;166:1214–1222. doi: 10.4049/jimmunol.166.2.1214. [DOI] [PubMed] [Google Scholar]
- Yu M, Moreno JL, Stains JP, Keegan AD. Complex regulation of tartrate-resistant acid phosphatase (TRAP) expression by interleukin 4 (IL-4): IL-4 indirectly suppresses receptor activator of NF-kappaB ligand (RANKL)-mediated TRAP expression but modestly induces its expression directly. J Biol Chem. 2009;284:32968–32979. doi: 10.1074/jbc.M109.001016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwerina J, Hayer S, Tohidast-Akrad M, Bergmeister H, Redlich K, Feige U, Dunstan C, Kollias G, Steiner G, Smolen J, Schett G. Single and combined inhibition of tumor necrosis factor, interleukin-1, and RANKL pathways in tumor necrosis factor-induced arthritis: Effects on synovial inflammation, bone erosion, and cartilage destruction. Arthritis Rheum. 2004;50:277–290. doi: 10.1002/art.11487. [DOI] [PubMed] [Google Scholar]