

Abstract
Microglia cells have been implicated, to some extent, in the pathogenesis of all of the common neurodegenerative disorders involving protein aggregation such as Alzheimer’s disease, Parkinson’s disease and Amyotrophic Lateral Sclerosis. However, the precise role they play in the development of the pathologies remains unclear and it seems that they contribute to the pathological process in different ways depending on the specific disorder. A better understanding of their varied roles is essential if they are to be the target for novel therapeutic strategies.
Keywords: Microglia, protein aggregates neuroinflammation, cytokines, neurotoxicity, Alzheimer’s, Parkinson’s, ALS
Introduction
Microglia are the resident immune cells of the central nervous system and, contrary to the description of them as being “resting” when in their highly ramified form, they are constantly monitoring the environment for any signs of change (1, 2). In this role they are inevitably activated to some extent by any brain pathology and this is particularly evident in those neurodegenerative disorders which involve protein aggregation events such as Alzheimer’s and Parkinson’s disease. However, what remains unclear is the precise nature of that activation and whether it is similar in different diseases. One of the key questions is, do they simply react to external stimuli to phagocytose and remove protein aggregates or do they have an active role in the development and progression of protein aggregate pathology? Furthermore, while they might intuitively have a role in dealing with extracellular protein aggregates in the brain parenchyma do they also react to intracellular protein aggregations such as neurofibrillary tangles and Lewy bodies? In this short review we will address these and a number of other questions related to the neuropathology of the main protein aggregation disorders.
Alzheimer’s disease (AD)
The potential roles of microglia in neuroinflammation and the progression of pathology have been studied to varying degrees in all of the common neurodegenerative disorders but the most extensive literature relates to AD. Two papers published back-to-back in 1989 were instrumental in highlighting the potential role of inflammatory cytokines in driving AD pathology. Griffin and colleagues showed that there was a 30 fold increase in the number of interleukin 1(IL-1) immunoreactive microglia in AD and Down’s syndrome brains as compared to age-matched controls (3). Furthermore, Goldgaber and colleagues showed in human umbilical vein endothelial cells that IL-1 upregulates amyloid precursor protein (APP) gene expression (4). Processing of APP gives rise to the Aβ peptide that is the main constituent of the senile plaques found in the AD brain (5). Taken together these two observations raised the possibility that, rather than having a purely reactive phagocytic role, microglia could drive the formation of Aβ plaques. Since then there has been a lot of interest in exactly how microglial phenotype correlates with plaque type, progression and distribution (6, 7). In terms of plaque type there appears to be a particular association between IL-1 immunoreactive microglia and diffuse Aβ plaques whereas the dense plaque cores, presumed to be the end-stage of plaque formation, do not show any such association (8). There is also correlation between microglia with a neuroinflammatory phenotype and the anatomical distribution of neuritic plaques (9) suggesting perhaps a role for cytokines in the formation of this type of plaque (10). These observations were brought together by Sue Griffin when she postulated the existence of a “cytokine cycle” whereby glial cells in a positive feedback cycle of cytokine release drive the development of AD pathology (11) (Fig 1). Such a sustained pro-inflammatory activation can have significant neurotoxic effects (12).
Fig 1.
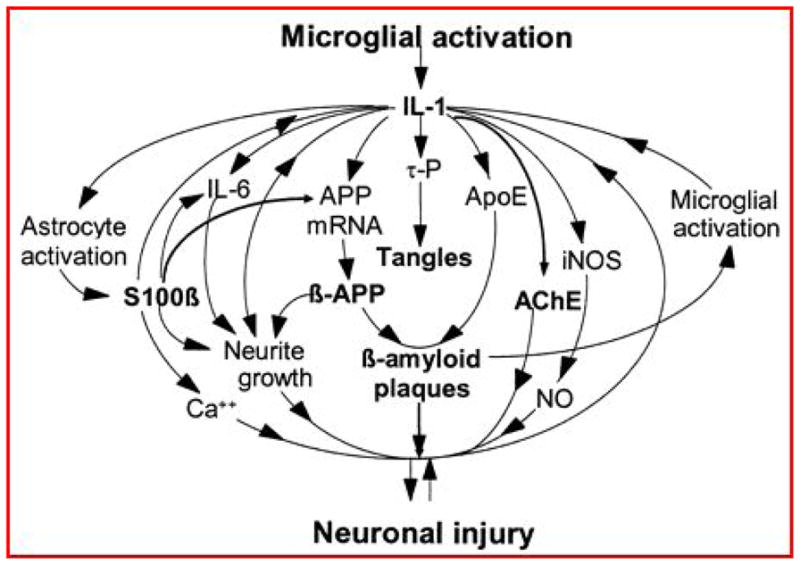
The cytokine cycle [6] which postulates a key role for microglial activation and cytokine release in the formation of protein aggregate pathology and ultimately neuronal injury.
An alternative view is that the microglial cells do not drive pathological change at all but are in fact themselves impaired. More specifically, it is suggested that the microglial cells associated with the plaques are not activated but are actually dystrophic or senescent (13). Streit and colleagues carried out a detailed morphological examination of microglia in a series of AD brains at varying Braak stages. They reported an association of ramified “non-activated” microglial cells with Aβ plaques and an association of dystrophic damaged, rather than functionally active, microglia with the various forms of tau pathology (14). On this basis the microglia could be considered as neuroprotective and if there is an age-related diminishment in this protective capacity the neurons will become vulnerable. A potential result of this would be the development of neurofibrillary tangle pathology. There have been a number of studies showing such an association between tangle distribution and activated microglia (7, 15, 16). In one of these studies the distribution of microglia in relation to that of neurofibrillary tangles was examined in AD and elderly control cases and it was found that microglia are most extensive in areas with significant tangle pathology (in AD and control cases). The conclusion drawn was that the regional distribution of microglia might provide the template for the development of the tangles (16).
Further evidence that microglia may play an active role in causing neuronal death, rather than reacting to it, comes from in vivo imaging studies in animals. Using two-photon imaging in transgenic mice expressing the Cx3cr1 gene, tagged with green fluorescent protein, it has been shown that microglia are very mobile and are constantly remodelling their processes, presumably as part of their active monitoring role (17). When there is any perturbation of the parenchyma this remodelling becomes more focussed and the microglia move towards the damaged area of tissue. However, using this technique in a triple transgenic AD mouse model it has been shown that microglia are recruited early and not just after the death of neurons (18). Furthermore when the triple transgenics were crossed with Cx3cr1 knockout mice the neuronal loss was prevented. Cx3cr1 is a chemokine receptor which binds to fractalkine and is important in neuron-microglia interactions.
While there is significant evidence that microglia can contribute to the initiation and/or progression of AD pathology it’s also clear that they can have beneficial effects. In animal models of AD it has been shown that impairment of microglial phagocytic activity accelerates pathology progression (19) whereas exogenous supplementation of microglia favours clearance of Aβ plaques (20). Similarly, post mortem follow up of patients who took part in a clinical trial of Aβ peptide immunisation revealed evidence of extensive phagocytic clearance of Aβ from the cortex (21).
The basis for these apparently contradictory roles of microglia is that there is heterogeneity in the activation process. What defines a microglial phenotype and how the cell is going to react to a stimulus depends on the nature of that stimulus and what other factors are present locally. It is clear that there are alternatives to the classical activation pathway whereby microglia exposed to Aβ peptide secrete inflammatory cytokines. Based on work primarily on peripheral macrophages alternative activation pathways have been identified whereby microglia can be stimulated to secrete anti-inflammatory cytokines and trophic factors (22–24). Some of these alternative activation states of microglia in the brain have been observed in AD and mouse models of AD (25). It is this mixed functional phenotype of the microglia in the AD brain, which is still not well understood, that provides the biggest challenge in terms of possible therapeutic intervention. Is there a normal physiological balance between the alternative activation states that we need to try and correct in AD? Can individual microglia move between different activation states? Are the microglial cells in specific activated state at different stages of the disease and hence, is the timing of any intervention important? (26, 27).
Parkinson’s disease (PD)
As is seen in AD, there is a neuroinflammatory component to the pathology of PD. In the substantia nigra, the locus classicus of PD pathology, the loss of dopaminergic cells is associated with the presence of activated microglia cells (28–30). This specific association between differential susceptibility of cells in the substantia nigra and the number of reactive microglia is supported by rodent studies in which the inflammatory toxin, LPS, was injected in to different brain regions and the numbers of microglia found in the midbrain were significantly higher (31). Interferon-γ (IFN-γ) is increased in the plasma of patients with PD (32) and has been implicated in the development of nigrostriatal degeneration (33). Based on these observations IFN-γ deficient mice were treated with MPTP, which kills the dopaminergic cells in the substantia nigra, and it was observed that there were fewer microglial cells present and less dopaminergic cell loss in these animals than in the wild type animals. This would support the concept of a detrimental effect of microglial activation in the substantia nigra of PD patients. The inflammatory component of PD pathology has been extensively reviewed and one of the key potential pathological factors regarding microglia is that they express inducible nitric oxide synthase (iNOS) which can generate free radicals and potentially cause oxidative damage to dopaminergic cells (34).
While these various inflammatory mechanisms can be used to explain some of the dopamainergic cell death they don’t directly address the toxic effects of the alpha-synuclein (αSN) aggregates that characterise PD pathology. αSN has been shown to promote microglia activation (35, 36) and more specifically the monomeric form is more effective than the aggregates in promoting phagocytosis (37). This is similar to the findings in AD where the oligomeric forms of the Aβ peptide are thought to be more toxic. Perhaps unsurprisingly much of the research focus has been on the substantia nigra in PD but with the advent of αSN antibodies it’s become clear that there is a much wider, extra-nigral, distribution of αSN pathology throughout the brain in PD. It is these pathological αSN lesions that are probably responsible for many of the non-motor symptoms in PD and they are also associated with microglial activation and proliferation (38). In this respect there is a similar variety of microglial functional phenotype as is seen in AD (see Fig 2).
Fig 2.

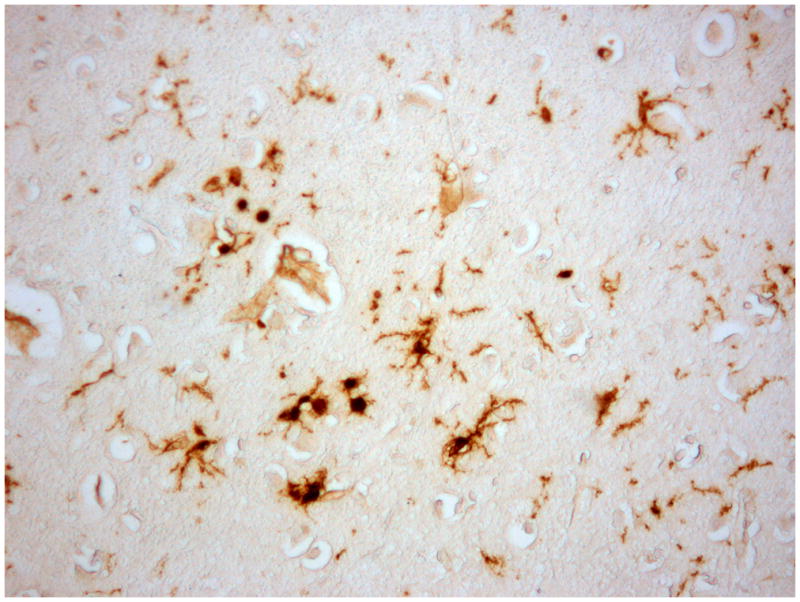

Microglial cells in a Parkinson’s disease brain demonstrating a variety of morphologies and displaying different phenotypic markers. A) Ionized calcium binding adaptor molecule 1 (iba1), a pan-microglial marker staining cells with multiple morphologies. B) Microglia with a more activated morphology, with retracted thicker processes immunostained with an antibody to CR3/43, an MHC II marker. C) Perivascular macrophages and some parenchymal microglial-like cells immunostained with CD163, a membrane-bound scavenger receptor.
There is a similar discussion to that in AD as to whether microglial cells initiate or react to pathology in PD. More recently it has been suggested that astrocytes may have more of a primary role and that microglia are reactive (39). More specifically αSN has been found to accumulate in astrocytes and this parallels the spread of pathology (40, 41). Via a mechanism that has yet to be determined this astrocytic accumulation of αSN is thought to initiate a microglial reaction. In support of this, astrocytic expression of mutated αSN has been shown to cause neurodegeneration in mice (42).
Amyotrophic Lateral Sclerosis (ALS)
In ALS there is a complex interplay between microglia and T cells that modify the rate of disease progression in a way that is not seen in other neurodegenerative disorders (43). Microglial activation has been observed around motoneurons in the primary motor cortex, brainstem motor nuclei and in the ventral horn of the spinal cord (44, 45). Microglial activation has also been seen in vivo in ALS patients using the PK11195 ligand in PET imaging (46). Based largely on observations in animal models of ALS the microglial cells seem to have neuroprotective and neurotoxic roles, slowing and accelerating disease progression respectively (47). This was nicely illustrated in a recent study in a mutant superoxide dismutase (mSOD1) animal model where the microglial phenotype apparently changed during the course of the disease. The microglia isolated at disease onset had a neuroprotective phenotype whereas those isolated from end-stage disease animals had a classical neuroinflammatory phenotype (48). Earlier studies in mSOD1 where the mutant protein was expressed in the microglia showed an enhanced neurotoxicity of these cells compared to wild type microglia not expressing the mutant (49, 50). Conversely a possible neuroprotective role for microglia in ALS patients is supported by the observation of insulin growth factor II (IGF-II) in these cells in post mortem tissue samples (51).
Summary
It appears that microglial cells have a role in the pathogenesis of AD, PD and ALS but their precise role appears to vary between these disorders. In AD there is evidence for a role in the initiation and progression of the disease. Alternatively, senescence and impaired function of the microglia may lead to loss of a neuroprotective function. In PD the microglial response does not seem to be quite so prolific and is perhaps more reactive and closely linked to astrocytic changes whereas in ALS the microglia expressing mutant protein seem to be directly toxic to motoneurons.
Acknowledgments
The author acknowledges support for work in this area from NIH grant AG12411
References
- 1.Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nature neuroscience. 2007;10(11):1387–94. doi: 10.1038/nn1997. Epub 2007/10/30. [DOI] [PubMed] [Google Scholar]
- 2.Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiological reviews. 2011;91(2):461–553. doi: 10.1152/physrev.00011.2010. Epub 2011/04/30. [DOI] [PubMed] [Google Scholar]
- 3.Griffin WS, Stanley LC, Ling C, White L, MacLeod V, Perrot LJ, et al. Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proceedings of the National Academy of Sciences of the United States of America. 1989;86(19):7611–5. doi: 10.1073/pnas.86.19.7611. Epub 1989/10/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goldgaber D, Harris HW, Hla T, Maciag T, Donnelly RJ, Jacobsen JS, et al. Interleukin 1 regulates synthesis of amyloid beta-protein precursor mRNA in human endothelial cells. Proceedings of the National Academy of Sciences of the United States of America. 1989;86(19):7606–10. doi: 10.1073/pnas.86.19.7606. Epub 1989/10/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kang J, Lemaire HG, Unterbeck A, Salbaum JM, Masters CL, Grzeschik KH, et al. The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature. 1987;325(6106):733–6. doi: 10.1038/325733a0. Epub 1987/02/19. [DOI] [PubMed] [Google Scholar]
- 6.D’Andrea MR, Cole GM, Ard MD. The microglial phagocytic role with specific plaque types in the Alzheimer disease brain. Neurobiology of aging. 2004;25(5):675–83. doi: 10.1016/j.neurobiolaging.2003.12.026. Epub 2004/06/03. [DOI] [PubMed] [Google Scholar]
- 7.Hayes A, Thaker U, Iwatsubo T, Pickering-Brown SM, Mann DM. Pathological relationships between microglial cell activity and tau and amyloid beta protein in patients with Alzheimer’s disease. Neuroscience letters. 2002;331(3):171–4. doi: 10.1016/s0304-3940(02)00888-1. Epub 2002/10/18. [DOI] [PubMed] [Google Scholar]
- 8.Griffin WS, Sheng JG, Roberts GW, Mrak RE. Interleukin-1 expression in different plaque types in Alzheimer’s disease: significance in plaque evolution. Journal of neuropathology and experimental neurology. 1995;54(2):276–81. doi: 10.1097/00005072-199503000-00014. Epub 1995/03/01. [DOI] [PubMed] [Google Scholar]
- 9.Sheng JG, Mrak RE, Griffin WS. Microglial interleukin-1 alpha expression in brain regions in Alzheimer’s disease: correlation with neuritic plaque distribution. Neuropathology and applied neurobiology. 1995;21(4):290–301. doi: 10.1111/j.1365-2990.1995.tb01063.x. Epub 1995/08/01. [DOI] [PubMed] [Google Scholar]
- 10.Sheng JG, Griffin WS, Royston MC, Mrak RE. Distribution of interleukin-1-immunoreactive microglia in cerebral cortical layers: implications for neuritic plaque formation in Alzheimer’s disease. Neuropathology and applied neurobiology. 1998;24(4):278–83. doi: 10.1046/j.1365-2990.1998.00122.x. Epub 1998/10/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Griffin WS. Inflammation and neurodegenerative diseases. The American journal of clinical nutrition. 2006;83(2):470S–4S. doi: 10.1093/ajcn/83.2.470S. Epub 2006/02/14. [DOI] [PubMed] [Google Scholar]
- 12.Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nature reviews Neuroscience. 2007;8(1):57–69. doi: 10.1038/nrn2038. Epub 2006/12/21. [DOI] [PubMed] [Google Scholar]
- 13.Streit WJ. Microglial senescence: does the brain’s immune system have an expiration date? Trends in neurosciences. 2006;29(9):506–10. doi: 10.1016/j.tins.2006.07.001. Epub 2006/07/25. [DOI] [PubMed] [Google Scholar]
- 14.Streit WJ, Braak H, Xue QS, Bechmann I. Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer’s disease. Acta neuropathologica. 2009;118(4):475–85. doi: 10.1007/s00401-009-0556-6. Epub 2009/06/11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sheffield LG, Marquis JG, Berman NE. Regional distribution of cortical microglia parallels that of neurofibrillary tangles in Alzheimer’s disease. Neuroscience letters. 2000;285(3):165–8. doi: 10.1016/s0304-3940(00)01037-5. Epub 2000/05/12. [DOI] [PubMed] [Google Scholar]
- 16.Kitazawa M, Yamasaki TR, LaFerla FM. Microglia as a potential bridge between the amyloid beta-peptide and tau. Annals of the New York Academy of Sciences. 2004;1035:85–103. doi: 10.1196/annals.1332.006. Epub 2005/02/01. [DOI] [PubMed] [Google Scholar]
- 17.Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308(5726):1314–8. doi: 10.1126/science.1110647. Epub 2005/04/16. [DOI] [PubMed] [Google Scholar]
- 18.Fuhrmann M, Bittner T, Jung CK, Burgold S, Page RM, Mitteregger G, et al. Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer’s disease. Nature neuroscience. 2010;13(4):411–3. doi: 10.1038/nn.2511. Epub 2010/03/23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.El Khoury J, Toft M, Hickman SE, Means TK, Terada K, Geula C, et al. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nature medicine. 2007;13(4):432–8. doi: 10.1038/nm1555. Epub 2007/03/14. [DOI] [PubMed] [Google Scholar]
- 20.Takata K, Kitamura Y, Yanagisawa D, Morikawa S, Morita M, Inubushi T, et al. Microglial transplantation increases amyloid-beta clearance in Alzheimer model rats. FEBS letters. 2007;581(3):475–8. doi: 10.1016/j.febslet.2007.01.009. Epub 2007/01/24. [DOI] [PubMed] [Google Scholar]
- 21.Nicoll JA, Barton E, Boche D, Neal JW, Ferrer I, Thompson P, et al. Abeta species removal after abeta42 immunization. Journal of neuropathology and experimental neurology. 2006;65(11):1040–8. doi: 10.1097/01.jnen.0000240466.10758.ce. Epub 2006/11/07. [DOI] [PubMed] [Google Scholar]
- 22.Cameron B, Landreth GE. Inflammation, microglia, and Alzheimer’s disease. Neurobiology of disease. 2010;37(3):503–9. doi: 10.1016/j.nbd.2009.10.006. Epub 2009/10/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Colton CA. Heterogeneity of microglial activation in the innate immune response in the brain. Journal of neuroimmune pharmacology : the official journal of the Society on NeuroImmune Pharmacology. 2009;4(4):399–418. doi: 10.1007/s11481-009-9164-4. Epub 2009/08/06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nature reviews Immunology. 2005;5(12):953–64. doi: 10.1038/nri1733. Epub 2005/12/03. [DOI] [PubMed] [Google Scholar]
- 25.Colton CA, Mott RT, Sharpe H, Xu Q, Van Nostrand WE, Vitek MP. Expression profiles for macrophage alternative activation genes in AD and in mouse models of AD. Journal of neuroinflammation. 2006;3:27. doi: 10.1186/1742-2094-3-27. Epub 2006/09/29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lue LF, Kuo YM, Beach T, Walker DG. Microglia activation and anti-inflammatory regulation in Alzheimer’s disease. Molecular neurobiology. 2010;41(2–3):115–28. doi: 10.1007/s12035-010-8106-8. Epub 2010/03/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schwartz M, Butovsky O, Bruck W, Hanisch UK. Microglial phenotype: is the commitment reversible? Trends in neurosciences. 2006;29(2):68–74. doi: 10.1016/j.tins.2005.12.005. Epub 2006/01/13. [DOI] [PubMed] [Google Scholar]
- 28.McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology. 1988;38(8):1285–91. doi: 10.1212/wnl.38.8.1285. Epub 1988/08/01. [DOI] [PubMed] [Google Scholar]
- 29.Croisier E, Moran LB, Dexter DT, Pearce RK, Graeber MB. Microglial inflammation in the parkinsonian substantia nigra: relationship to alpha-synuclein deposition. Journal of neuroinflammation. 2005;2:14. doi: 10.1186/1742-2094-2-14. Epub 2005/06/07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Beach TG, Sue LI, Walker DG, Lue LF, Connor DJ, Caviness JN, et al. Marked microglial reaction in normal aging human substantia nigra: correlation with extraneuronal neuromelanin pigment deposits. Acta neuropathologica. 2007;114(4):419–24. doi: 10.1007/s00401-007-0250-5. Epub 2007/07/20. [DOI] [PubMed] [Google Scholar]
- 31.Kim WG, Mohney RP, Wilson B, Jeohn GH, Liu B, Hong JS. Regional difference in susceptibility to lipopolysaccharide-induced neurotoxicity in the rat brain: role of microglia. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2000;20(16):6309–16. doi: 10.1523/JNEUROSCI.20-16-06309.2000. Epub 2000/08/10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mount MP, Lira A, Grimes D, Smith PD, Faucher S, Slack R, et al. Involvement of interferon-gamma in microglial-mediated loss of dopaminergic neurons. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2007;27(12):3328–37. doi: 10.1523/JNEUROSCI.5321-06.2007. Epub 2007/03/23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chakrabarty P, Ceballos-Diaz C, Lin WL, Beccard A, Jansen-West K, McFarland NR, et al. Interferon-gamma induces progressive nigrostriatal degeneration and basal ganglia calcification. Nature neuroscience. 2011;14(6):694–6. doi: 10.1038/nn.2829. Epub 2011/05/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hirsch EC, Hunot S. Neuroinflammation in Parkinson’s disease: a target for neuroprotection? Lancet neurology. 2009;8(4):382–97. doi: 10.1016/S1474-4422(09)70062-6. Epub 2009/03/20. [DOI] [PubMed] [Google Scholar]
- 35.Zhang W, Wang T, Pei Z, Miller DS, Wu X, Block ML, et al. Aggregated alpha-synuclein activates microglia: a process leading to disease progression in Parkinson’s disease. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2005;19(6):533–42. doi: 10.1096/fj.04-2751com. Epub 2005/03/26. [DOI] [PubMed] [Google Scholar]
- 36.Beraud D, Hathaway HA, Trecki J, Chasovskikh S, Johnson DA, Johnson JA, et al. Microglial Activation and Antioxidant Responses Induced by the Parkinson’s Disease Protein alpha-Synuclein. Journal of neuroimmune pharmacology : the official journal of the Society on NeuroImmune Pharmacology. 2012 doi: 10.1007/s11481-012-9401-0. Epub 2012/10/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Park JY, Paik SR, Jou I, Park SM. Microglial phagocytosis is enhanced by monomeric alpha-synuclein, not aggregated alpha-synuclein: implications for Parkinson’s disease. Glia. 2008;56(11):1215–23. doi: 10.1002/glia.20691. Epub 2008/05/02. [DOI] [PubMed] [Google Scholar]
- 38.Doorn KJ, Lucassen PJ, Boddeke HW, Prins M, Berendse HW, Drukarch B, et al. Emerging roles of microglial activation and non-motor symptoms in Parkinson’s disease. Progress in neurobiology. 2012;98(2):222–38. doi: 10.1016/j.pneurobio.2012.06.005. Epub 2012/06/27. [DOI] [PubMed] [Google Scholar]
- 39.Halliday GM, Stevens CH. Glia: initiators and progressors of pathology in Parkinson’s disease. Movement disorders : official journal of the Movement Disorder Society. 2011;26(1):6–17. doi: 10.1002/mds.23455. Epub 2011/02/16. [DOI] [PubMed] [Google Scholar]
- 40.Braak H, Sastre M, Del Tredici K. Development of alpha-synuclein immunoreactive astrocytes in the forebrain parallels stages of intraneuronal pathology in sporadic Parkinson’s disease. Acta neuropathologica. 2007;114(3):231–41. doi: 10.1007/s00401-007-0244-3. Epub 2007/06/20. [DOI] [PubMed] [Google Scholar]
- 41.Lee HJ, Suk JE, Patrick C, Bae EJ, Cho JH, Rho S, et al. Direct transfer of alpha-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. The Journal of biological chemistry. 2010;285(12):9262–72. doi: 10.1074/jbc.M109.081125. Epub 2010/01/15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gu XL, Long CX, Sun L, Xie C, Lin X, Cai H. Astrocytic expression of Parkinson’s disease-related A53T alpha-synuclein causes neurodegeneration in mice. Molecular brain. 2010;3:12. doi: 10.1186/1756-6606-3-12. Epub 2010/04/23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lewis CA, Manning J, Rossi F, Krieger C. The Neuroinflammatory Response in ALS: The Roles of Microglia and T Cells. Neurology research international. 2012;2012:803701. doi: 10.1155/2012/803701. Epub 2012/06/06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Troost D, Van den Oord JJ, Vianney de Jong JM. Immunohistochemical characterization of the inflammatory infiltrate in amyotrophic lateral sclerosis. Neuropathology and applied neurobiology. 1990;16(5):401–10. doi: 10.1111/j.1365-2990.1990.tb01276.x. Epub 1990/10/01. [DOI] [PubMed] [Google Scholar]
- 45.Ince PG, Shaw PJ, Slade JY, Jones C, Hudgson P. Familial amyotrophic lateral sclerosis with a mutation in exon 4 of the Cu/Zn superoxide dismutase gene: pathological and immunocytochemical changes. Acta neuropathologica. 1996;92(4):395–403. doi: 10.1007/s004010050535. Epub 1996/10/01. [DOI] [PubMed] [Google Scholar]
- 46.Turner MR, Cagnin A, Turkheimer FE, Miller CC, Shaw CE, Brooks DJ, et al. Evidence of widespread cerebral microglial activation in amyotrophic lateral sclerosis: an [11C](R)-PK11195 positron emission tomography study. Neurobiology of disease. 2004;15(3):601–9. doi: 10.1016/j.nbd.2003.12.012. Epub 2004/04/02. [DOI] [PubMed] [Google Scholar]
- 47.Henkel JS, Beers DR, Zhao W, Appel SH. Microglia in ALS: the good, the bad, and the resting. Journal of neuroimmune pharmacology : the official journal of the Society on NeuroImmune Pharmacology. 2009;4(4):389–98. doi: 10.1007/s11481-009-9171-5. Epub 2009/09/05. [DOI] [PubMed] [Google Scholar]
- 48.Liao B, Zhao W, Beers DR, Henkel JS, Appel SH. Transformation from a neuroprotective to a neurotoxic microglial phenotype in a mouse model of ALS. Experimental neurology. 2012;237(1):147–52. doi: 10.1016/j.expneurol.2012.06.011. Epub 2012/06/28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Clement AM, Nguyen MD, Roberts EA, Garcia ML, Boillee S, Rule M, et al. Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science. 2003;302(5642):113–7. doi: 10.1126/science.1086071. Epub 2003/10/04. [DOI] [PubMed] [Google Scholar]
- 50.Boillee S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, et al. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006;312(5778):1389–92. doi: 10.1126/science.1123511. Epub 2006/06/03. [DOI] [PubMed] [Google Scholar]
- 51.Kihira T, Suzuki A, Kubo T, Miwa H, Kondo T. Expression of insulin-like growth factor-II and leukemia inhibitory factor antibody immunostaining on the ionized calcium-binding adaptor molecule 1-positive microglias in the spinal cord of amyotrophic lateral sclerosis patients. Neuropathology : official journal of the Japanese Society of Neuropathology. 2007;27(3):257–68. doi: 10.1111/j.1440-1789.2007.00776.x. Epub 2007/07/25. [DOI] [PubMed] [Google Scholar]