

Abstract
Lumican regulates collagenous matrix assembly as a keratan sulfate proteoglycan in the cornea and is also present in the connective tissues of other organs and embryonic corneal stroma as a glycoprotein. In normal unwounded cornea, lumican is expressed by stromal keratocytes. Our data show that injured mouse corneal epithelium ectopically and transiently expresses lumican during the early phase of wound healing, suggesting a potential lumican functionality unrelated to regulation of collagen fibrillogenesis, e.g. modulation of epithelial cell adhesion or migration. An anti-lumican antibody was found to retard corneal epithelial wound healing in cultured mouse eyes. Healing of a corneal epithelial injury in Lum−/− mice was significantly delayed compared with Lum+/− mice. These observations indicate that lumican expressed in injured epithelium may modulate cell behavior such as adhesion or migration, thus contributing to corneal epithelial wound healing.
Rapid re-epithelialization is essential for restoration of homeostasis in injured tissues; impaired healing of injured epithelium increases the risks of infection and further damage underlying tissues (1, 2). The cornea provides an ideal model to evaluate interactions of migrating epithelial cells and the extracellular matrix of the underlying basement membrane during wound healing because epithelial injuries of the avascular corneal tissue heal in a bloodless wound field. Various specific proteins such as vinculin (3), keratins (4), CD44 hyaluronan receptors (5), and gelatinases and metalloproteinase inhibitors (6, 7) are up-regulated during corneal epithelial wound healing. These proteins are believed to modulate cell adhesion or migration.
Lumican belongs to the family of small leucine-rich proteoglycans (SLRPs)1 that includes keratocan, mimecan, decorin, biglycan, fibromodulin, epiphycan, and osteoadherin. In the cornea, lumican, keratocan, and mimecan are modified with keratan sulfate glycosaminoglycan chains comprising the keratan sulfate proteoglycans (KSPG) of the stromal extracellular matrix (8-13). In normal unwounded mouse cornea, lumican mRNA is expressed in stromal keratocytes (14). Lumican KSPG is a key regulator of collagen fibrillogenesis, a process critical to corneal transparency. Mice lacking lumican show an age-dependent corneal opacity and a high proportion of abnormally thick collagen fibers in the corneal stroma (15).
Lumican is also widely present as a non- or low-sulfated glycoprotein in connective tissues of many other organ systems, e.g. skeleton, heart, kidney, and lung (14, 16-18). During mouse embryonic ocular development, lumican is synthesized by keratocytes; detected as a glycoprotein, not as a KSPG (19); and also transiently expressed by the corneal epithelium, neural retina, and epidermis (14). These observations suggest that epithelial tissues possess the capacity to express lumican under certain conditions.
Several studies have demonstrated that SLRP proteins can modulate cellular behaviors, i.e. cell migration and proliferation during embryonic development, tissue repair, and tumor growth, in addition to their extracellular matrix functions as regulators of tissue hydration and collagen fibrillogenesis (20-22). For example, decorin is one of the SLRP proteins with well characterized functions for modulating cellular behaviors. Decorin protein alters the cell cycle process in neoplastic cells both by modulating the activities of growth factors and by direct interaction with a cell-surface receptor (23, 24). Cultured vascular endothelial cells start to express decorin after the formation of tube-like structures and also up-regulate biglycan expression during repair after a damage (25, 26). Under some pathological conditions, the corneal epithelium shows an increase in decorin immunoreactivity compared with normal corneal epithelium (27). Macrophages, on the other hand, express the cell-surface receptor for lumican. These cells bind the low-sulfated glycoprotein form of lumican, but not lumican modified with keratan sulfate chains, suggesting that non- or low-sulfated lumican might provide a scaffold for macrophages invading injured corneal stroma (28). These observations prompted us to hypothesize that lumican, like decorin, may have biological functions for modulating corneal cell behavior such as adhesion, migration, or proliferation during tissue morphogenesis or wound healing.
In this study, employing in situ hybridization and immunocytochemistry, we first showed that migrating corneal epithelial cells ectopically and transiently express lumican during wound healing. To examine the hypothesis that lumican actively modulates epithelial wound healing, we then examined the effects of an anti-lumican antibody on closure of a corneal epithelial defect and the healing of corneal epithelial defects in lumican-null mice. Our results suggest that lumican may play a role in epithelial cell migration or adhesion, thus contributing to corneal epithelial wound healing.
MATERIALS AND METHODS
Animal Experiments for Histology
Experimental mice (n = 52) were anesthetized by intraperitoneal injection of pentobarbital (70 mg/kg of body weight). The central corneal epithelium (3 mm in diameter) was demarcated with a trephine and subsequently removed using a No. 69 Beaver Blade® under a stereomicroscope as previously reported (29, 30). Neomycin ointment was topically applied to prevent bacterial infection. The animals were then killed at specific intervals of healing (1, 2, 4, or 8 h and 1, 2, 3, 5, 7, 14, 21, or 28 days). Each eye was removed, fixed in 4% paraformaldehyde in 0.1 m phosphate buffer (pH 7.4) for 48 h, embedded in paraffin, and processed for histology.
In Situ Hybridization of Lumican mRNA
Paraffin sections 5 μm thick were deparaffinized and processed for in situ hybridization with sense and antisense riboprobes of mouse lumican and mouse keratocan as previously reported (12, 31). Finally, the sections were counterstained with 0.5% neutral red and dehydrated through a series of ethanol, mounted, and observed under a light microscope.
Preparation of an Epitope-specific Polyclonal Anti-lumican Antibody
To prepare the polyclonal antibody, a synthetic oligopeptide sequence (YYDYDIPLFMYGQISPNC) deduced from mouse lumican cDNA was conjugated to keyhole limpet hemocyanin (32). The polyclonal antibodies were raised in rabbits as described previously (32). Anti-lumican antibodies were purified with an affinity column prepared by conjugating the oligopeptide to Sulfolink® (Pierce) using the procedures recommended by the manufacturer.
Western Blotting to Characterize the Anti-lumican Antibody
Mouse corneal KSPGs and recombinant mouse lumican expressed in Escherichia coli were prepared as described previously (33), and the core proteins of the KSPGs were deglycosylated by treatment with N-glycanase (9). Two μg of total protein was subjected to SDS-polyacrylamide gel electrophoresis and electrotransferred to polyvinylidene difluoride membranes (9). Lumican was detected by immunostaining with the anti-lumican antibody (10 μg/ml) using an indirect method as described previously (32).
Immunohistochemistry
To locate lumican protein, paraffin sections from normal and injured corneas healed for various periods of times were subjected to immunohistochemistry with the epitope-specific anti-lumican antibody (10 μg/ml) and nonimmune rabbit IgG (control) as described previously (34).
Transmission Electron Microscopy
Corneas were fixed in 2.0% glutaraldehyde in 0.1 m phosphate buffer for 24 h at 4 °C. The samples were then post-fixed in 2.0% osmium tetroxide, rinsed in 0.1 m phosphate buffer, and embedded in Epon 812 (Quetol 812, Nissin EM, Tokyo, Japan). Ultrathin sections were stained with uranyl acetate and lead citrate and observed by transmission electron microscopy (35).
In Vitro Wound Healing of the Corneal Epithelia of Organ-cultured Mouse Eyes
To examine whether the anti-lumican antibody inhibits corneal epithelial wound healing, we developed an in vitro wound healing model using cultured mouse eyes. A central corneal epithelial defect (2 mm in diameter) was produced in both eyes of 38 anesthetized wild-type mice under a stereomicroscope. Our preliminary immunohistochemical examination with a rat monoclonal anti-laminin antibody (X50, BIODESIGN International, Kennebunkport, ME) showed the presence of the uninterrupted epithelial basement membrane immediately after the epithelial scraping (data not shown). The animals were killed immediately after the epithelial débridement. Each eyeball was enucleated and cultured in Dulbecco’s modified Eagle’s medium (Life Technologies, Inc.) supplemented with 1.4% fetal calf serum and 50 μg/ml gentamycin in 10% CO2 and 90% air at 37 °C with the anti-lumican antibody (40 μg/ml) or normal rabbit IgG (40 μg/ml; Sigma). Both the antibody and control IgG were dialyzed against phosphate-buffered saline for 24 h before application. After incubation for 48 h, each eye was stained with fluorescein and photographed using a stereomicroscope. The healing of epithelial defects was categorized into three groups: 1) healed, 2) resurfaced with regenerated epithelium and punctate epithelial staining, and 3) not healed with remaining defects. Distribution of individuals in these groups was statistically analyzed using the χ2 test.
Gene Targeting and Creation of Mice Lacking the Lumican Gene (Lum)
To examine the in vivo role of lumican in corneal epithelial wound healing, we prepared lumican-null mice via gene-targeting techniques. The lumican gene-targeting construct contains 4.1 kb of 5′-homology (SacI/XbaI fragment), the 2.9-kb pgk-hprt cassette, 1.8 kb of 3′-homology (XhoI/BamHI fragment), and a 2.0-kb pMC-tk cassette in the pBluescript vector. The XbaI/XhoI fragment containing 481 base pairs of exon 2 was deleted and replaced with a pgk-hprt cassette. The targeting vector was transfected into hprt-negative E14TG2a ES cells derived from mouse strain 129/Sv by electroporation using a Bio-Rad Gene-Pulser. A targeted ES cell clone (frequency of 1:186) was identified by Southern blot hybridization and used to generate chimeric mice in our Gene Targeting Core Facility (University of Cincinnati). C57BL/6J blastocysts injected with 10–12 ES cells were implanted in pseudopregnant F1(CBA × C57BL/6) foster mothers (Jackson ImmunoResearch Laboratories, Inc.). Chimeric mice, identified by agouti coat color, were mated with C57BL/6J mice. Agouti coat-colored offspring were tested for the presence of the targeted locus by polymerase chain reaction (PCR) and Southern hybridization as described previously (12). To distinguish between individuals with none, one, or two copies of the mutant gene, we designed three primers in a PCR for detection of the wild type (primer 1, 5′-TACTTCAAGCGCTTCAC-3′; and primer 2, antisense 5′-CAAGTTCATTGACCTCCAGG-3′; 190 base pairs) and mutant (primers 2 and 3, antisense 5′-CGAGACTAGTGAGACGTGCT-3′ of the hprt minigene; 381 base pairs), respectively. Northern hybridization, in situ hybridization, and immunohistochemistry were used to determine the phenotypes of littermates using the procedures described above.
Healing of Corneal Epithelial Defects in Lumican-deficient Mice
Age-matched littermates were used as controls. Two-month-old Lum+/− (n = 22) and Lum−/− (n = 16) mice were anesthetized and subjected to 2-mm corneal epithelial débridement as described above. Our preliminary immunohistochemistry results showed the presence of the non-interrupted epithelial basement membrane immediately after epithelial scraping in both Lum+/− and Lum−/− mice as described above. The injured corneas were stained with fluorescein and photographed with a stereomicroscope every 24 h, beginning immediately after wounding, to evaluate the re-epithelialization and to detect any sign of infection. Healing of epithelial defects was graded and statistically analyzed in a manner similar to the in vitro wound healing experiment described above.
RESULTS
Characterization of Polyclonal Anti-Lumican Antibody
We prepared epitope-specific anti-lumican antibody as described under “Methods and Methods.” Western blot immune analysis was used to characterize an affinity-purified rabbit polyclonal antibody directed against the N-terminal oligopeptide of mouse lumican (YYDYDIPLFMYGQISPNC). This sequence is not found in keratocan or other members of the SLRP family (12, 27). Fig. 1 demonstrates that the antibodies reacted with recombinant lumican prepared from the expression clone of E. coli(lane 1) and a 41-kDa KSPG core protein isolated from mouse corneas after deglycosylation with N-glycanase (lane 2).
Fig. 1. Western blot characterization of the anti-lumican antibody.
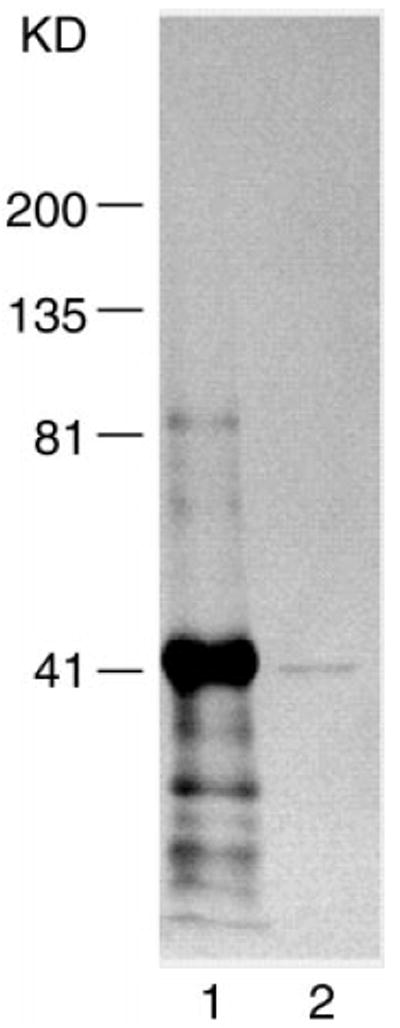
Recombinant mouse lumican and mouse KSPG) core protein were separated by SDS-polyacrylamide gel electrophoresis and transferred to a polyvinylidene difluoride membrane. The membrane was immunostained with the affinity-purified polyclonal anti-lumican antibody (10 μg/ml). The antibody reacted to recombinant lumican (lane 1) and deglycosylated mouse KSPG core protein (~41 kDa) (lane 2).
Phenotypic Changes in Mice Lacking Lumican
Fig. 2A summarizes the strategy used to ablate the lumican gene in mice via gene-targeting techniques. A targeting construct containing the human hypoxanthine phosphoribosyltransferase and herpes simplex virus thymidine kinase genes was prepared as described under “Materials and Methods.” The genotypes of the lumican knockout mice were determined by PCR and Southern blot analysis (Fig. 2, B and C). Northern hybridization, in situ hybridization, and immunohistochemistry revealed no expression of lumican mRNA and the absence of lumican protein antigens in Lum−/− mice (Fig. 2, D and E). This phenotype of the lumican-null mice is indicative of a loss of lumican expression rather than the presence of a dominant-negative mutation. The homozygous mutant mice were born alive in the expected mendelian ratio and were fertile. The skin of adult Lum−/− mice was fragile as evidenced by the disruption of skin when experimental animals were killed by cervical dislocation. The back skin hairs of adult Lum−/− mice were disarranged. It is of interest to note that the male lumican-null mice appear to produce a smaller number of offspring compared with female Lum−/− mice when they are mated with wild-type mice. The reason for the phenomenon is not known. However, it could be secondary to the fragile skin phenotype.
Fig. 2.

A, targeted disruption of the mouse lumican gene (Lum) and phenotyping the lumican knockout mice. Panel 1, shown is a restriction enzyme map of the Lum gene. Panel 2, a 5.0-kb SacI/BamHI fragment was used to generate the replacement targeting vector. A 481-base pair XbaI/XhoI fragment from the exon 2 was deleted and replaced with a pgkHPRTpA cassette in the antisense orientation with respect to the Lum gene. In addition, a pMC-tk cassette was placed on the 3′-end of the targeting vector. Panel 3, shown is the targeted allele after homologous recombination. Panel 4, shown are the expected sizes of restriction fragments detected by a 5′- or 3′-probe. B, autoradiography of Southern blot hybridization. DNA from one litter of a Lum-targeted heterozygous intercross was digested with SpeI or XbaI and separated by 0.8% agarose gel electrophoresis. The DNA was then subjected to Southern blot hybridization with a 5′-probe (SpeI digestion). The 5.0- and 7.0-kb bands represent wild-type and mutant alleles, respectively. Hybridization with 3′-probes yielded similar results (not shown). C, genotyping of offspring from the heterozygous mating by PCR. PCR products amplified from the DNA were resolved by 4% NuSieve/Sea Kem (3:1) agarose gel electrophoresis. D, Northern hybridization of lumican mRNA from eyes of lumican-deficient mice. Lumican mRNA was not detected in Lum−/− mice with 32P-labeled mouse lumican probes. 28 S and 18 S ribosomal RNAs indicate the quality and quantity of RNA samples. E, immunohistochemistry (panels 1 and 2) and in situ hybridization (panels 3 and 4) of lumican protein and mRNA in a Lum+/− or Lum−/− mouse. In situ hybridization and immunohistochemistry were performed to further characterize the phenotype of the mice. The corneal stroma showed immunoreactivity for lumican (panel 1), and keratocytes expressed lumican mRNA (panel 3) in a Lum+/− mouse, whereas the stroma had no lumican protein (panel 2) and mRNA (panel 4) in a Lum−/− mouse. pgk, phosphoglycerate kinase I gene; pr, promoter; hprt, hypoxanthine phosphoribosyltransferase; pA, polyadenylation signal sequence; tk, thymidine kinase gene.
Fig. 3 demonstrates that abnormal thick collagen fibers were seen in the posterior corneal stromata of lumican-null mice 4 months old, whereas the anterior stromal collagen fibers appeared normal (data not shown). These larger collagen fibrils were not observed in the stromata of younger lumican-null mice. The corneal haze as recognized with a stereomicroscope was concurrent with the presence of a disorganized collagenous matrix in lumican-null mice. This phenotype is consistent with the observations reported by Chakravarti et al. (15).
Fig. 3. Electron micrographs of corneal stromata.

Corneas of 4-month-old Lum+/− and Lum−/− mice were subjected to transmission electron microscopy. A, collagenous matrix in the posterior corneal stroma from a Lum+/− mouse. Collagen fibrils are uniform in size and consistent in inter-fibril distances. B, irregular large collagen fibrils from lateral fusion in the posterior stroma of a Lum−/− mouse. Bar is equal to 100 and 50 nm in the panels and insets, respectively.
Clinical and Histological Observations of Eyes after Epithelial Débridement in Wild-type Mice
A corneal epithelial defect (3 mm in diameter) was created as described under “Materials and Methods.” One day after injury, the central corneal stroma was still exposed, and a healing epithelium was observed in the periphery. Polymorphonuclear leukocytes were seen in the healing stroma at day 1 after injury. By day 3, the defect was covered by regenerated epithelia in all corneas examined (data not shown).
In Situ Hybridization Detection of Lumican mRNA
As shown in Fig. 4A, lumican mRNA was detected in the stromal keratocytes, but not in epithelial cells of uninjured corneas. Injured corneal epithelium expressed lumican mRNA from 8 h until 3 days after the epithelial débridement (Fig. 4, C–E and G–I). Seven days after injury, corneal epithelial defects were healed with a stratified multicellular epithelium that did not yield positive hybridization signals (Fig. 4J). No signals were seen with sense probes (Fig. 4, B and F). Neither lens epithelial cells nor neural retina hybridized to the lumican antisense riboprobes (data not shown). Keratocan mRNA was not detected in either normal or migrating epithelium, but was present in keratocytes (data not shown).
Fig. 4. In situ hybridization detection of lumican mRNAs in normal and healing mouse corneas.
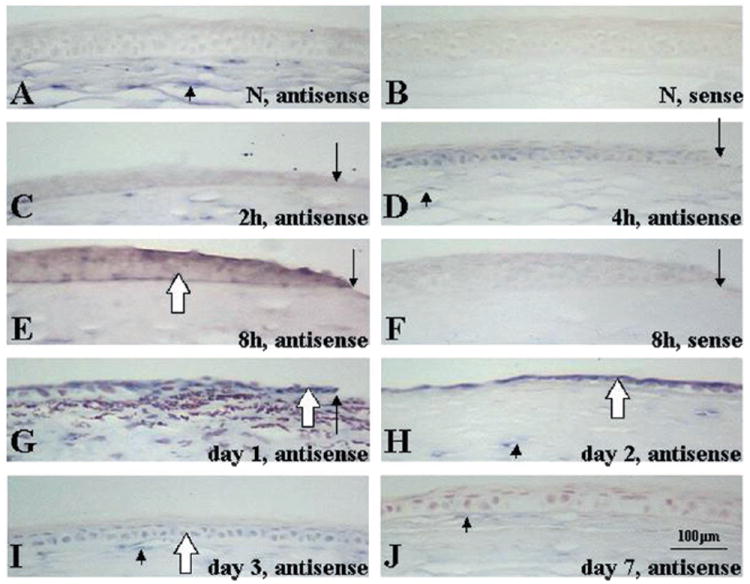
An epithelial defect (3 mm in diameter) was created in corneas of anesthetized mice. The animals were killed after specific intervals of healing, and corneas were processed for in situ hybridization with sense or antisense lumican riboprobes. In a normal mouse cornea, lumican mRNAs were expressed by keratocytes (A, arrowhead). No signals were seen with sense probes (B). Expression patterns of lumican mRNAs in healing mouse corneas after 3-mm-diameter epithelial débridement are shown (C–J). Two and 4 h after injury, no signals for lumican mRNA were observed in injured epithelium (C and D). Signals for lumican mRNAs were detected in injured corneal epithelia (open arrows) and in keratocytes (arrowheads) at 8 h (E) and at 1 (G), 2 (H), and 3 (I) day(s) after epithelial débridement, whereas no signals were seen with sense probes (F). At day 7, regenerated epithelium was negative for lumican mRNA, whereas keratocytes were positive (J). A–F, without counterstaining; G–J, counterstained with 0.5% neutral red. The arrows indicate the edge of the injured or healing epithelium. Bar = 100 μm.
Immunohistochemistry
In tissue sections, the anti-lumican antibody stained normal corneal stroma (Fig. 5A) as well as sclera and dermal connective tissue of the eyelid (data not shown). Lumican staining in the corneal stroma was more intense in the posterior than in the anterior stroma. The anti-lumican antibody did not react to the epithelium or basement membrane of uninjured cornea (Fig. 5A) or other ocular tissues, e.g. neural retina, lens epithelial cells, and the epidermis of the eyelid (data not shown). After wounding, faint lumican intracellular immunoreactivity was seen in migrating epithelium 1 day after the injury (Fig. 5C). The healing epithelia at days 2 and 3 were strongly labeled by the antibodies (Fig. 5, D and E). At day 3, lumican immunoreactivity was observed mainly in the cytoplasm of basal cells of regenerated epithelium. The antibodies also reacted to the basement membrane underlying the basal epithelial cells of injured corneas healed for 2 and 3 days. After day 7, the epithelium was negative for lumican protein (Fig. 5F).
Fig. 5. Immunolocalization of lumican protein in mouse corneas with an anti-lumican antibody.
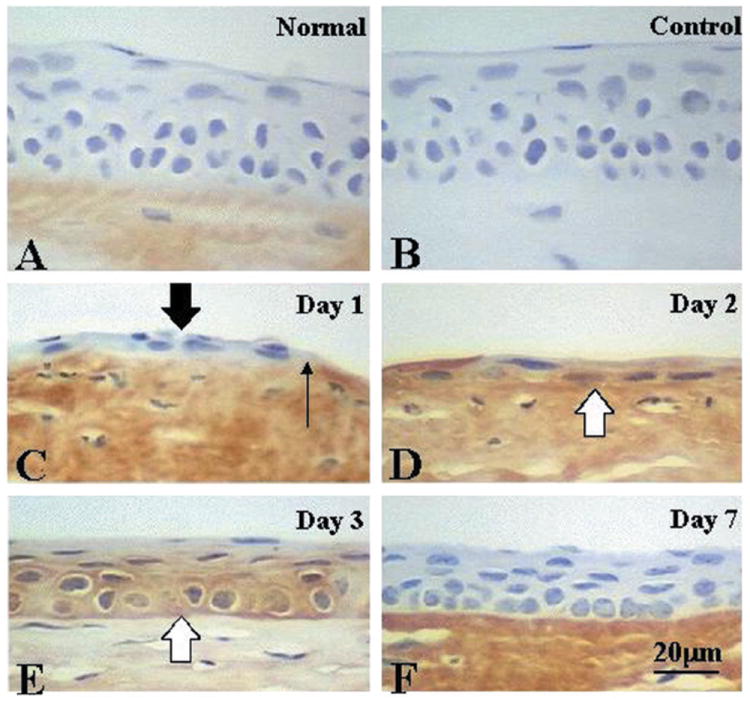
An epithelial defect (3 mm in diameter) was created in corneas of anesthetized mice. The animals were killed after specific intervals of healing, and corneas were processed for immunohistochemistry with the anti-lumican antibody (10 μg/ml). In a normal mouse cornea, lumican protein was detected in the corneal stroma (A). No staining was seen in the control (B). Localization of lumican protein in healing mouse corneas after an epithelial débridement is shown in C–F. Very faint lumican immunoreactivity was detected in healing corneal epithelium (thick arrow) at day 1 (C) and was obviously observed in healing epithelia (open arrows) at days 2 (D) and 3 (E). Regenerated epithelium at day 7 (F) was negative for lumican protein. A–F were counterstained with hematoxylin. The thin arrow indicates the edge of the healing epithelium. Bar = 20 μm.
Effect of the Anti-lumican Antibody on Epithelial Healing
Epithelial defects (2 mm in diameter) closed in ~48 h in the in vitro wound healing model. As shown in Table I, the anti-lumican antibody (40 μg/ml) significantly inhibited epithelial healing as compared with control normal rabbit IgG (40 μg/ml). Preliminary observations revealed that the addition of mitomycin C (0.1 μg/ml) to the culture medium did not retard the closure of the epithelial defect, but did inhibit epithelial proliferation as judged by incorporation of bromodeoxyuridine (data not shown). These observations are consistent with the notion that lumican may have a role in epithelial cell migration during wound healing.
Table I. Wound healing of a corneal epithelial defect in organ-cultured mouse eyes.
Results are from organ cultures of mouse eyes with a 2-mm-diameter epithelial defect in the presence of the anti-lumican antibody (40 μg/ml) or normal rabbit IgG (40 μg/ml) for 48 h. Healed indicates completely resurfaced; punctate, resurfaced by regenerated epithelium with punctate staining; and defect, not healed with remaining defects.
Percent defected area remaining (ranges): #, averages of 1.63% (1.0–2.3) and 14.1% (1.0–64.0), respectively.
p < 0.005 compared with control IgG using the χ2 test.
Healing of Epithelial Defects in Lumican-deficient Mice
Fig. 6 and Table II summarize the wound healing of a 2-mm-diameter central corneal epithelial defect in lumican-null mice. One day after injury, the number of corneas completely resurfaced was significantly higher in Lum+/− mice than in Lum−/− mice. Two days after injury, 19 of 22 injured eyes completely re-epithelialized and became transparent in Lum+/− mice, whereas 8 of 16 eyes healed in Lum−/− mice. At day 3, 5 of 16 corneas of Lum−/− mice still had epithelial defects, whereas all epithelial defects in the corneas of Lum+/− mice had healed. At day 5, the epithelial defects had healed in Lum−/− mice.
Fig. 6. Wound healing of a corneal epithelial defect in Lum+/− (panels 1–3) and Lum−/− (panels 4–9) mice.

An epithelial defect (2 mm in diameter) was created at the center of the corneas (panels 1, 4, and 7). After 1 day, no epithelial defect was detected in Lum+/− mice (panel 2), and a central non-resurfaced area stained with fluorescein was seen in each Lum−/− mouse (panels 5 and 8). At day 2, no epithelial staining was observed in Lum+/−(panel 3) or Lum−/−(panel 6) mice, whereas punctate fluorescein staining (arrowheads) was detected in the regenerated central epithelium of many Lum−/− mice (panel 9). Bar = 1 mm.
Table II. Wound healing of a corneal epithelial defect in lumican-deficient mice.
A 2-mm epithelial defect was created at the center of the cornea of a Lum+/− or Lum−/− mouse with a No. 69 Beaver Blade® and was allowed to heal as described under “Materials and Methods.”
| Genotype | No. of corneas examined | Outcome | No. of corneas
|
||||
|---|---|---|---|---|---|---|---|
| Day 1a | Day 2b | Day 3 | Day 4 | Day 5 | |||
| +/− | 22 | Healed | 13 | 19 | 22 | 22 | 22 |
| Punctate | 3 | 3 | 0 | 0 | 0 | ||
| Defect | 6c | 0 | 0 | 0 | 0 | ||
| −/− | 16 | Healed | 0 | 8 | 11 | 14 | 16 |
| Punctate | 1 | 5 | 3 | 2 | 0 | ||
| Defect | 15d | 3e | 2f | 0 | 0 | ||
Healed indicates completely resurfaced; punctate, resurfaced by regenerated epithelium with punctate staining; defect, not healed with remaining defects. All injured corneas of Lum−/− mice healed within 5 days, whereas those of heterozygous Lum+/− healed in 3 days.
p < 0.005 and p < 0.05, respectively; statistically significant using the χ2 test.
Percent defected area remaining in defect group corneas (ranges): averages of 5.9% (0.25–25), 18.7% (1.0–56.3), 7.5% (4.0–12.3), and 3.3% (0.3–6.3), respectively.
DISCUSSION
The evidence available suggests that SLRP proteins are key regulatory molecules of collagen fibrillogenesis (36). Null mutants of SLRP family proteins, i.e. decorin, biglycan, fibromodulin, and lumican, are manifested by malfunctions of connective tissues associated with abnormal extracellular matrix, i.e. fragile skin, cloudy cornea, and thick collagen fibrils (15, 37-39). Our lumican-null mice have phenotypes that closely resemble those described by Chakravarti et al. (15). It is of interest that the results of the present study imply that lumican, in addition to serving as a regulator of collagen fibrillogenesis, may modulate epithelial cell migration during corneal wound healing. We observed an ectopic and transient expression of lumican mRNA in healing mouse corneal epithelium as early as 8 h after an epithelial débridement (Fig. 4). An obvious accumulation of lumican protein occurred in the basal epithelial cells and the basement membrane of injured epithelia after 2–3 days of healing (Fig. 5). The observation is consistent with the notion that lumican is secreted by the epithelial cells. The up-regulated lumican synthesis can account for the intracellular immunoreactivity observed in the healing epithelium. The addition of anti-lumican antibodies to the culture medium retarded closure of an epithelial defect in healing wounds in vitro (Table I), and Lum−/− mice lacking lumican showed delayed re-epithelialization of corneal epithelial defects in vivo (Fig. 6 and Table II). These observations are consistent with the hypothesis that lumican expressed in injured corneal epithelium modulates corneal epithelial wound healing. Interestingly, keratocan, another member of SLRP family closely related to lumican, was not expressed by injured corneal epithelium, but only by stromal keratocytes. This finding indicates that these two KSPG proteins are transcriptionally regulated differently, albeit both are major KSPG constituents of the corneal stroma extracellular matrix.
Several other studies have presented results consistent with epithelial expression of lumican. In chick, KSPG precursor protein synthesis by organ culture of corneal epithelia amounts to 7.2% of the protein synthesis of organ-cultured whole corneas (40). Moreover, the basal and suprabasal cells of the hyperproliferative corneal epithelium of a Corn1 mouse (41), a mutant mouse characterized by hyperplasia of the central corneal epithelium associated with corneal neovascularization, express lumican mRNA.2 In the adult cornea, lumican exists as KSPG; however, in non-corneal tissues as well as in embryonic and wounded corneas, lumican is found as a low- or non-sulfated glycoprotein (14, 18, 42). Rat corneal epithelium was previously shown to transiently up-regulate glycoprotein synthesis as a result of wounding (43, 44). This glycoprotein may represent the lumican induction described in the present study. Our data do not directly elucidate the mechanism by which lumican may modulate epithelial cell adhesion or migration. Recently, a novel bone KSPG core protein of the SLRP family, osteoadherin, was found to be distributed in bovine fetal rib growth plate and in newly deposited bone in vivo (45). It has been demonstrated that osteoadherin can mediate cell attachment via binding by αvβ3 integrin in vitro (45). A divalent cation-dependent lumican cell-surface receptor has been identified in macrophages, implying possible binding of integrin to lumican. Binding of cells to lumican, however, is strongly inhibited by the presence of keratan sulfate chains on lumican (28). It is possible that lumican transiently expressed by injured epithelium might interact with integrin receptors of epithelial cells; thus, it may facilitate healing of epithelial defects. However, the presence of such cell-surface receptor(s) of lumican in corneal epithelial cells is to be proven in future studies. Our unpublished observations3 by transmission electron microscopy revealed that there is no apparent difference in the ultrastructure of the basement membrane and hemidesmosomes of unwounded corneas and of those of 1 or 2 months after an epithelial débridement in Lum+/− and Lum−/− mice. These observations are consistent with the notion that impaired epithelial wound healing was not caused by structural abnormalities of the denuded corneal surface due to the absence of lumican in Lum−/− mice.
Our finding that lumican may have a role in modulating corneal wound healing of epithelial defects is consistent with an emerging body of data showing that SLRP proteins not only serve as regulators of collagenous extracellular matrix assembly, but also have biological roles involving direct interactions with cells. These include mediation of cell migration and proliferation (20-24). Laser surgery to correct refractive errors is a widely practiced procedure, after which rapid re-epithelialization is required to avoid complications such as infection or induction of scar tissue. The findings reported here suggest the possible therapeutic use of lumican protein in topical administration after such surgery or in the treatment of corneal or skin wounds.
Footnotes
This work was supported by Grants EY10556 and EY11845 from the National Institutes of Health and the Ohio Lion’s Eye Research Foundation (Columbus, OH) (to W. W.-Y. K); Grant EY09368 from the National Institutes of Health and Grant KS-96-GS-2 from the American Heart Association, Kansas Affiliate (to J. L. F); the Ministry of Education, Culture, and Science of Japan, the Nippon Eye Bank Association Fund (Tokyo, Japan), and a Bausch-Lomb overseas research fellowship award (Tokyo) (to S. S).
The abbreviations used are: SLRPs, small leucine-rich proteoglycans; KSPG, keratan sulfate proteoglycan; kb, kilobase(s); PCR, polymerase chain reaction
I.-J. Wong, S. Saika, C.-Y. Liu, C. W.-C. Kao, R. S. Smith, P. M. Nishina, J. P. Sundberg, and W. W.-Y. Kao, unpublished observations.
S. Saika, C.-Y. Lin, and W. W.-Y. Kao, unpublished observations.
References
- 1.Brown DL, Kao WW-Y, Greenhalgh DG. Surgery (St Louis) 1997;121:372–380. doi: 10.1016/s0039-6060(97)90306-8. [DOI] [PubMed] [Google Scholar]
- 2.Wilson SE. J Refract Surg. 1997;13:171–175. doi: 10.3928/1081-597X-19970301-15. [DOI] [PubMed] [Google Scholar]
- 3.Zieske JD, Bukusoglu G, Gipson IK. J Cell Biol. 1989;109:571–576. doi: 10.1083/jcb.109.2.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yu FX, Gipson IK, Guo Y. Invest Ophthalmol Visual Sci. 1995;36:1997–2007. [PubMed] [Google Scholar]
- 5.Yu FX, Guo J, Zhang Q. Invest Ophthalmol Visual Sci. 1998;39:710–717. [PubMed] [Google Scholar]
- 6.Inoue M, Kratz G, Haegerstrand A, Stahle-Backdahl M. J Invest Dermatol. 1995;104:479–483. doi: 10.1111/1523-1747.ep12605917. [DOI] [PubMed] [Google Scholar]
- 7.Saarialho-Kere UK, Kovacs SO, Pentland AP, Olerud JE, Welgus HG, Parks WC. J Clin Invest. 1993;92:2858–2866. doi: 10.1172/JCI116906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Corpuz LM, Funderburgh JL, Funderburgh ML, Bottomley GS, Prakash S, Conrad GW. J Biol Chem. 1996;271:9759–9763. doi: 10.1074/jbc.271.16.9759. [DOI] [PubMed] [Google Scholar]
- 9.Funderburgh JL, Conrad GW. J Biol Chem. 1990;265:8297–8303. [PubMed] [Google Scholar]
- 10.Funderburgh JL, Corpuz LM, Roth MR, Funderburgh ML, Tasheva ES, Conrad GW. J Biol Chem. 1997;272:28089–28095. doi: 10.1074/jbc.272.44.28089. [DOI] [PubMed] [Google Scholar]
- 11.Funderburgh JL, Funderburgh ML, Brown SJ, Vergnes JP, Hassell JR, Mann MM, Conrad GW. J Biol Chem. 1993;268:11874–11880. [PubMed] [Google Scholar]
- 12.Liu C-Y, Shiraishi A, Kao CW-C, Converse RL, Funderburgh JL, Corpuz LM, Conrad GW, Kao WW-Y. J Biol Chem. 1998;273:22584–22588. doi: 10.1074/jbc.273.35.22584. [DOI] [PubMed] [Google Scholar]
- 13.Scott JE. Biochemistry. 1996;35:8795–8799. doi: 10.1021/bi960773t. [DOI] [PubMed] [Google Scholar]
- 14.Ying S, Shiraishi A, Kao CW-C, Converse RL, Funderburgh JL, Swiergiel J, Roth MR, Conrad GW, Kao WW-Y. J Biol Chem. 1997;272:30306–30313. doi: 10.1074/jbc.272.48.30306. [DOI] [PubMed] [Google Scholar]
- 15.Chakravarti S, Magnuson T, Lass JH, Jepsen KJ, LaMantia C, Carroll H. J Cell Biol. 1998;141:1277–1286. doi: 10.1083/jcb.141.5.1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Funderburgh JL, Funderburgh ML, Mann MM, Conrad GW. J Biol Chem. 1991;266:24773–24777. [PubMed] [Google Scholar]
- 17.Funderburgh JL, Caterson B, Conrad GW. J Biol Chem. 1987;262:11634–11640. [PubMed] [Google Scholar]
- 18.Grover J, Chen XN, Korenberg JR, Roughley PJ. J Biol Chem. 1995;270:21942–21949. doi: 10.1074/jbc.270.37.21942. [DOI] [PubMed] [Google Scholar]
- 19.Cornuet PK, Blochberger TC, Hassell JR. Invest Ophthalmol Visual Sci. 1994;35:870–877. [PubMed] [Google Scholar]
- 20.Fullwood NJ, Davies Y, Nieduszynski IA, Marcyniuk B, Ridgway AE, Quantock AJ. Invest Ophthalmol Visual Sci. 1996;37:1256–1270. [PubMed] [Google Scholar]
- 21.Iozzo RV. Crit Rev Biochem Mol Biol. 1997;32:141–174. doi: 10.3109/10409239709108551. [DOI] [PubMed] [Google Scholar]
- 22.Wight TN, Kinsella MG, Qwarnstrom EE. Curr Opin Cell Biol. 1992;4:793–801. doi: 10.1016/0955-0674(92)90102-i. [DOI] [PubMed] [Google Scholar]
- 23.Iozzo RV, Moscatello DK, McQuillan DJ, Eichstetter I. J Biol Chem. 1999;274:4489–4492. doi: 10.1074/jbc.274.8.4489. [DOI] [PubMed] [Google Scholar]
- 24.Moscatello DK, Santra M, Mann DM, McQuillan DJ, Wong AJ, Iozzo RV. J Clin Invest. 1998;101:406–412. doi: 10.1172/JCI846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jarvelainen HT, Iruela-Arispe ML, Kinsella MG, Sandell LJ, Sage EH, Wight TN. Exp Cell Res. 1992;203:395–401. doi: 10.1016/0014-4827(92)90013-x. [DOI] [PubMed] [Google Scholar]
- 26.Jarvelainen HT, Kinsella MG, Wight TN, Sandell LJ. J Biol Chem. 1991;266:23274–23281. [PubMed] [Google Scholar]
- 27.Funderburgh JL, Hevelone ND, Roth MR, Funderburgh ML, Rodrigues MR, Nirankari VS, Conrad GW. Invest Ophthalmol Visual Sci. 1998;39:1957–1964. [PubMed] [Google Scholar]
- 28.Funderburgh JL, Mitschler RR, Funderburgh ML, Roth MR, Chapes SK, Conrad GW. Invest Ophthalmol Visual Sci. 1997;38:1159–1167. [PubMed] [Google Scholar]
- 29.Kao WW-Y, Kao CW-C, Kaufman AH, Kombrinck KW, Converse RL, Good WV, Bugge TH, Degen JL. Invest Ophthalmol Visual Sci. 1998;39:502–508. [PubMed] [Google Scholar]
- 30.Kao WW-Y, Liu C-Y, Converse RL, Shiraishi A, Kao CW-C, Ishizaki M, Doetschman T, Duffy J. Invest Ophthalmol Visual Sci. 1996;37:2572–2584. [PubMed] [Google Scholar]
- 31.Delorenzi NJ, Sculsky G, Gatti CA. Int J Biol Macromol. 1996;19:15–20. doi: 10.1016/0141-8130(96)01094-x. [DOI] [PubMed] [Google Scholar]
- 32.Moyer PD, Kaufman AH, Zhang Z, Kao CW-C, Spaulding AG, Kao WW-Y. Differentiation. 1996;60:31–38. doi: 10.1046/j.1432-0436.1996.6010031.x. [DOI] [PubMed] [Google Scholar]
- 33.Funderburgh JL, Funderburgh ML, Hevelone ND, Stech ME, Justice MJ, Liu C-Y, Kao WW-Y, Conrad GW. Invest Ophthalmol Visual Sci. 1995;36:2296–2303. [PubMed] [Google Scholar]
- 34.Ishizaki M, Zhu G, Haseba T, Shafer SS, Kao WW-YY. Invest Ophthalmol Visual Sci. 1993;34:3320–3328. [PubMed] [Google Scholar]
- 35.Saika S, Shimezaki M, Ohkawa K, Okada Y, Tawara A, Ohnishi Y, Ooshima A, Kimura M, Kurumatani N, Naka H, Awata T, Kao WW-Y. Curr Eye Res. 1998;17:1049–1057. doi: 10.1076/ceyr.17.11.1049.5227. [DOI] [PubMed] [Google Scholar]
- 36.Iozzo R, Murdoch AD. FASEB J. 1996;10:598–614. [PubMed] [Google Scholar]
- 37.Danielson KG, Baribault H, Holmes DF, Graham H, Kadler KE, Iozzo RV. J Cell Biol. 1997;136:729–743. doi: 10.1083/jcb.136.3.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Svensson L, Aszodi A, Reinholt FP, Fassler R, Heinegard D, Oldberg A. J Biol Chem. 1999;274:9636–9647. doi: 10.1074/jbc.274.14.9636. [DOI] [PubMed] [Google Scholar]
- 39.Xu T, Bianco P, Fisher LW, Longenecker G, Smith E, Goldstein S, Bonadio J, Boskey A, Heegaard AM, Sommer B, Satomura K, Dominguez P, Zhao C, Kulkarni AB, Robey PG, Young MF. Nat Genet. 1998;20:78–82. doi: 10.1038/1746. [DOI] [PubMed] [Google Scholar]
- 40.Schrecengost PK, Blochberger TC, Hassell JR. Arch Biochem Biophys. 1992;292:54–61. doi: 10.1016/0003-9861(92)90050-7. [DOI] [PubMed] [Google Scholar]
- 41.Smith RS, Hawes NL, Kuhlmann SD, Heckenlively JR, Chang B, Roderick TH, Sundberg JP. Invest Ophthalmol Visual Sci. 1996;37:397–404. [PubMed] [Google Scholar]
- 42.Funderburgh JL, Funderburgh ML, Mann MM, Conrad GW. J Biol Chem. 1991;266:14226–14231. [PubMed] [Google Scholar]
- 43.Gipson IK, Kiorpes TC. Dev Biol. 1982;92:259–262. doi: 10.1016/0012-1606(82)90170-1. [DOI] [PubMed] [Google Scholar]
- 44.Gipson IK, Kiorpes TC, Brennan SJ. Dev Biol. 1984;101:212–220. doi: 10.1016/0012-1606(84)90131-3. [DOI] [PubMed] [Google Scholar]
- 45.Wendel M, Sommarin Y, Heinegard D. J Cell Biol. 1998;141:839–847. doi: 10.1083/jcb.141.3.839. [DOI] [PMC free article] [PubMed] [Google Scholar]