

Abstract
Inherited epidermolysis bullosa is a rare condition that often present at birth with skin blisters and erosions. They are associated with defective cohesion of the dermis and epidermis. There are 3 principal types: Simple, junctional and dystrophic. The severity of the condition is quite variable. The most severe forms are incompatible with life. The most common types in our country are the severe ones such as the Hallopeau -Siemens subtype. Hands and mucosal areas can develop synechia. We report here a case of dystrophic epidermolysis bullosa in a 27-year-old woman whose finger lesion was managed surgically. This treatment consisted of complete removal of constrictions and adhesions, accompanied by use of a Hueston flap and skin graft to repair the tissue deficit. The patient's clinical course required several repeat operations. This surgery allowed the possible total loss of hand function to be delayed but the inevitable progression of the illness made the treatment somewhat disappointing. Psychosocial implications are very significant in our setting.
KEY WORDS: Congenital, epidermolysis bullosa, fingers
INTRODUCTION
Inherited epidermolysis bullosa is a rare congenital skin condition characterized by appearance of blisters and mucosal or cutaneous erosions either spontaneously or after microtrauma. These lesions are associated with abnormal fragility of the attachment of dermis to the epidermis.
The severity of epidermolysis bullosa is quite variable, ranging from minimal forms to handicapping or even lethal forms, such as the Herlitz type.
We report here a clinical case of inherited epidermolysis bullosa, which was particularly localized to the hands, with a summary of its epidemiological and therapeutic aspects.
CASE REPORT
A.N. is a 27-year-old woman. She was seen in consultation in April 2008 in the Orthopaedic Surgery Department for a stricture of her left 4th finger.
She reported that the condition was observed at birth with spontaneous appearance of diffuse cutaneous blisters that ruptured. They left crusts and then skin scars.
She came from a family of 7 children in which 4 girls had this condition. One of them died when she was 14 years old. Another affected sister presented with a minimal form of the disease with only oral cavity involvement. The third affected sister is handicapped with reduced mobility. Two brothers and a sister are free of disease. The father who has remarried has a daughter who is not affected. The family tree summarizes the family history [Figure 1].
Figure 1.
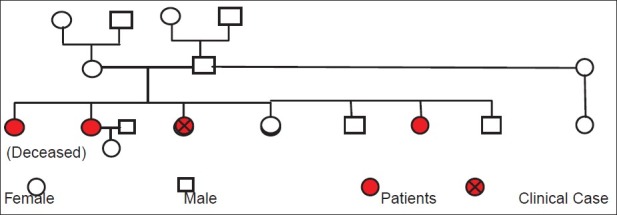
Family Tree
There is no suggestion of parental consanguinity: the parents originally are from the same village. No known family history is found on either side of the family.
The patient's hands showed generalized erythematoid, crusting blisters and cutaneous atrophy. The skin was ichthyotic with sparing of the face and plantar surfaces of hands and feet. Nail dystrophy was seen. There was a stricture of the left 4th finger and thumb adduction. This stricture caused very severe pain. She also suffered from dysphagia which prevented good nutrition and therefore she was quite thin. She developed conjunctival involvement in the last month for which she is being treated. In summary, this patient showed signs of dystrophic epidermolysis bullosa of the Hallopeau-Siemens subtype.
Surgical treatment was limited to the hands; her cutaneous lesions were treated by dermatologists. Surgical treatment began in January 2009 with removal of the stricture ring from the left 4th finger and repair of tissue loss by a double Hueston flap. This was completed with thin skin graft taken from the hypothenar eminence [Figures 2a and b]. Healing was achieved in 3 weeks. Recurrence occurred 2 months later in November 2010. She was operated again in January 2011.
Figure 2.
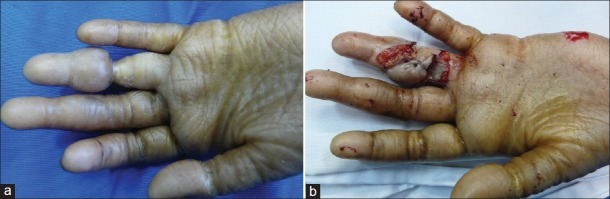
Synechia, left 4th finger (a): Preoperative appearance (b): Immediate postoperative appearance
Her left 5th finger was operated similarly as in February 2009 and closed by a Hueston flap with a superior pedicle. It healed in 2 to 3 weeks but recurred in June 2011. She was re-operated in September 2011 [Figures 3a and b].
Figure 3.
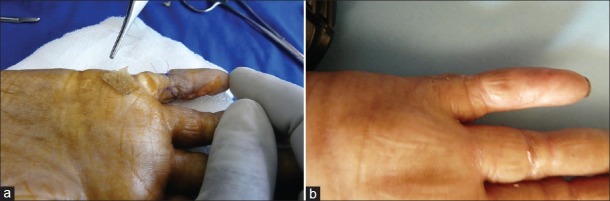
Synechia, left 5th finger (a): Operative appearance (b): Appearance after healing
At the present time this young woman has presented with stricture of the right 5th finger that is very painful [Figures 4a and b]. She underwent the same surgical procedure in March 2012. Postoperative care is in progress. She has been operated on 5 times between January 2009 and March 2012.
Figure 4.
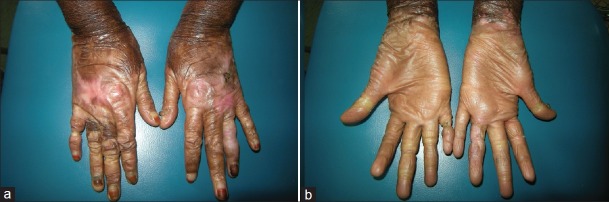
Current appearance of hands (a): Dorsal side (b): Palmar side (Note the stricture of the right 5th finger before surgery)
DISCUSSION
There is little data in the literature concerning this condition in Africa. Winship[1] in South Africa in 1986 reported case prevalence of one case in a population of 354,145. Its prevalence in the United States is estimated at 8 cases per 1 million population.[2] More recently in Dakar, Senegal, Bazolo reported that 15 cases were collected in 25 years in the Dermatology Department.[3]
The classification of the National Epidermolysis Bullosa Registry[4,5] is complicated and is based on the level of the blister's cleavage plane. It distinguishes 3 types of epidermolysis bullosa (EB) according to the level of the detachment:
-
-
Simplex (EBS), where the detachment is within the epidermis.
-
-
Junctional (EBJ), with detachment at the level of the epidermal–dermal junction
-
-
Dystrophic or dermolytic (EBD), with detachment under the basement membrane.
Dystrophic epidermolysis bullosa of Hallopeau Siemens is the most severe form. It is generalized and transmitted genetically as a recessive. Diagnosis is often made of birth with the blisters leaving diffuse skin erosions.[6] Risk of death is significant. Moreover, it is often associated with severe mucosal involvement and skin appendage conditions. Clinical course is marked by complications with severe oesophageal stenosis, conjunctival involvement and the possibility of squamous cell carcinoma arising in the cutaneous lesions.[7] However, the locomotor consequences of this condition can cause the patients to suffer greater isolation. This is because the scars become dystrophic with appearance of synechia, pseudo-syndactyly and retraction of the first web contracture. This can progress to the development of “mitten hand” deformities. Areas of friction and areas exposed to microtrauma such as the hands are the most affected by these deformities.[6] In our setting we note the risk of keloid formation.
Surgical treatment is disappointing since clinical progression of the condition appears inevitable. However, it is the only therapeutic choice available to treat the deformities.[7,8] Recurrences can soon occur and were seen within 2 years on the average in our patient. However, this surgery avoids amputation of the fingers. Complete synechia leading to “mitten hand” deformities are often difficult to treat.[9,10]
For many authors[7,10–12] skin grafts are useless in these patients even in severe cases because of the poor quality of the skin. They recommend “directed healing” after immobilization by Kirschner pins. In these cases healing was achieved in approximately 4 weeks. This treatment is less traumatic and less stressful for the patient.[7] There is no consensus concerning the use of skin substitutes such as Biobrane™[9] or keratinocyte cultures[13] in the literature. Moreover, their high cost limits their use.
The importance of postoperative functional rehabilitation must be stressed. It improves postoperative results and delays the recurrences.[14] Bernardis[6] insists that “surgery on request” cannot be always offered.
Psychosocial consequences of this condition are significant, especially if there are several cases in the same family.[7] These children are often ostracized from society and parents are made to feel guilty. This gives rise to mystical explanations and turning toward traditional practitioners. This further delays diagnosis and treatment.
CONCLUSION
Inherited epidermolysis bullosa is a rare condition. It is characterized by cutaneous manifestations at the level of the epidermal–dermal junction. It has 3 distinct types. The Hallopeau-Siemens subtype involves mainly the hands. Treatment of these lesions is surgical.
Multidisciplinary care is important and includes surgeons, dermatologists, rehabilitation specialists, nutritionists, dentists, otolaryngologists, genetic specialists, paediatricians, ophthalmologists, psychologists, and others.
Psychosocial implications are very significant in our country.
The authors declare that they have no conflict of interest and that no external sources of funding were obtained for the conduct of this study.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Winship I. Epidermolysis bullosa in South Africa. S Afr Med J. 1986;69:743–6. [PubMed] [Google Scholar]
- 2.Fine JD. Epidermolysis Bullosa: A Genetic Disease of Altered Cell Adhesion and Wound Healing and the Possible Clinical Utility of Topically Applied Thymosin β4. Ann N Y Acad Sci. 2007;1112:396–406. doi: 10.1196/annals.1415.017. [DOI] [PubMed] [Google Scholar]
- 3.Bazolo N’Dossani. Etat Médecine, Dakar: UCAD; 2009. Epidermolyses bulleuses héréditaires: Etude rétrospective sur 25 ans, N 111 Th. [Google Scholar]
- 4.Fine JD, Eady RA, Bauer EA, Briggaman RA, Bruckner- Tudermanl, Christiano A, et al. Revised classification system for inherited epidermolysis bullosa: Report of the second international consensus meeting on diagnosis and classification of epidermolysis bullosa. J Am Acad Dermatol. 2000;42:1051–66. [PubMed] [Google Scholar]
- 5.Fine JD, Bauer EA, Briggaman RA, Carter DM, Eady RA, Esterly NB, et al. Revised clinical and laboratory criteria for subtypes of inherited epidermolysis bullosa.A consensus report by the Subcommittee on Diagnosis and Classification of the National Epidermolysis Bullosa Registry. J Am Acad Dermatol. 1991;24:119–35. doi: 10.1016/0190-9622(91)70021-s. [DOI] [PubMed] [Google Scholar]
- 6.Bernardis C, Box R. Review Surgery of the hand in recessive dystrophic epidermolysis bullosa. Dermatol Clin. 2010;28:335–41. doi: 10.1016/j.det.2010.01.013. [DOI] [PubMed] [Google Scholar]
- 7.Diedrichson J, Talanow D, Safi A. Epidermolysis bullosa dystrophica (Hallopeau-Siemens syndrome) of the hand -surgical strategy and results. Handchir Mikrochir Plast Chir. 2005;37:316–22. doi: 10.1055/s-2005-872849. [DOI] [PubMed] [Google Scholar]
- 8.Bensouda S, Belgass J, Belkirane L, El Arabi S, Msefer S. L’épidermolyse bulleuse congénitale à propos d’un cas clinique. Journal de l’ordre des dentistes du Québec J Dent Que. 2006;43:519–26. http://www.odq.qc.ca/Publications/JournaldelOrdre/tabid/293/language/en-US/Default.aspx . [Google Scholar]
- 9.Jutkiewicz J, Noszczyk BH, Wrobel M. The use of Biobrane for hand surgery in Epidermolysis bullosa. J Plast Reconstr Aesthet Surg. 2010;63:1305–11. doi: 10.1016/j.bjps.2009.06.038. [DOI] [PubMed] [Google Scholar]
- 10.Siepe P, Roessing C, Safi A. Dystrophic epidermolysis bullosa: Surgical treatment of advanced hand deformities. Handchir Mikrochir Plast Chir. 2002;34:307–13. doi: 10.1055/s-2002-36305. [DOI] [PubMed] [Google Scholar]
- 11.Tian F, Li B, Tian LJ. Treatment of severe hand deformities caused by epidermolysis bullosa. Orthopedics. 2011;34:e780–3. doi: 10.3928/01477447-20110922-37. [DOI] [PubMed] [Google Scholar]
- 12.Marín-Bertolín S, Amaya Valero JV, Neira Giménez C, Marquina Vila P, Amorrortu- Velayos J. Surgical management of hand contractures and pseudosyndactyly in dystrophic epidermolysis bullosa. Ann Plast Surg. 1999;43:555–9. doi: 10.1097/00000637-199911000-00017. [DOI] [PubMed] [Google Scholar]
- 13.Campiglio GL, Pajardi G, Rafanelli G. A new protocol for the treatment of hand deformities in recessive dystrophic epidermolysis bullosa (13 cases) Ann Chir Main Memb Super. 1997;16:91–100. doi: 10.1016/s0753-9053(97)80025-7. [DOI] [PubMed] [Google Scholar]
- 14.Formsma SA, Maathuis CB, Robinson PH, Jonkman MF. Postoperative hand treatment in children with recessive dystrophic epidermolysis bullosa. J Hand Ther. 2008;21:80–4. doi: 10.1197/j.jht.2007.10.001. [DOI] [PubMed] [Google Scholar]