

Abstract
HIV-Tat protein has been implicated in the pathogenesis of HIV-1 neurological complications (i.e., neuroAIDS), but direct demonstrations of the effects of Tat on behavior are limited. GT-tg mice with a doxycycline (Dox)-inducible and brain-selective tat gene coding for Tat protein were used to test the hypothesis that the activity of Tat in brain is sufficient to impair learning and memory processes. Western blot analysis of GT-tg mouse brains demonstrated an increase in Tat antibody labeling that seemed to be dependent on the dose and duration of Dox pretreatment. Dox-treated GT-tg mice tested in the Barnes maze demonstrated longer latencies to find an escape hole and displayed deficits in probe trial performance, versus uninduced GT-tg littermates, suggesting Tat-induced impairments of spatial learning and memory. Reversal learning was also impaired in Tat-induced mice. Tat-induced mice additionally demonstrated long-lasting (up to one month) deficiencies in novel object recognition learning and memory performance. Furthermore, novel object recognition impairment was dependent on the dose and duration of Dox exposure, suggesting that Tat exposure progressively mediated deficits. These experiments provide evidence that Tat protein expression is sufficient to mediate cognitive abnormalities seen in HIV-infected individuals. Moreover, the genetically engineered GT-tg mouse may be useful for improving our understanding of the neurological underpinnings of neuroAIDS-related behaviors.
Keywords: HIV-1, neuroAIDS, Tat, learning, memory, mouse
1. Introduction
The activity of HIV-1 and/or the accessory proteins produced by HIV-1 infection of brain, such as Tat, may be responsible for the persistence of HIV-related neuropathology and subsequent cognitive and psychomotor slowing [1–4]. For instance, exogenous administration of the HIV accessory protein gp120 into rodent brains impaired learning in the Morris water maze [5,6] and Barnes maze [7], and induced apoptosis in exposed brain regions [8]. It has been suggested that Tat may also play a crucial role in the neurotoxicity and cognitive impairment evident in neuroAIDS [9]. Epidemiological reports indicate that clade-specific variation among HIV-1 strains linked to mutations within the neurotoxic epitope of the tat gene lead to differences in the prevalence of HIV-associated dementia [10] and human neuronal toxicity [11,12]. HIV-Tat protein directly damages [13,14] and kills cells [15,16]. In vitro, Tat protein induced CA1 hippocampal and entorhinal cell dysfunction, and the incubation of slices of the hippocampal-entorhinal cortex or CA1 hippocampus with Tat1–86 suppressed long-term potentiation (LTP) [17,18]. Consistent with this finding, the suppression of LTP was positively correlated with performance errors committed by rats in the eight-arm radial arm maze after intracerebroventricular (i.c.v.) administration of recombinant Tat protein [18].
However, to date, few behavioral studies have examined the contribution of Tat protein to learning and memory deficits. In addition to spatial learning and memory impairment in an eight-arm radial maze [18], infusion of Tat protein into the hippocampus of neonatal rats impaired the subsequent learning and memory performance of both preweanling and adult animals [19]. Likewise, exogenous hippocampal administration of Tat to adult rats undergoing withdrawal from ethanol resulted in significant increases in the time to find the hidden platform in the later trials of the Morris water maze task [20]. Accordingly, we tested the hypothesis that Tat activity is sufficient to impair learning and memory performance. Our studies employed the GT-tg bigenic mouse [15], which possesses a gene that codes for Tat1–86 protein specifically integrated into glial fibrillary acidic protein (GFAP)-containing astrocytes, producing brain-specific expression. In this model, Tat protein expression is induced by the activation of a tetracycline-on promoter site with administration of doxycycline (Dox). All mice in this study were tested in either the Barnes maze [21,22] or a novel object recognition (NOR) assay [23,24] to determine whether Tat expression was sufficient to impair different types of learning and memory performance.
2. Materials and Experimental Methods
2.1 Animals and housing
Adult male GT-tg bigenic [15] and C57BL/6J wild-type (Jackson Labs, Bar Harbor, ME) mice, 8–14 weeks of age, were used in all experiments. Mice were housed and cared for in the Northeastern University animal facility in accordance with the 1996 National Institutes of Health Guide for the Care and Use of Laboratory Animals, as approved by the Institutional Animal Care and Use Committee. The creation and development of the GT-tg mouse and genotype confirmation of the inducible and brain-targeted HIV-1 Tat protein were described previously [15]. GT-tg mice were engineered to express the Tat1–86 gene upon the Dox-mediated activation of a brain-specific promoter (GFAP in astrocytes). Breeding pairs of GT-tg bigenic mice, previously back-crossed 7 generations onto the C57BL/6J line, were used to establish a colony for this study. The C57BL/6J strain of mice were used to determine whether there were differences in behavior displayed in uninduced GT-tg mice as compared to the parent strain of mouse.
2.2 Chemicals
Doxycycline hyclate (Dox; see below), obtained from Sigma-Aldrich (St. Louis, MO), was dissolved in 0.9% saline prior to injection.
2.3 Induction of brain-targeted Tat with Dox treatment
To induce Tat1–86 protein expression, GT-tg bigenic mice were administered Dox via a single daily intraperitoneal (i.p.) injection (100 mg/kg, dissolved in 0.9% saline in a volume of 0.3 ml/30 mg body weight) for 1, 3, 5, or 7 days as indicated. (Characterization of Dox dose-effect relationships on Tat expression are detailed in the supplemental material). The 100 mg/kg, i.p., daily dose for 7 days maximal dosing was selected based on previously demonstrated efficacy of Tat induction (JJ He, personal communication and [16]). C57BL/6J mice used in behavioral experiments were administered the maximal Dox dosing to control for any possible effects of Dox on behavior. Vehicle (0.9% saline) treatment of GT-tg mice for 7 days was included as a direct control for Dox-treated GT-tg mice. Vehicle-treated GT-tg mice are referred to as “uninduced” throughout the manuscript.
2.4 Western blot
Whole brains were isolated from GT-tg mice pretreated with vehicle or Dox (100 mg/kg, i.p. up to 7 days) and homogenized for Western blot analysis. Brains were also obtained from C57BL/6J mice lacking the Tat transgene that had been pretreated with vehicle or Dox (100 mg/kg, i.p.) for 7 days as a control. Prepared aliquots of homogenated protein (100 μg/lane) were then resolved by gel electrophoresis on 10% Bis-Tris gels (at pH 6.4) using NuPAGE MES SDS running buffer and NuPAGE antioxidant using methods described previously [25]. After blocking with 5% nonfat dry milk powder (NFDM) in 0.1% Tris-buffered saline-Tween (TBS-T) for 1 h, resolved, nitrocellulose bound proteins were incubated overnight at 4°C in 5% NFDM/0.1% TBS-T containing the primary antibodies for β-actin (0.02 μg/ml, Cell Signaling Technologies) and Tat protein (1:2000 of the rabbit polyclonal antibody ab43014, Abcam, Cambridge, MA). After washing, membranes were incubated with a horseradish peroxidase-conjugated goat antirabbit IgG (diluted 1:5000 in 5% NFDM/0.1% TBS-T) and visualized with LumiGlo enhanced chemiluminescence agent (Cell Signaling Technologies, Ipswich, MA) detected with a Biospectrum AC imaging system (UVP, Upland, CA).
Western blots were performed in triplicate for each of three brain samples, and βactin signal intensity bands and bands corresponding to Tat protein weights in GT-tg mice were quantified and statistically analyzed as described in section 2.7.1.
Note that further Western blot analyses were performed with additional tissue samples, conditions of Tat induction and alternative antibodies for Tat protein. Full details may be found in the supplemental material.
2.5 Barnes Maze
The Barnes maze assesses spatial learning and memory and is based on the natural tendency of mice to explore and escape through holes [22,26]. The paradigm as performed here was carried out in stages over 5 days, and included habituation (on day 1), acquisition (days 1–4), probe testing (day 4) and reversal testing (on day 5). The task requires the use of fixed and distinct spatial cues within the testing room to navigate to one of 40 evenly-spaced, 5-cm diameter escape holes positioned evenly around the perimeter of a brightly lit 105 cm circular surface. An escape chamber (15 × 8 × 3 cm) was located beneath the escape hole and remained fixed throughout the acquisition period. Mice were habituated by allowing them to freely explore (but not escape) the maze for 1 min, and then by placing the mice directly into the escape chamber (not yet attached to the escape hole), for 30 s.
After habituation, mice were trained to locate an assigned escape hole/chamber (i.e., acquisition trials). The acquisition period consisted of two trials/day for four days with a 15-min inter-trial interval (ITI). Each 3-min trial began by placing a mouse enclosed in a cylinder in the center of the maze, ensuring that they were randomly oriented at trial initiation. Data collection began when a static radio noise was played as the cylinder was removed to free the mouse; the static noise was played to motivate mice to escape the maze (i.e., in addition to the bright light [27]). Mice not finding the escape hole within 3 min were guided toward and allowed to enter the hole. When mice entered the escape hole, the static noise was terminated and the mice were kept in the escape chamber for 15 s. In between each trial, the maze was wiped with 70% ethanol to remove odor cues.
During these acquisition trials, escape latencies and maze errors (see below) were recorded. A mouse was considered to have completed an escape when its last foot left the maze surface. Errors were deemed as nose or head deflections into a hole not leading to the escape box. Reference memory errors were recorded when a mouse made a nose and head deflection into a non-escape hole, whereas working memory errors were recorded when a mouse made a nose and head deflection into an non-escape hole already visited during that same trial. In addition, the type of search strategy employed by mice on each trial was recorded as serial (searching the holes in a clockwise or counterclockwise order), random (hole searches separated by crossing through the center of the maze or unorganized search), or direct search (moving directly to escape hole or within the two adjacent holes before visiting the escape) [28].
At the end of day 4, a probe trial was performed. During this trial, the escape chamber was removed and mice explored the maze for 90 s [29]. The variables recorded during this trial were 1) success at finding the former escape location and 2) time spent in the target quadrant (where the escape hole was located). Reversal learning was tested on day 5 by moving the escape hole 135 degrees from the previous location and conducting four trials. Latency to escape through the new location was recorded.
On the last day of testing the mice performed one 3-min trial in which they were required to locate an escape tunnel in a novel location made visible with a contrasting object placed behind the tunnel; this trial assessed whether any vision problems existed that might confound task performance. All mice in the study successfully found the visually-cued escape location.
2.6 Novel object recognition (NOR) assay
Learning and memory performance was examined using the NOR assay [23,24,30]. The NOR task for rats and mice is based on their trait to spontaneously explore their environment; unaffected animals will spend more time exploring a novel object than a familiar one [31,32]. As this task does not require motivation such as food or water deprivation, it minimizes external, physical stressors [31]. The NOR task probes both learning and memory function, as the final output of the assay is affected by both. The paradigm was carried out over three phases as described in Carey et al. [24]. Each phase lasted 10 min with a 10 min inter-trial interval (ITI), which was found to be optimal in previous experiments using untreated-C57BL/6J mice [24]. During each trial, mice freely explored the environment before being returned to their home cages. During phase 1, objects A and B (e.g., pair of dice) were centered across from each other in a rectangular cage (16 × 24 × 12 cm), 2 cm away from the walls. In phase 2, object B was moved laterally to 1 cm from the edge of the cage whereas object A remained fixed. In phase 3, after completion of the training phases, object B (the die) was replaced with a novel object (a marble). Note that phase 2 served as a control, to demonstrate that subsequent performance in phase 3 was based on novel object recognition, rather than simple changes in object location. In contrast, phase 3 testing assessed both learning and memory, as mice will typically recognize and reject previously encountered objects to spend more time with the novel object [24]. Use of marbles and dice as novel objects was randomized across the study. For each phase, the time mice spent attending to each object was recorded with a stopwatch. Object attending was defined as the duration of time a mouse spent in physical contact with the object, using any body part other than the tail, or whenever it was within 0.5 cm of the object, facing it, and engaged in active exploration (e.g., sniffing or manipulating). Data are presented as a percent recognition index (RI) for object B: RI = (time attending to object B/time attending to object A+B)*100. In addition to described Dox-treatment comparisons, separate groups of mice were tested at 48 h or one week after the completion of Dox treatment. An additional group of mice treated with the maximal dose of Dox for the maximum duration (100 mg/kg, i.p., 7 days) was tested one month after the completion of Tat induction.
2.7 Statistical analyses
2.7.1 Western blot
β-actin antibody labeling was measured at the expected weight of 45 kDa, whereas Tat protein labeling was quantified from the band found at 19 kDa, which has been suggested to be the observed weight of expressed Tat protein in the GT-tg mouse model (J.J. He, personal communication). All bands were quantified with ImageJ 1.62 software (National Institutes of Health (NIH)). Band values were then calculated as percent of C57BL/6J control (saline-treated) sample labeling, and plotted as average values ± SEM across 7–9 experiments. Statistical differences in labeling were assessed using a one-way ANOVA test with Tukey’s HSD post hoc testing using Prism 5.0 software (GraphPad Software, La Jolla, CA). Additional bands labeled with Tat-selective antibodies and identified elsewhere as Tat protein by others (14 kDa [33] and 23 kDa [34]) were analyzed as described in the Supplemental material.
2.7.2 General Behavioral Analysis
Data from all behavioral experiments were analyzed using SPSS 16 statistical package for the social sciences (Chicago, Illinois, USA) and are presented as mean ± standard error of the mean, unless noted otherwise. Significance was set at p ≤ 0.05.
2.7.3 Barnes maze
Latency was analyzed with a two-way repeated measures ANOVA with day (acquisition, day 1–4) or trial (reversal, trial 1–4) as the within-subjects repeated measure and group as the between-subjects variable. The number of errors was analyzed with one-way ANOVA. Tukey’s Honestly Significant Difference (HSD) post hoc test was used to further elucidate all significant ANOVA effects. Search strategies were analyzed with Mann-Whitney U. Probe trial success was analyzed with Fisher’s exact test and is presented as proportions and 95% confidence intervals. Time in the target quadrant during the probe trial was analyzed with a one-sample t-test comparing the time spent in the quadrant to chance (22.5 s).
2.7.4 Novel object recognition
An omnibus repeated measures ANOVA was run with phase as the within subjects repeated measure, and time of testing, Dox dose, and duration of administration as the between subjects variables. Data were analyzed for two comparisons of interest: between group differences in phase 3 RIs and/or within group differences between phase 1 and phase 3 RIs, where an unaffected mouse will show a significant increase in RI from phase 1 to phase 3. Significant interactions and main effects were further analyzed with subsequent one-way ANOVA and/or Tukey’s HSD post hoc testing.
3. Results
3.1 HIV-1 Tat protein is expressed in GT-tg mouse brain in a manner dependent on the degree of exposure to doxycycline
HIV-1 Tat expression in this model has been previously monitored and confirmed to be brain-specific by RT-PCR of Tat mRNA [15], but this is a proxy for Tat protein expression and not a direct measurement. To attempt a more direct demonstration of Dox-induced Tat expression, we performed Western blots on whole homogenized brains isolated from GT-tg mice pretreated with vehicle (0.9% saline, i.e., uninduced) for 7 days or Dox (100 mg/kg, i.p. for 1, 3, 5 or 7 days). C57BL/6J mice were also pretreated with vehicle or Dox (100 mg/kg, i.p.) for 7 days as a control for the effects of Dox treatment. β-actin antibody labeled a single band of similar intensity among all samples, suggesting equivalent amounts of protein in all samples (Fig. 1a, upper image, (F7,56 = 0.850, p = 0.55; one-way ANOVA with Tukey’s HSD post hoc test). At the suggested observed weight of Tat protein (19 kDa, Fig. 1a, lower image), antibody labeling intensity differed with the presence and duration of Dox administered (Fig. 1b; F6,33 = 8.455, p < 0.0001; one-way ANOVA with Tukey’s HSD post hoc test). Although Tat antibody demonstrated a modest degree of non-selective binding by labeling proteins of similar weight in samples of control C57BL6/J mouse brains (Fig. 1a), this labeling did not differ from the amounts of protein labeled in uninduced GT-tg mice (Fig. 1b, p > 0.05, Tukey’s HSD post hoc test). Tat antibody labeling was statistically greater in brain samples taken from GT-tg mice treated with Dox for 3, 5 or 7 days as compared to samples from saline-treated GT-tg mice (Fig. 1b, p < 0.01, Tukey’s HSD post hoc test). Additional testing demonstrated Tat expression increases that were dependent on the Dox dose of used (see supplemental Fig. 1). Together, these results suggest that Tat protein expression is dependent on the dose and duration of Dox exposure.
Fig. 1. Doxycycline-induced HIV-1 Tat protein expression in GT-tg mouse brain.

(a) Representative Western blots from whole brain. The β-actin antibody labeled a single band (upper panel) corresponding to the weight of the β-actin protein of similar intensity across all samples. By contrast, the Tat antibody labeled a number of proteins non-selectively (lower panel), but only demonstrated a difference in labeling intensity that corresponded to the presence and duration of Dox administered.
(b) Summary graph of the quantified whole brain Western blots. All band intensities are plotted as percent control (saline-treated C57BL/6J) labeling. Dox treatment resulted in an increase in Tat-antibody labeling in GT-tg mice dependent on the duration of pretreatment. Labeling intensity differences corresponded to the presence and duration of Dox administered at 19 kDa, which has been suggested to be the observed weight of expressed Tat protein in the GT-tg mouse.
Similar increases in Tat antibody labeling were also observed in a band of a weight attributed to Tat protein (14 kDa [33]; see supplemental Fig. 2). Although the Tat antibody nonspecifically labeled protein at 14 kDa in the control C57BL/6J mice (which lack Tat protein), the increase over control labeling was statistically significant after Dox treatment.
3.2 Barnes Maze Testing Results
3.2.1 Strain differences in Barnes maze performance
A repeated measures ANOVA indicated an overall group difference in latencies to escape the Barnes maze during acquisition trials (average of all trials, saline-treated C57BL/6J 130 ± 7 s; Dox-treated C57BL/6J 114 ± 5 s; uninduced GT-tg, 100 ± 6 s; F2,90 = 4.55, p < 0.05). The escape latencies of the saline- and Dox- treated C57BL/6J mice did not differ on any day (p = 0.96, p = 0.84, p = 0.16, p = 0.84, days 1–4, respectively, Tukey’s HSD). However, uninduced GT-tg bigenic mice exhibited a shorter escape latency than either saline- or Dox-treated C57BL/6J mice on days 1 (p = 0.06 (trend) and p < 0.01, respectively) and 2 (p < 0.05 versus both C57BL/6J groups).
These data suggest the presence of subtle strain differences between the GT-tg mice and the C57BL/6J mice in spatial acquisition performance that preclude a direct comparison of the GT-tg mice to the C57BL/6J mice for this particular task. However, these data do suggest that Dox-treatment itself did not affect task performance in the absence of the Tat gene, as Dox-treated C57BL/6J mice did not exhibit differences in latency versus saline-treated C57BL/6J mice.
3.2.2 Tat-induced mice demonstrated impaired Barnes maze acquisition versus uninduced GT-tg littermates
GT-tg mice were induced to express Tat protein by administering Dox (100 mg/kg, i.p.) daily for 5 or 7 days prior to being tested in the Barnes maze. For the sake of clarity, these mice will be referred to as 5 day and 7 day Tat-induced mice, respectively. Performance was compared to uninduced (0.9% saline, i.p., 7 days) GT-tg littermates. A two-way repeated measures ANOVA indicated an overall group difference in escape latencies over the four acquisition days, with the average differences of the 5 day Tat-induced mice (148 ± 8 s) and the 7 day Tat-induced mice (133 ± 6 s) significantly longer than that of uninduced littermates (100 ± 6 s, (F2,89 = 14.99, p < 0.001). Post hoc analysis of effects on individual acquisition days (Fig. 2a), indicated that there were no differences in escape latencies on any of the four acquisition days between the 5 day and 7 day Tat-induced mice (p = 0.53, p = 0.22, p = 0.99, p = 0.65, days 1–4, respectively). However, the 5 day Tat-induced mice demonstrated longer latencies to escape from the maze compared to uninduced GT-tg mice on days 1, 2, and 4 (p < 0.05), but not on day 3 (p = 0.20). Similarly, the 7 day Tat-induced mice demonstrated longer latencies to escape from the maze compared to uninduced mice on days 1 and 4 (p < 0.01), but not days 2 and 3 (p = 0.54, p = 0.09, respectively).
Fig. 2. Tat-induced mice demonstrated longer escape latencies and committed more Barnes maze acquisition errors versus uninduced GT-tg mice.
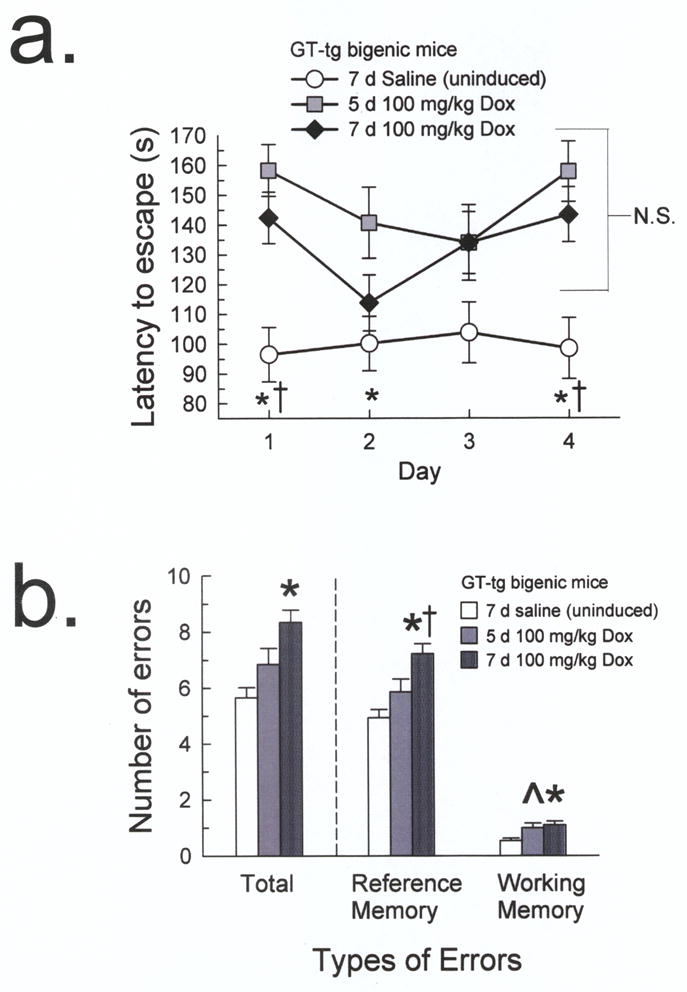
(a) The 5 day Tat-induced mice (100 mg/kg Dox, i.p.; gray squares) demonstrated longer escape latencies compared to uninduced GT-tg mice (0.9% saline, i.p., 7 days; white circles) on days 1, 2, and 4, while the 7 day Tat-induced mice (100 mg/kg Dox, i.p.; black diamonds) demonstrated longer latencies on days 1 and 4. (n = 9–18; n.s. = not significant, * = different from 5 day Tat-induced mice, † = different from 7 day Tat-induced mice, p < 0.05, Tukey’s HSD).
(b) The 7 day Tat-induced mice (dark gray patterned bars) committed more total, reference memory, and working memory errors than the uninduced mice (white bars). The 5 day Tat-induced mice (gray bars) showed a trend toward committing more working memory errors than the uninduced mice. (n= 8–16; ^ = trend from uninduced mice, p = 0.06, * = different from uninduced mice, † = different from 5 day Tat-induced mice, p < 0.05, Tukey’s HSD)
There also was an overall group difference in the total number of errors made over acquisition testing, excluding trial 1 (F2,362 = 11.03, p < 0.001, one-way ANOVA, Fig. 2b). Post-hoc analyses indicated that the 7 day Tat-induced mice made significantly more errors (8.3 ± 0.5) than uninduced mice (5.6 ± 0.4, p < 0.001, Tukey’s HSD). However, while the 5 day Tat-induced mice did not commit significantly more errors (6.6 ± 0.6) than the uninduced mice (p = 0.21), the number or errors committed also did not differ from the 7 day Tat-induced mice (p = 0.09).
A significant overall group difference was found in the number of reference memory errors made (F2,360 = 12.11, p <0.001, one-way ANOVA; Fig. 2b). Post hoc analyses indicated that the 7 day Tat-induced mice made significantly more reference memory errors (7.1 ± 0.4) on average than the uninduced mice (4.8 ± 0.3, p < 0.001, Tukey’s HSD). However, the 5 day Tat-induced mice did not commit more reference memory errors (5.7 ± 0.5) than the uninduced mice (p = 0.23) and committed significantly fewer errors than the 7 day Tat-induced mice (p < 0.05). Although all mice committed fewer working memory errors overall than reference memory errors, there was a group difference in the number of working memory errors made (F2,361 = 6.70, p = 0.001). The 7 day Tat-induced mice committed significantly more working memory errors on average (1.1 ± 0.1) than uninduced mice (0.5 ± 0.1, p < 0.01, Tukey’s HSD). The 5 day Tat-induced mice did not commit significantly more working memory errors (0.9 ± 0.2) than the 7 day Tat-induced mice (p = 0.82), but did show a trend toward committing more working memory errors than uninduced mice (p = 0.06).
The 5 day and 7 day-Tat induced mice demonstrated different search strategy patterns compared to the uninduced mice, who favored the direct search strategy (vs. 5-day Tat induced mice, U = 2230, p < 0.01; vs. 7-day Tat-induced mice, U = 5171.5, p < 0.05, Mann-Whitney U). The 5 and 7 day Tat-induced mice did not show different search strategy patterns (U = 2583, p = 0.20), with both groups favoring the random strategy.
3.2.3 Tat induced mice demonstrated probe trial performance deficits
Uninduced GT-tg mice spent significantly more time than chance would predict in the target quadrant (36.5 ± 5.2 s vs. 22.5 s; p < 0.05, one-sample t-test). In contrast, the 5 day and 7 day Tat-induced mice did not spent more time in the target quadrant than chance (33.8 ± 11.3 s, p = 0.35 and 34.6 ± 6.9 s, p = 0.10, respectively). Moreover, the uninduced mice demonstrated significantly greater success (74%) at finding the acquisition escape hole versus 5 day Tat-induced mice (13%, p < 0.01, Fisher’s exact test). The 7 day Tat-induced mice displayed a trend toward being less successful at finding the escape than the uninduced mice (41%, p = 0.09), although the frequency of escape did not differ from that of the 5 day Tat-induced mice (p = 0.21).
3.2.4 Tat-induced mice were slower to learn a new escape location during a Barnes maze reversal learning task
A two-way repeated measures ANOVA indicated an overall group difference in escape latency (F2,42 = 4.86, p < 0.05), with the 4-trial average latency of 5 day Tat-induced mice (148 ± 8 s) and 7 day Tat-induced mice (128 ± 7 s, p = 0.19, Tukey’s HSD) was significantly longer than in uninduced GT-tg littermates (102 ± 7 s). Post-hoc analysis of individual trials (Fig. 3) indicated that 5 day and 7 day Tat-induced mice did not perform differently on any of the four trials (p = 0.85, p = 0.62, p = 0.37, p = 0.80, trials 1–4, respectively). However, compared to uninduced mice, the 5 day Tat-induced mice demonstrated a trend to longer latencies to escape from the maze on trial 1 (p = 0.11) that was significant on trials 2 and 3 (p < 0.01 and p < 0.05), and reached the performance of uninduced mice only in trial 4 (p = 0.43). Likewise, the 7 day Tat-induced mice demonstrated increased latencies to escape from the maze compared to uninduced mice on trials 1 and 2 (both p < 0.05), but not trials 3 and 4 (p = 0.30 and p = 0.99).
Fig. 3. Tat-induced mice required more trials to learn a new escape location during a Barnes maze reversal learning task.
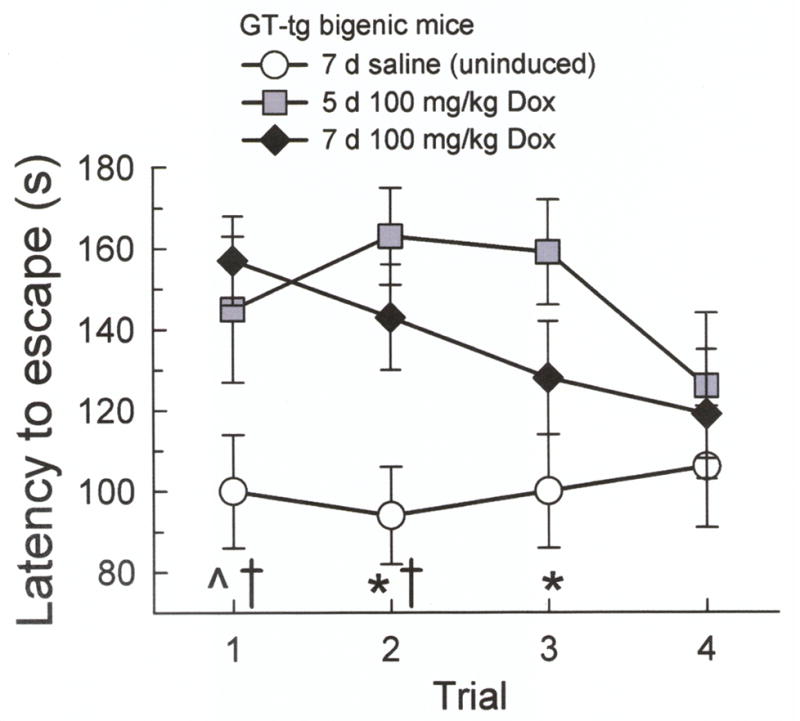
Escape latencies did not differ between the GT-tg mice induced to express Tat with Dox (100 mg/kg, i.p.) for 5 days (gray squares) or 7 days (black diamonds). The 5 day Tat-induced mice demonstrated longer escape latencies compared to uninduced GT-tg mice (0.9% saline, i.p., 7 days) (white circles) on trials 2 and 3, and showed a trend on trial 1. The 7 day Tat-induced mice demonstrated longer escape latencies compared to uninduced mice on trials 1 and 2. (n = 9–19; ^ = trend from 5 day Tat-induced mice, p = 0.11, * = different from 5 day Tat-induced mice, † = different from 7 day Tat-induced mice, p < 0.05, Tukey’s HSD).
3.3 Novel Object Recognition Testing Results
3.3.1 Tat protein expression impaired novel object recognition
There was no group difference in NOR performance in Dox-treated C57BL/6J mice tested either 2 or 7 days after Dox treatment (phase 1, F1,42 = 0.05, p = 0.83; phase 2, F1,42 = 0.01, p = 0.92; phase 3, F1,42 = 1.79, p = 0.19, one-way ANOVA). Uninduced GT-tg mice tested 2 or 7 days after saline treatment also did not demonstrate NOR performance differences (phase 1, F1,51 = 0.84, p = 0.36; phase 2, F1,51 = 2.03, p = 0.16; phase 3, F1,51 = 2.43, p = 0.13, one-way ANOVA). Therefore, both time points were collapsed into a single group for each strain of mice.
Analysis identified a main effect of phase of task (F2,706 = 11.78, p < 0.001; omnibus repeated measures ANOVA), Dox dose (F2,353 = 3.71, p < 0.05; Fig. 4a), and induction duration (F3,353 = 4.16, p < 0.01; Fig. 4b). Also noted were an interaction of phase × duration (F46,706 = 5.78, p < 0.001) and phase × dose (F4,706 = 4.42, p < 0.01). No main effect of testing time (F2,353 = 0.86, p = 0.42) or phase × time interaction effect (F64,706 = 0.71, p = 0.60) was detected, suggesting that task performance after Tat protein induction remained consistent (i.e., did not recover). Post-hoc analysis of these main effects and interactions using one-way ANOVA indicated that there were no overall group performance differences in phase 1 (F14,353 = 1.22, p = 0.26) or phase 2 (F14,353 = 1.23, p = 0.25). There was however, an overall group difference in phase 3 performance (F14,353 = 6.05, p < 0.001).
Fig. 4. Tat-induced GT-tg mice exhibited phase 3 novel object recognition deficits.

GT-tg mice were administered the maximum Dox dose (100 mg/kg, i.p.) for 1, 3, 5, or 7 days (a, gray bars) or 25, 50, or 100 mg/kg Dox for 7 days (b, gray bars). Groups were tested 2 days (solid bars) or 7 days (hatched bars) after induction.
(a) Tat-induced GT-tg mice demonstrated a Dox duration-dependent deficit in phase 3 novel object recognition. The 5 and 7 day Tat-induced mice showed differences in phase 3 RI when compared to C57BL/6J mice and uninduced littermates. (n = 17–53 mice/bar; * = different from matching phase 1 response, † = different from phase 3 response of C57BL/6J and uninduced mice, p ≤ 0.05, Tukey’s HSD).
(b) Tat-induced GT-tg mice demonstrated a duration-dependent deficit in phase 3 novel object recognition. Mice induced for 7 days with 50 or 100 mg/kg Dox were impaired in phase 3 NOR. Mice induced with 25 mg/kg Dox and tested one week after induction also demonstrated impairment in phase 3. (n = 17–53 mice/bar; * = different from matching phase 1 response, † = different from phase 3 response of C57BL/6J and uninduced mice, p ≤ 0.05, Tukey’s HSD).
3.3.2 Tat induced GT-tg mice demonstrated novel object recognition deficits that were dependent on the duration of Dox induction
Both the 5- and 7-day Dox-treated mice exhibited phase 3 RI differences versus C57BL/6J mice and uninduced littermates during testing at both 2 and 7 days’ post induction (p <0.01 and p < 0.05, respectively, Tukey’s HSD; Fig. 4a). Importantly, the 5- and 7-day Dox-treated mice did not show an increase in NOR performance when compared to phase 1 RI values when tested at either 2 or 7 days after induction (p = 0.99). By contrast, the RI values of 1-day Dox-treated mice did not differ when comparing C57BL/6J or uninduced mice at 2 days (p = 0.99) or 7 days (p = 0.99), and showed significant phase 3 RI increases compared to phase 1 (p = 0.05, both time points tested).
3.3.3 Tat induced GT-tg mice demonstrated a Dox dose-dependent deficit in phase 3 novel object recognition
GT-tg mice treated daily with the lowest dose of Dox (25 mg/kg) exhibited normal NOR learning when tested 2 days after the completion of induction (p < 0.01; Fig. 4b), a result similar to that found in C57BL/6J and uninduced mice (p = 0.99). However, prolonged Tat protein exposure impaired the RI between phase 1 to phase 3 at 7 days testing (p = 0.99). By contrast, GT-tg mice treated with 50 mg/kg Dox demonstrated impaired NOR both at 2 and 7 days (p = 0.99, Tukey’s HSD). Interestingly, although NOR was significantly different from C57BL/6J and uninduced littermates in the group tested 48 h after the completion of induction (p < 0.01), mice so treated demonstrated mild improvement when tested at one week after Tat induction, at which time they did not differ from controls (vs. C57BL/6J, p = 0.14, and vs. uninduced, p = 0.25).
3.3.4 Tat protein expression induced lasting suppression of novel object recognition
We examined the duration of the NOR deficits induced by maximal Tat induction (Fig. 5). Maximally induced mice were tested either within 48 h, one week, or one month after the completion of Tat induction. These groups were compared to uninduced GT-tg mice (0.9% saline, i.p., 7 days) and C57BL/6J mice pretreated with Dox (100 mg/kg, i.p., 7 days). At all time points, Tat-induced mice spent less time on the novel object in phase 3 than C57BL/6J mice (p < 0.05, Tukey’s HSD) and their uninduced littermates (p < 0.05). There was no NOR difference between C57BL/6J and uninduced mice (p = 0.99). Furthermore, while uninduced and C57BL/6J mice spent more time on the novel object in phase 3 versus phase 1 (both p < 0.01, Tukey’s HSD), Tat-induced mice did not show increased RI values in phase 3 compared to matching group performance in phase 1 at any time point (p = 0.99), suggesting a deficit in novel object recognition.
Fig. 5. Tat-induced GT-tg mice exhibited persistent suppression of phase 3 novel object recognition performance.

Mice were induced to express Tat with 100 mg/kg Dox for 7 days and tested 2 days (light gray thatched bars), 7 days (dark gray thatched bars), or 30 days (black bars) after the completion of induction. Phase 3 NOR of the C57BL/6J and uninduced mice was not different. Tat-induced mice tested at all time points spent less time on the novel object in phase 3 than controls and showed no increase in phase 3 RIs from phase 1. (n = 17–53 mice/bar; * = significant increase from phase 1 response, † = different from phase 3 response of C57BL/6J and uninduced mice, p < 0.05, Tukey’s HSD).
4. Discussion
The present study demonstrated that mice expressing increased Tat protein levels exhibited impaired task performance on the Barnes maze including total, reference, and working memory errors, as well as abnormal NOR task performance. In the Barnes maze, Tat expressing mice exhibited longer latencies to find an escape hole compared to uninduced littermates.
Overall, these data are in agreement with earlier studies. A single acute i.c.v. administration of Tat protein was shown to impair spatial learning and memory in an eight-arm radial maze [18]. Further, infusion of Tat1–72 protein into the CA1 region of the rat hippocampus increased time to find the hidden platform in the Morris water maze task during ethanol withdrawal [20]. While these studies utilized acute exogenously administered Tat proteins over short durations, the common spatial learning deficits identified in each of the three studies suggest that Tat expression is sufficient to induce learning and memory performance deficits. The demonstration of impaired learning and memory performance months after i.c.v. administration of Tat protein [19] is also consistent with the prolonged duration of detrimental effects by exposure to Tat protein observed in the present study.
Furthermore, in our study the 5-day and 7-day Tat-induced mice committed more working memory errors during acquisition than uninduced mice. The working memory errors reported here are consistent with the results of the Li et al. study [18], as the type of errors the Tat-injected mice made in that study were based on a similar behavior. Moreover, the 5- and 7-day Tat-induced mice favored the random search strategy during acquisition training, while the uninduced mice favored the direct search strategy. Use of the direct search strategy suggests knowledge of the escape location, whereas the random search strategy (or lack of a strategy) suggests that mice may not retain knowledge of the escape hole location [28]. Along these lines, 5-day Tat-induced mice also demonstrated Barnes maze probe trial deficits. Together, these results constitute strong evidence for a clear impairment of spatial memory in the Tat-induced mice. This finding is further consistent with clinical studies, where HIV infiltration often is identified in the hippocampus and adjacent parts of the entorhinal and temporal cortices (see [35] for review). The present Barnes maze findings of deficits in spatial learning and memory are also consistent with data collected from other animal models of HIV-infection showing impaired spatial ability [18,36–39].
Our study extends findings from prior spatial learning experiments by showing that Tat-induced mice demonstrated impaired reversal learning in the Barnes maze, characterized by an inability to learn a new escape location. However, by trial 4, the 5 day Tat-induced mice performed comparably to uninduced mice. We do not interpret this finding as reflecting a “recovery” of performance, as our NOR data suggest that Tat-induced mice exhibit persistent learning impairments. Rather, we believe that this result suggests that the mice exposed to Tat protein were slower to learn a new escape location than their uninduced littermates. This difficulty in learning a new escape location is similar to the cognitive inflexibility often observed in patients suffering from progressive neurodegenerative diseases [40], and is consistent with the cognitive deficits associated with prolonged HIV-1 infection [2,41]. Additional studies comparing Tat-induced structural deficits in mouse brain regions to brain areas found to be abnormal in imaging studies with HIV-positive humans, specifically in the dorsal frontal cortex [42,43] and hippocampus [44], may provide further insight into these findings.
It should be noted the Tat-induced GT-tg mice were not compared to C57BL/6J mice in the Barnes maze. It was demonstrated that the C57BL/6J mice showed significant learning differences during acquisition training when compared to the uninduced GT-tg mice, therefore precluding direct comparison in Barnes maze experiments. The C57BL/6J mice used as controls here are direct ancestors of mice born at a breeding colony from Jackson Laboratories, whereas the GT-tg mice were created and back-crossed in the laboratory of Dr. Johnny He. Although the GT-tg mice have been back-crossed 7 generations onto the C57BL/6 line, it is possible that there is some divergence in phenotype from the C57BL/6J strain, resulting in subtle differences in spatial acquisition due to the ancestry of the mice. Although this effect highlights the importance of making direct comparisons between the uninduced GT-tg mouse and the parent strain of mice in the future, it does not negate the Barnes maze findings reported here.
Furthermore, significant deficits in other measures of learning and memory performance were observed, suggesting Tat protein-induced deficits extend beyond spatial learning. NOR deficits were dependent on Dox dose and duration (and thus, presumably, Tat) exposure, and persisted for up to one month after Tat-induction. Interestingly, a very low Dox dose (25 mg/kg/d for 7 days) did not produce significant NOR impairment when mice were tested 2 days after induction completion, but NOR impairment was observed in a separate group of Dox-treated mice tested 7 days after induction. These data suggest that Tat effects may not only persist but progress over time even after induction is terminated. Viral proteins such as Tat have been shown to induce progressive damage [45], consistent with our present findings. Together, these data suggest that the learning and memory deficits we found are related to the amount of Tat produced, to the duration of Tat exposure, and may persist (and even progress) after induction is terminated.
In contrast to the Barnes maze results, no differences in NOR performance were found between C57BL/6J and uninduced GT-tg mice. Although spatial learning and memory performance strongly varies between mouse strains [46,47], novel object recognition is thought to be relatively consistent between mouse strains [48]. This contrast may speak to the different sub-systems of learning and working memory represented by the two tasks used in this study. Reports suggest that different types of learning and memory are subserved by distinct brain regions [49,50]. The hippocampus has been demonstrated to be a major brain structure involved in spatial learning [51], whereas the ability of animals to recognize a novel object has been attributed to the activity of the perirhinal cortex in the medial temporal lobe [52]. While backcrossed 7 generations onto the C57BL/6J line, the differences in baseline spatial memory performance demonstrated by the GT-tg bigenic mice could conceivably arise from subtle neuroanatomical differences (e.g., in hippocampus) remaining between the two strains. Administration of exogenous Tat protein into the hippocampi of neonate rats impaired spatial memory performance into adulthood [19], suggesting Tat exposure during development produces prolonged disability of spatial memory. Although the extent of Tat-induced neurotoxic effects within the perirhinal cortex remains uncharacterized, induction of Tat protein was reported to result in significant neurotoxicity and neuronal death in the hippocampus and cerebral cortex of GT-tg bigenic mice [16]. Overall, the existing neuroanatomical evidence seems consistent with the learning and memory deficits we observed, but further investigation of the structural differences between C57BL/6J and uninduced GT-tg mice and characterization of Tat-induced neurotoxicity and neuroanatomical integrity throughout the brain is needed.
It should be further noted that assays of learning and memory depend on the integrity of systems mediating attention. It is conceivable that the deficits in novel object recognition may be due to Tat-mediated deficits in attention. Although attentional deficits are associated with the cognitive decline of HIV patients [53], the effect of Tat protein on attention has not been studied in animal models. The novel object recognition test may be configured differently to examine attention [54], but the present study did not examine this effect. Further study is required to determine the impact of attention on Tat-induced deficits in learning and memory.
Of interest, Tat protein possesses multiple means to induce cellular dysfunction, potentially mediating the observed learning and memory impairments. Extracellular Tat protein also activates cell surface growth factor receptors [55] modulating signal transduction including the activation of PKC [56] and MAP kinases [55,57] associated with LTP and synaptic plasticity, the neuronal basis for hippocampal-dependent learning (see [58,59] for reviews). Tat exposure in vitro can result in CA1 hippocampal and entorhinal cell dysfunction by suppressing LTP [17,18]. Intriguingly, Tat was recently shown in vitro to upregulate histone deacetylase-2 (HDAC2), a regulator of gene expression and chromatin remodeling [60]. As HDAC2 overexpression was shown to downregulate CREB and CaMKII genes essential to synaptic plasticity, the authors speculated that this upregulation may be the mechanism by which Tat protein induces HIV-associated neurocognitive dysfunction [60]. Additionally, Tat protein is known to upregulate a number of mediators of inflammation, including tumor necrosis factor-alpha [61] and cytokines such as interleukin-1β (IL-1β; [62]). Tat-induced upregulation of cytokines was shown to occur in hippocampus after infusion into that structure [61]. As cytokines have been demonstrated to suppress spatial learning and memory performance [63,64,65], the neuroinflammatory effects of Tat protein could conceivably contribute to the behavioral deficits observed here. Finally, in vitro research suggests that Tat produces neurotoxicity [66] correlated with the induction of oxidative stress and astrocytosis [67,68] that impairs neuronal function and survival [68], particularly the dysfunction of pyramidal hippocampal neurons [69,70]. Rat studies in vivo using direct injections of Tat into the brain have confirmed multiple neurohistopathological manifestations [66] modeling aspects of HIV-associated dementia [71,72], including learning and memory deficits [18,19,20]. Together, these findings suggest future experiments examining signaling, neuroinflammatory and neurotoxicity effects of Tat on learning and memory performance would be valuable in establishing the biological mechanisms and brain structural targets mediating learning and memory deficits in Tat-expressing mice and in neuroAIDS patients.
5. Conclusion
In summary, we have shown that brain-selective Tat protein expression induces spatial, reversal, and novel object recognition learning and memory impairments, which persist and even progress after termination of Tat induction. Tat expression appears to be sufficient to mediate spatial learning and memory deficits, and thus may mediate some of the cognitive deficits observed in HIV-infected individuals.
Supplementary Material
Research Highlights.
Mice expressing HIV-Tat protein show deficits in spatial learning and memory
Mice expressing HIV-Tat protein demonstrate demonstrate perseveration
Mice expressing HIV-Tat protein demonstrate impairment of novel object recognition
Acknowledgments
We thank Johnny He for the gift of the GT-tg transgenic breeder mice, Andrew Heusser for building the Barnes maze, and Elisa Mizrachi for assistance with the Western blot analysis. This research is supported by F31 NS064872-01 from NINDS to ANC, R03 DA16415 from NIDA, R01 MH085607 from NIMH and funds from the State of Florida, Executive Office of the Governor’s Office of Tourism, Trade, and Economic Development to JPM.
Abbreviations
- ANOVA
analysis of variance
- Dox
doxycycline
- GFAP
glial fibrillary acidic protein
- HIV
human immunodeficiency virus
- HSD
Honestly Significant Difference
- i.c.v
intracerebroventricular
- i.p
intraperitoneal
- ITI
inter-trial interval
- LTP
long-term potentiation
- NOR
novel object recognition
- RI
recognition index
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Frankel AD, Young JA. HIV-1 fifteen proteins and an RNA. Ann Rev Biochem. 1998;67:1–25. doi: 10.1146/annurev.biochem.67.1.1. [DOI] [PubMed] [Google Scholar]
- 2.Power C, McArthur JC, Nath A, Wehrly K, Mayne M, Nishio J, Langelier T, Johnson RT, Chesebro B. Neuronal death induced by brain-derived human immunodeficiency virus type 1 envelope genes differs between demented and nondemented AIDS patients. J Virol. 1998;72:9045–9053. doi: 10.1128/jvi.72.11.9045-9053.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brack-Werner R. Astrocytes: HIV cellular reservoirs and important participants in neuropathogenesis. AIDS. 1999;13:1–22. doi: 10.1097/00002030-199901140-00003. [DOI] [PubMed] [Google Scholar]
- 4.Johnston JB, Zhang K, Silva C, Shalinsky DR, Conant K, Ni W, Corbett D, Yong VW, Power C. HIV-1 Tat neurotoxicity is prevented by matrix metalloproteinase inhibitors. Ann Neurol. 2001;49:230–241. doi: 10.1002/1531-8249(20010201)49:2<230::aid-ana43>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 5.Glowa JR, Panlilio LV, Brenneman DE, Gozes I, Fridkin M, Hill JM. Learning impairment following intracerebral administration of the HIV envelope protein gp120 or a VIP antagonist. Brain Res. 1992;570:49–53. doi: 10.1016/0006-8993(92)90562-n. [DOI] [PubMed] [Google Scholar]
- 6.D’Hooge R, Franck F, Mucke L, De Deyn PP. Age-related behavioural deficits in transgenic mice expressing the HIV-1 coat protein gp120. Eur J Neurosci. 1999;11:4398–4402. doi: 10.1046/j.1460-9568.1999.00857.x. [DOI] [PubMed] [Google Scholar]
- 7.Sánchez-Alavez M, Criado J, Gómez-Chavarín M, Jiménez-Anguiano A, Navarro L, Díaz-Ruiz O, Galicia O, Sánchez-Narváez F, Murillo-Rodríguez E, Henriksen SJ, Elder JH, Prospéro-García O. HIV- and FIV-derived gp120 alter spatial memory, LTP, and sleep in rats. Neurobiol Dis. 2000;7:384–394. doi: 10.1006/nbdi.2000.0302. [DOI] [PubMed] [Google Scholar]
- 8.Nosheny RL, Bachis A, Aden SA, De Bernardi MA, Mocchetti I. Intrastriatal administration of human immunodeficiency virus-1 glycoprotein 120 reduces glial cell-line derived neurotrophic factor levels and causes apoptosis in the substantia nigra. J Neurobiol. 2006;66:1311–1321. doi: 10.1002/neu.20288. [DOI] [PubMed] [Google Scholar]
- 9.Rappaport J, Joseph J, Croul S, Alexander G, Del Valle L, Amini S, Khalili K. Molecular pathway involved in HIV-1-induced CNS pathology: role of viral regulatory protein, Tat. J Leuko Biol. 1999;65:458–465. doi: 10.1002/jlb.65.4.458. [DOI] [PubMed] [Google Scholar]
- 10.Ranga U, Shankarappa R, Siddappa NB, Ramakrishna L, Nagendran R, Mahalingam M, Mahadevan A, Jayasuryan N, Satishchandra P, Shankar SK, Prasad VR. Tat protein of human immunodeficiency virus type 1 subtype C strains is a defective chemokine. J Virol. 2004;78:2586–2590. doi: 10.1128/JVI.78.5.2586-2590.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mishra M, Vertivel S, Siddappa NB, Ranga U. Clade-specific differences in neurotoxicity of human immunodeficiency Virus-1 B and C Tat of human neurons: Significance of dicysteine C30C31 motif. Ann Neurol. 2008;63:366–376. doi: 10.1002/ana.21292. [DOI] [PubMed] [Google Scholar]
- 12.Rao VR, Sas AR, Eugenin EA, Siddappa NB, Bimonte-Nelson H, Berman JW, Ranga U, Tyor WR, Prasad VR. HIV-1 clade-specific differences in the induction of neuropathogenesis. J Neurosci. 2008;28:10010–10016. doi: 10.1523/JNEUROSCI.2955-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aksenov M, Hasselrot U, Bansal A, Wu G, Nath A, Anderson C, Mactutus CF, Booze R. Oxidative damage induced by the injection of HIV Tat protein in the rat striatum. Neurosci Letts. 2001;305:5–8. doi: 10.1016/s0304-3940(01)01786-4. [DOI] [PubMed] [Google Scholar]
- 14.Aksenov MY, Hasselrot U, Wu G, Nath A, Anderson C, Mactutus CF, Booze RM. Temporal relationships between HIV-1 Tat-induced neuronal degeneration, OX-42 immunoreactivity, reactive astrocytosis, and protein oxidation in the rat striatum. Brain Res. 2003;987:1–9. doi: 10.1016/s0006-8993(03)03194-9. [DOI] [PubMed] [Google Scholar]
- 15.Kim BO, Liu Y, Ruan Y, Xu ZC, Schantz L, He JJ. Neuropathologies in transgenic mice expressing HIV-1 Tat protein under the regulation of the astroctye-specific glial fibrillary acidic protein promoter and doxycycline. Am J Pathology. 2003;162:1693–1707. doi: 10.1016/S0002-9440(10)64304-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zou W, Kim BO, Zhou BY, Liu Y, Messing A, He JJ. Protection against Human Immunodeficiency Virus Type 1 Tat neurotoxicity by Ginkgo biloba extract EGb 761 involving Glial Fibrillary Acidic Protein. Am J Pathol. 2007;171:1923–1935. doi: 10.2353/ajpath.2007.070333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Behnisch T, Francesconi W, Sanna PP. HIV secreted protein Tat prevents long-term potentiation in the hippocampal CA1 region. Brain Res. 2004;1012:187–189. doi: 10.1016/j.brainres.2004.03.037. [DOI] [PubMed] [Google Scholar]
- 18.Li ST, Matsushita M, Moriwaki A, Saheki Y, Lu YF, Tomizawa K, Wu HY, Terada H, Matsui H. HIV-1 Tat inhibits long-term potentiation and attenuates spatial learning. Ann Neurol. 2004;55:362–371. doi: 10.1002/ana.10844. [DOI] [PubMed] [Google Scholar]
- 19.Fitting S, Booze RM, Mactutus CF. Neonatal intrahippocampal injection of the HIV-1 proteins gp120 and Tat: Differential effects on behavior and the relationship to stereological hippocampal measures. Brain Res. 2008;1232:139–154. doi: 10.1016/j.brainres.2008.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Self RL, Smith KJ, Butler TR, Pauly JR, Prendergast MA. Intra-cornu ammonis 1 administration of the human immunodeficiency virus-1 protein trans-activator of transcription exacerbates the ethanol withdrawal syndrome in rodents and activates N-methyl-D-aspartate glutamate receptors to produce persisting spatial learning deficits. Neuroscience. 2009;163:868–876. doi: 10.1016/j.neuroscience.2009.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barnes CA, Markowska AL, Ingram DK, Kametani H, Spangler EL, Lemken VJ, Olton DS. Acetyl-1-carnitine 2: Effects on learning and memory performance of aged rats in simple and complex mazes. Neurobiol Aging. 1990;11:499–506. doi: 10.1016/0197-4580(90)90110-l. [DOI] [PubMed] [Google Scholar]
- 22.Bach ME, Hawkins RD, Osman M, Kandel ER, Mayford M. Impairment of spatial but not contextual memory in CaMKII mutant mice with a selective loss of hippocampal LTP in the range of the theta frequency. Cell. 1995;81:905–915. doi: 10.1016/0092-8674(95)90010-1. [DOI] [PubMed] [Google Scholar]
- 23.Save E, Poucet B, Foreman N, Buhot M-C. Object exploration and reactions to spatial and nonspatial changes in hooded rats following damage to parietal cortex or hippocampal formation. Behav Neurosci. 1992;106:447–456. [PubMed] [Google Scholar]
- 24.Carey AN, Lyons A, Shay CF, Dutten O, McLaughlin JP. Endogenous kappa opioid activation mediates stress-induced deficits in learning and memory. J Neurosci. 2009;29:4293–4300. doi: 10.1523/JNEUROSCI.6146-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McLaughlin JP, Myers LC, Zarek PE, Caron MG, Lefkowitz RJ, Czyzyk TA, Pintar JE, Chavkin C. Prolonged kappa opioid receptor phosphorylation mediated by G-protein receptor kinase underlies sustained analgesic tolerance. J Biol Chem. 2004;279:810–1808. doi: 10.1074/jbc.M305796200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dawood MY, Lumley LA, Robison CL, Saviolakis GA, Meyerhoff JL. Accelerated Barnes maze test in mice for assessment of stress effects on memory. Ann N Y Acad Sci. 2004;1032:304–307. doi: 10.1196/annals.1314.047. [DOI] [PubMed] [Google Scholar]
- 27.Sarkisyan G, Hedlund PB. The 5-HT7 receptor is involved in allocentric spatial memory information processing. Behav Brain Res. 2009;202:26–31. doi: 10.1016/j.bbr.2009.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barnes CA. Memory deficits associated with senescence: a neurophysiological and behavioral study in the rat. J Comp Physiol Psychol. 1979;93:74–104. doi: 10.1037/h0077579. [DOI] [PubMed] [Google Scholar]
- 29.Sunyer B, Sudarshan P, Hoger H, Lubec G. Barnes maze, a useful task to assess spatial reference memory in the mice. Nat Protoc. 2007 doi: 10.1038/nprot.2007.390. [DOI] [Google Scholar]
- 30.Genoux D, Haditsch U, Knobloch M, Michalon A, Storm D, Mansuy IM. Protein phosphatase 1 is a molecular constraint on learning and memory. Nature. 2002;418:970–975. doi: 10.1038/nature00928. [DOI] [PubMed] [Google Scholar]
- 31.Ennaceur A, Delacour J. A new one-trial test for neurobiological studies of memory in rats. Behav Brain Res. 1988;31:47–59. doi: 10.1016/0166-4328(88)90157-x. [DOI] [PubMed] [Google Scholar]
- 32.Frick KM, Gresack JE. Sex differences in the behavioral response to spatial and object novelty in adult C57BL/6 mice. Behav Neurosci. 2003;117:1283–1291. doi: 10.1037/0735-7044.117.6.1283. [DOI] [PubMed] [Google Scholar]
- 33.Pugliese A, Vidotto V, Beltramo T, Petrini S, Torre D. A review of HIV-1 Tat protein biological effects. Cell Biochem Funct. 2005;23:223–227. doi: 10.1002/cbf.1147. [DOI] [PubMed] [Google Scholar]
- 34.Fitting S, Xu R, Bull C, Buch SK, El-Hage N, Nath A, Knapp PE, Hauser KF. Interactive comorbidity between opioid drug abuse and HIV-1 Tat. Am J Pathology. 2010;177:1397–1410. doi: 10.2353/ajpath.2010.090945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anthony IC, Bell JE. The Neuropathology of HIV/AIDS. Int Rev Psychiatry. 2008;20:15–24. doi: 10.1080/09540260701862037. [DOI] [PubMed] [Google Scholar]
- 36.Sei Y, Arora PK, Skolnick P, Paul IA. Spatial learning impairment in a murine model of AIDS. FASEB J. 1992;6:3008–3013. doi: 10.1096/fasebj.6.11.1644264. [DOI] [PubMed] [Google Scholar]
- 37.Zink WE, Anderson E, Boyle J, Hock L, Rodriguez-Sierra J, Xiong H, Gendelman HE, Persidsky Y. Impaired spatial cognition and synaptic potentiation in a murine model of human immunodeficiency virus type 1 encephalitis. J Neurosci. 2002;22:2096–2105. doi: 10.1523/JNEUROSCI.22-06-02096.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Griffin WC, 3rd, Middaugh LD, Cook JE, Tyor WR. The severe combined immunodeficient (SCID) mouse model of human immunodeficiency virus encephalitis: deficits in cognitive function. J Neurovirol. 2004;10:109–115. doi: 10.1080/13550280490428333. [DOI] [PubMed] [Google Scholar]
- 39.Vigorito M, LaShomb AL, Chang SL. Spatial learning and memory in HIV-1 transgenic rats. J Neuroimmune Pharmacol. 2007;2:319–328. doi: 10.1007/s11481-007-9078-y. [DOI] [PubMed] [Google Scholar]
- 40.Traykov L, Baudic S, Thibaudet MC, Rigaud AS, Smagghe A, Boller F. Neuropsychological deficit in early subcortical vascular dementia: comparison to Alzheimer’s disease. Dement Geriatr Cogn Disord. 2002;14:26–32. doi: 10.1159/000058330. [DOI] [PubMed] [Google Scholar]
- 41.Tardieu M. HIV-I-related central nervous system diseases. Curr Opin Neurol. 1999;12:377–381. doi: 10.1097/00019052-199908000-00002. [DOI] [PubMed] [Google Scholar]
- 42.Ernst T, Chang L, Jovicich J, Ames N, Arnold S. Abnormal brain activation on functional MRI in cognitively asymptomatic HIV patients. Neurology. 2002;59:1343–1349. doi: 10.1212/01.wnl.0000031811.45569.b0. [DOI] [PubMed] [Google Scholar]
- 43.Chang L, Arnold S, Tomasi D, Caparelli EC, Carasig D, Zimmerman L, Cloak C, Ernst T. Decreased brain activation and deactivation during visual attention in HIV patients. Proc Intl Soc Mag Reson Med. 2003;11:10. [Google Scholar]
- 44.Castelo JM, Sherman SJ, Courtney MG, Melrose RJ, Stern CE. Altered hippocampal-prefrontal activation in HIV patients during episodic memory encoding. Neurology. 2006;66:1688–1695. doi: 10.1212/01.wnl.0000218305.09183.70. [DOI] [PubMed] [Google Scholar]
- 45.Nath A, Conant K, Chen P, Scott C, Major EO. Transient exposure to HIV-1 Tat protein results in cytokine production in macrophages and astrocytes. A hit and run phenomenon. J Biol Chem. 1999;274:17098–11702. doi: 10.1074/jbc.274.24.17098. [DOI] [PubMed] [Google Scholar]
- 46.Nguyen PV, Abel T, Kandel ER, Bourtchouladze R. Strain-dependent differences in LTP and hippocampus-dependent memory in inbred mice. Learn Mem. 2000;7:170–179. doi: 10.1101/lm.7.3.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wright JW, Alt JA, Turner GD, Krueger JM. Differences in spatial learning comparing transgenic p75 knockout, New Zealand Black, C57BL/6, and Swiss Webster mice. Behav Brain Res. 2004;153:453–458. doi: 10.1016/j.bbr.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 48.Sik A, van Nieuwehuyzen P, Prickaerts J, Blokland A. Performance of different mouse strains in an object recognition task. Behav Brain Res. 2003;147:49–54. doi: 10.1016/s0166-4328(03)00117-7. [DOI] [PubMed] [Google Scholar]
- 49.Bussey TJ, Dias R, Amin E, Muir JL, Aggleton JP. Perirhinal cortex and place-object conditional learning in the rat. Behav Neurosci. 2001;115:776–785. doi: 10.1037//0735-7044.115.4.776. [DOI] [PubMed] [Google Scholar]
- 50.Broadbent NJ, Squire LR, Clark RE. Spatial memory, recognition memory, and the hippocampus. Proc Natl Acad Sci. 2004;101:14515–14520. doi: 10.1073/pnas.0406344101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nadel L, Hardt O. Update on memory systems and processes. Neuropsychopharmacology. 2011;36:251–273. doi: 10.1038/npp.2010.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Murray EA, Richmond BJ. Role of perirhinal cortex in object perception, memory and associations. Curr Opin Neurobiol. 2001;11:188–193. doi: 10.1016/s0959-4388(00)00195-1. [DOI] [PubMed] [Google Scholar]
- 53.Durvasula RS, Hinkin CH. Neuropsychological dysfunction among HIV infected drug abusers. Am J Infect Dis. 2006;2:67–73. [PMC free article] [PubMed] [Google Scholar]
- 54.Silvers JM, Harrod SB, Mactutus CF, Booze RM. Automation of the novel object recognition task for use in adolescent rats. J Neurosci Methods. 2007;166:99–103. doi: 10.1016/j.jneumeth.2007.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li JCB, Lee DCW, Cheung BKW, Lau ASY. Mechanisms for HIV Tat upregulation of IL-10 and other cytokine expression: Kinase signaling and PKR-mediated immune response. FEBS Lett. 2005;579:3055–3062. doi: 10.1016/j.febslet.2005.04.060. [DOI] [PubMed] [Google Scholar]
- 56.Borgatti P, Zauli G, Cantley LC, Capitani S. Extracellular HIV-1 Tat protein induces a rapid and selective activation of protein kinase C (PKC)-alpha, and -epsilon and -zeta isoforms in PC12 cells. Biochem Biophys Res Commun. 1998;242:332–337. doi: 10.1006/bbrc.1997.7877. [DOI] [PubMed] [Google Scholar]
- 57.Rusnati M, Urbinati C, Musulin B, Ribatti D, Albini A, Noonan D, Marchisone C, Waltenberger J, Presta M. Activation of endothelial cell mitogen activated protein kinase ERK(1/2) by extracellular HIV-1 Tat protein. Endothelium. 2001;8:65–74. doi: 10.3109/10623320109063158. [DOI] [PubMed] [Google Scholar]
- 58.Teyler TJ. Long-term potentiation and memory. Int J Neurol. 1987–1988;21–22:163–171. [PubMed] [Google Scholar]
- 59.Sarvey JM, Burgard EC, Decker G. Long-term potentiation: studies in the hippocampal slice. J Neurosci Methods. 1989;28:109–124. doi: 10.1016/0165-0270(89)90016-2. [DOI] [PubMed] [Google Scholar]
- 60.Saiyed ZM, Gandhi N, Agudelo M, Napuri J, Samikkannu T, Reddy PV, Khatavkar P, Yndart A, Saxena SK, Nair MP. HIV-1 Tat upregulates expression of histone deacetylase-2 (HDAC2) in human neurons: Implication for HIV-associated neurocognitive disorder (HAND) Neurochem Int. 2011;58:656–664. doi: 10.1016/j.neuint.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pu H, Tian J, Flora G, Lee YW, Nath A, Hennig B, Toborek M. HIV-1 Tat protein upregulates inflammatory mediators and induces monocyte invasion into the brain. Mol Cell Neurosci. 2003;24:224–237. doi: 10.1016/s1044-7431(03)00171-4. [DOI] [PubMed] [Google Scholar]
- 62.Yang Y, Wu J, Lu Y. Mechanism of HIV-1 Tat induction of interleukin-1β from human monocytes: involvement of phospholipase C/Protein kinase C signaling cascade. J Med Virol. 2010;82:735–746. doi: 10.1002/jmv.21720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Goshen I, Kreisel T, Ounallah-Saad H, Renbaum P, Zalzstein Y, Ben-Hur T, Levy-Lahad E, Yirmiya R. A dual role for interleukin-1 in hippocampal-dependent memory processes. Psychoneuroendocrinology. 2007;32:1106–1115. doi: 10.1016/j.psyneuen.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 64.Tanaka S, Hondo H, Kanda K, Ashino T, Nakamachi T, Sekikawa K, Iwakura Y, Shioda S, Numazawa S, Yoshida T. Involvement of interleukin-1 in lipopolysaccaride- induced microglial activation and learning and memory deficits. J Neurosci Res. 2011;89:506–514. doi: 10.1002/jnr.22582. [DOI] [PubMed] [Google Scholar]
- 65.Mederios R, Figueiredo CP, Pandolfo P, Duarte FS, Prediger RD, Passos GF, Calixto JB. The role of TNF-alpha signaling pathway on COX-2 upregulation and cognitive decline induced by beta-amyloid peptide. Behav Brain Res. 2010;209:165–173. doi: 10.1016/j.bbr.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 66.Bansal AK, Mactutus CF, Nath A, Maragos W, Hauser KF, Booze RM. Neurotoxicity of HIV-1 proteins gp120 and Tat in the rat striatum. Brain Res. 2000;879:42–49. doi: 10.1016/s0006-8993(00)02725-6. [DOI] [PubMed] [Google Scholar]
- 67.Toborek M, Lee YW, Pu H, Malecki A, Flora G, Garrido R, Hennig B, Bauer HC, Nath A. HIV-Tatprotein induces oxidative and inflammatory pathways in brain endothelium. J Neurochem. 2003;84:169–179. doi: 10.1046/j.1471-4159.2003.01543.x. [DOI] [PubMed] [Google Scholar]
- 68.Zhou BY, Liu Y, Kim B, Xiao Y, He JJ. Astrocyte activation and dysfunction and neuron death by HIV-1 Tat expression in astrocytes. Mol Cell Neurosci. 2004;27:296–305. doi: 10.1016/j.mcn.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 69.Aksenova MV, Aksenov MY, Mactutus CF, Booze RM. Cell culture models of oxidative stress and injury in the central nervous system. Curr Neurovascular Res. 2005;2:73–89. doi: 10.2174/1567202052773463. [DOI] [PubMed] [Google Scholar]
- 70.Askenov MY, Aksenova MV, Nath A, Ray PD, Mactutus CF, Booze RM. Cocaine-mediated enhancement of Tat toxicity in rat hippocampal cell cultures: the role of oxidative stress and D1 receptor. Neurotoxicity. 2006;27:217–228. doi: 10.1016/j.neuro.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 71.Rappaport J, Joseph J, Croul S, Alexander G, Del Valle L, Amini S, Khalili K. Molecular pathway involved in HIV-1-induced CNS pathology: role of viral regulatory protein, Tat. J Leukoc Biol. 1999;65:458–465. doi: 10.1002/jlb.65.4.458. [DOI] [PubMed] [Google Scholar]
- 72.Bruce-Keller AJ, Chauhan A, Dimayuga FO, Gee J, Keller JN, Nath A. Synaptic transport of human immunodeficiency virus-Tat protein causes neurotoxicity and gliosis in rat brain. J Neurosci. 2003;23:8417–8422. doi: 10.1523/JNEUROSCI.23-23-08417.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.