

Abstract
Copy number variation (CNV) analysis has had a major impact on the field of medical genetics, providing a mechanism to identify disease-causing genomic alterations in an unprecedented number of diseases and phenotypes. CNV analysis is now routinely used in the clinical diagnostic laboratory, and has led to a significant increase in the detection of chromosomal abnormalities. These findings are used for prenatal decision making, clinical management and genetic counseling. Although a powerful tool to identify genomic alterations, CNV analysis may also result in the detection of genomic alterations that have unknown clinical significance or reveal unintended information. This highlights the importance of informed consent and genetic counseling for clinical CNV analysis. This review examines the advantages and limitations of CNV discovery in the clinical diagnostic laboratory, as well as the impact on the clinician and family.
Keywords: Comparative genomic hybridization, CGH, copy number variation, CNV, genetic counseling, microarray, single nucleotide polymorphism, SNP
Introduction
The adoption of copy number variation (CNV) analysis by the clinical diagnostic laboratory has had a major effect on the field of medical genetics. It has helped refine genotype-phenotype relationships in known disorders and has led to the discovery of new syndromes [1]. Systematic CNV analysis in large populations has begun to reveal the frequency and the effect of this variation in the human genome.
CNV within the genome is widely recognized as a source of disease. CNVs that involve genomic fragments containing one or more dosage-sensitive genes can result in genetic disorders and complex diseases, including autism, cancer, immune deficiency, and neurodegenerative and neuropsychiatric disorders [2-8]. However, apparently healthy individuals also have a significant number of CNVs within the human genome that seem to have no association with adverse phenotypic outcomes [8-18].
The term copy number variation refers to a difference in the dosage of genes or genomic fragments when compared with a reference human genome. It was originally used to describe genomic fragments that ranged in size from at least one kilobase to several million bases and had a variable copy number [12,19]. Higher resolution CNV analysis has revealed the existence of increasingly smaller CNVs (100 to 1,000 bp) in the human genome [20,21]. CNVs usually result from structural genomic alterations such as a deletion (loss), a duplication (gain), an insertion (usually a gain) or unbalanced translocations/inversions that may lead to either loss or gain of sequences near the breakpoints [12].
Both recurrent and unique (non-recurrent) CNVs are described and each class of CNV is mediated by a different rearrangement mechanism. Recurrent CNVs are usually flanked by low copy number repeats or segmental duplications. This allows for recombination between large, identical blocks of sequences (duplicons) by a process referred to as non-allelic homologous recombination [22-24]. Non-recurrent CNV formation is less well understood, although breakpoint analysis in several non-recurrent CNVs has suggested the involvement of non-homologous end joining, and replication-based mechanisms such as fork stalling and template switching [25-28].
Microarray-based CNV detection has been universally adopted in clinical diagnostics owing to the comprehensive nature of genome-wide analysis [29]. Despite the great advances made in CNV detection technologies, there are still several limitations to genome-wide CNV analyses that may affect their clinical utility. In this review we discuss the advantages and limitations of CNV discovery in the clinical diagnostic laboratory with specific emphasis on the impact of CNV analysis on the clinician in both the prenatal and the postnatal setting.
Variation in healthy individuals and in disease
The role of CNVs in genetic syndromes has long been recognized, with recurrent microdeletion/microduplications detected in syndromes such as Prader-Willi [30], Smith-Magenis [31] and Williams-Beuren [32]. However, initial studies utilizing microarray-based analysis focused on phenotypically normal individuals and identified large CNVs that did not appear to be associated with a genetic disease [9,10]. The recent expansion of microarray-based CNV analysis has led to a better appreciation of the extent of CNV-based variation within the genomes of apparently healthy individuals. These initial studies using microarrays with limited coverage predicted as many as ten CNVs per individual [9,10,16], but, as the resolution of the detection technology has improved, the number of CNVs detected within an individual's genome has continued to grow. Thus, more recent studies have estimated that individuals are hemizygous for approximately 30 to 50 deletions larger than 5 kb [11] and the frequency of CNVs may be greater than 100 per individual [12,15]. The accumulating CNV data from healthy controls has led to the establishment of public databases such as the Database of Genomic Variants and NCBI's Database of Genomic Structural Variation (Box 1).
There are a number of ways by which CNVs can result in a disease phenotype. In the most common scenario, a deletion or duplication alters the genomic copy number of dosage sensitive gene(s) [33,34]. Alternatively, a deletion either within or encompassing a gene sensitive to haploinsufficiency can have the same effect as a disruptive point mutation within the gene. Further, CNVs may result in monogenetic diseases by altering the expressions of genes flanking the CNV due to the disruption of regulatory elements [35,36]. CNVs have also been shown to be a source of mutation in autosomal recessive disorders, in which a deletion of one allele of the gene in combination with a point mutation on the other allele results in the disease phenotype. Recently, three individuals with unexplained intellectual disabilities (IDs) were found to have Cohen syndrome, an autosomal recessive disorder, after CNV analysis. All three patients had a CNV that deleted the COH1 gene and a second pathogenic point mutation was subsequently identified on the other allele [37]. All of these possibilities can lead to difficulty in differentiating a pathogenic from a benign CNV, further complicating CNV data interpretation.
Over the past decade, based solely upon the increased clinical use of microarray-based CNV analysis, the list of recurrent and non-recurrent CNVs associated with disease phenotypes has continued to grow. This has led to the discovery of many new microdeletion and microduplication syndromes, such as 1q21.1 microdeletions [38-40] and 15q13.3 microdeletion/microduplication [41-45]. These novel syndromes, combined with the ever-expanding literature on rare, one-off CNVs associated with disease phenotypes, highlight the significant involvement of CNVs as a causative mutation in genetic diseases. Clinically relevant CNVs can be found in databases such as DECIPHER, ECARUCA and the International Standards for Cytogenomic Arrays Database (Box 1). As clinical laboratories adopt CNV analysis, these resources will become invaluable for the clinician to discriminate pathogenic from non-disease associated CNVs.
CNV detection by high-resolution microarray
A number of technologies can detect copy number gains and losses throughout the human genome and their resolution is continuously increasing. These are either targeted assays for specific genomic regions or known disease genes using techniques such as real-time PCR [46-48], multiplex amplifiable probe hybridization [49,50], multiplex ligation-dependent probe amplification [50-52] or genome-wide assays using high-density microarrays [24,29], and more recently high-throughput sequencing (HTS) technologies [53,54].
High-resolution microarray-based CNV analysis provides a method to detect structural genomic alterations. It is useful for uncovering microdeletions and microduplications as well as novel CNVs that are undetectable by standard karyotype analysis or fluorescence in situ hybridization (FISH) [55]. CNV analysis is typically performed using two types of microarray: either array-based comparative genomic hybridization (aCGH) or SNP-based microarrays (SNP-arrays) (Table 1). aCGH and SNP-arrays are both efficient tools for researchers and clinicians. To decide which is most suitable for a certain application, several factors need to be considered, including resolution desired and ability to customize probe content.
Table 1.
Array-based comparative genomic hybridization versus single nucleotide polymorphism array
| aCGH | SNP-array | |
|---|---|---|
| Probes | Traditionally, the probes are long oligonucleotides (50 to 70 bases) solely designed for copy number analysis. Newer platforms have added SNP-based probes for limited regions of the genome mainly to detect UPD (for example, Agilent CGH + SNP arrays) | SNP-based oligonucleotides designed for SNP genotyping. Copy number can be inferred by additional analysis. Newer platforms have added long oligonucleotides designed solely for copy number analysis to improve resolution (for example, Affymetrix SNP 6.0, Illumina 1M) |
| Experimentation | Genomic DNA from the patient and a control are labeled with different fluorophores and hybridized to the same array. The differences in signal intensity between the patient and control samples, which is converted to a log2 ratio, indicates a difference of copy number | In SNP-arrays the patient sample is hybridized to the microarray by itself. Copy number is determined by comparing the signal intensity data obtained from the patient sample with a collection of controls previously analyzed and incorporated into the analysis software |
| Resolution | Probes can be tiled to provide very high resolution. More flexibility and customization of probe content and density. Single exon resolution possible | The placement of probes is dependent on location of SNPs limiting resolution. Less flexibility in way of customization of probe content |
| Applications | Can only be used for copy number analysis. Will not detect UPD or consanguinity unless SNP probes are specifically added. Will also detect low-level mosaicism of CNVs | Will detect UPD, consanguinity, as well as copy number variations. Will also detect low-level mosaicism of CNVs |
aCGH, array-based comparative genomic hybridization; CNV, copy number variation; SNP, single nucleotide polymorphism; SNP-array, SNP-based microarray; UPD, uniparental disomy.
Both aCGH and SNP-arrays can detect low-level mosaicism, which would be missed by traditional cytogenetic testing and may provide a more accurate measurement of mosaicism level [56-58]. Although mosaicism can also be identified through FISH analysis, typically it has to be suspected before FISH analysis is performed. Thus, microarray-based analysis is especially useful in cases when mosaicism is not clinically suspected and therefore would not have been screened for by FISH with an increased number of metaphase counts. Furthermore, SNP-arrays have an additional advantage that they may help determine whether the mosaic cell line originated from a meiotic or mitotic event [59,60].
In addition to determining copy number alterations, the genotype information provided by SNP-arrays allows the identification of copy number neutral loss of heterozygosity (LOH). This helps in identifying regions that are homozygous due to segmental uniparental disomy or parent of common origin effect, both of which can result in a disease phenotype if a disease gene within the segment is mutated or silenced by imprinting [61,62]. LOH has also been successfully used to identify candidate genes, especially in families with known consanguinity, as segments of homozygosity by descent may indicate a region containing a gene with a homozygous mutation [63-65].
Limitations
Despite the proven utility of microarryas in CNV detection, there are noted limitations in the clinical application. Pathogenic duplications are less commonly identified through clinical microarray-based testing than pathologic deletions. This may be due partially to technical limitations of identifying small duplications [66,67], although duplications are also associated with milder phenotypes, which may not result in CNV analysis, and duplications are often inherited from one parent and may be assumed to be benign as a result [68]. It is likely that this contributed to the dogma that deletion of genomic material is more likely to result in disease, and may have also introduced a bias that has probably led to an underestimation of copy number gains.
Further, microarray-based analysis will not detect genomic alterations that do not result in changes in the amount of genetic material (copy neutral alterations), such as balanced translocations and inversions. Inversions can be directly associated with disease such as the inversion in the F8C gene, which is found in approximately 50% of individuals with hemophilia A [69,70]. Such disease-causing inversions will not be identified through CNV analysis. Inversions can also predispose to genomic rearrangements. Inversions have been noted at an increased frequency in parents of children with Williams-Beuren syndrome [71] and in the mothers of children with Angelman syndrome [72].
As discussed above, an important disease mechanism in any CNV is the possible interruption of a critical gene. This is true of both small CNV and large genomic alterations. Balanced translocations can also result in a disease phenotype if the breakpoints of translocation reside within a coding region or ultraconserved element. Although the mechanism for disease may be similar, due to the neutral copy number change associated with a balance translocation, the etiology for disease will go unrecognized by microarray-based CNV analysis. In a study of 36,325 patients with idiopathic ID, 0.78% of individuals were found to carry copy-number-neutral genomic alterations. These deleterious genomic alterations were identified through chromosome analysis and were not detected through microarray-based testing [73]. Important disease mechanisms may go undiagnosed and may be underestimated if only microarray-based CNV analysis is performed. Other strategies, such as karyotype analysis, may be needed to rule out translocations and/or inversions as the underlying mechanism of disease etiology.
Examples of the utility and impact of clinical CNV analysis
Improvements in traditional cytogenetic techniques have helped identify a number of cytogenetic anomalies associated with ID [74,75]. Yet in comparison with traditional cytogenetic analysis, CNV arrays have a significantly increased diagnostic yield [76,77] and studies using microarray testing have identified pathogenic CNVs in approximately 10% to 20% of individuals with idiopathic ID [73,78-80]. The clinical laboratory has quickly adopted microarray-based CNV analysis as the recommended first tier of testing for individuals with non-syndromic ID, autism spectrum disorders (ASDs), and multiple congenital anomalies (MCA) [29,81].
The use of microarray testing in individuals with ASD and MCA has significantly increased the rate of identification of an underlying genetic etiology. In a study of 852 subjects with ASD, an underlying diagnosis was established in 0.46% of cases through fragile × testing, 2.23% through karyotype analysis, and 7.0% through microarray analysis [82]. Similar to the studies of individuals with ID and ASD, detection of genomic alterations in MCA through microarray analysis was significantly higher than in traditional cytogenetic analysis [83,84]. Establishing a genetic diagnosis is important as it can lead to appropriate referrals for therapy, surveillance for other organ involvement, end the diagnostic odyssey and provide accurate information for genetic counseling [85,86].
The use of CNV analysis has extended beyond the diagnosis of ID, ASD and MCA, allowing for a broader understanding of known disorders and the identification of new syndromes. Before microarray-based analysis, the diagnosis of microdeletion or duplication syndromes required either a visible submicroscopic deletion, such as in Miller-Dieker syndrome [87], or disease-specific FISH analysis. Recognition of disparate phenotypes within a specific syndrome remains a difficult challenge for the clinician. The unbiased nature of CNV analysis has allowed the diagnosis of known microdeletion and duplication syndromes with a wide phenotypic spectrum, which may have otherwise gone undiagnosed [88]. One example is Potocki-Lupski syndrome, which results from a duplication of 17p11.2 and is characterized by developmental delay and variable congenital anomalies. Although Potocki-Lupski syndrome was clinically characterized previously [89], the use of microarray-based testing has greatly improved the characterization of phenotypic spectrum and molecular analysis of the duplications found in the syndrome [90].
With the increased utilization of CNV analysis, new microdeletion and duplication syndromes have been identified. For example, screening of more than 10,000 patients with developmental disabilities through CNV analysis revealed seven patients with a similar phenotype (dysmorphic features, midline defects, seizures and developmental delay) and a microdeletion at 1q41q42 [91]. Isolated cases were previously reported with a similar microdeletion [92,93], although microarray-based CNV analysis provided a means to appreciate the phenotype and insight into pathophysiology of the recurrent microdeletion [94-97].
Clinical CNV analysis typically includes known microdeletion and duplication syndromes, although it can also be designed to include single gene disorders. Exonic or multiexonic CNVs within dosage-sensitive genes represent a significant percentage of mutations in monogenetic disorders [98]. As a result, exonic arrays (microarrays designed to evaluate CNV within transcribed regions) have been designed to target the exon of a single gene, exons within a group of specific disorders, or genome-wide exonic coverage [99,100]. Intragenic deletions/duplications have been identified in a number of autosomal dominant, X-linked recessive and X-linked dominant disorders, many of which would have been missed by non-exon targeted arrays and traditional sequencing methods [21].
Although CNV analysis is often used to evaluate a non-discrete phenotype such as ID, the high frequency of CNVs within monogenic disorders allows researchers to use microarray-based platforms for candidate gene selection. CHARGE syndrome has a distinct phenotype, follows an autosomal dominant inheritance pattern, and, until recently, had an unknown genetic etiology. A number of strategies were used to identify a candidate gene [101] and aCGH analysis in a patient with CHARGE syndrome revealed a microdeletion containing the CHD7 gene [102]. Similar strategies have been used to identify candidate genes in other diseases, such as NBPF23 in neuroblastoma [103], TCTE3 in congenital diaphragmatic hernia [104], TUSC3 in non-syndromic ID [105], and MTUS1 in familial breast cancer [106].
Challenges in interpretation of CNV analysis
CNV analysis is now routinely utilized for the diagnosis of an individual or family and the results are used for genetic counseling and clinical management [107]. Findings from clinical CNV analysis can often provide results that are unintended or difficult to interpret. The ability to detect CNVs has far outpaced our ability to discern their role in disease.
Initially, clinical CNV analysis was designed to limit the number of ambiguous findings and maximize the relevancy of results. This included limiting genomic coverage to known deletion and duplication syndromes [108-110], as well as using probes, such as bacterial artificial chromosomes (BACs), that could be confirmed through traditional cytogenetic and molecular techniques [111,112]. Although there may still be a role for such targeted arrays, the majority of clinical laboratories have adopted platforms with increasing genomic coverage, which increased detection of both deleterious CNVs and variants of unknown significance.
As discussed above, CNVs can result in disease pathology through multiple mechanisms. This makes the interpretation of novel CNVs difficult, and there appears to be wide variability in reporting the clinical significance of CNV results. In one study, 13 CNVs (from both BAC and oligo arrays) were sent to 11 different clinical laboratories. The laboratories designated each CNV as one of the following: normal, likely benign, uncertain clinical significance or abnormal. In none of the 13 cases was there unanimous agreement over the clinical significance of the CNV [113]. A number of guidelines are available to aid clinical laboratories in reporting the clinical significance of CNVs [111-113], and these include consideration of known contiguous gene syndromes, the size, dose and inheritance pattern of the CNV, genomic content within the CNV, and comparison of the medical literature and CNV databases.
As for any sequence or genomic variant, it is important to determine whether a CNV is inherited or de novo [114], and many clinical CNV analyses utilize parental samples or testing of relatives when possible [115]. In general, microduplications are more likely to be inherited than microdeletions [116], although simply being inherited does not indicate that a CNV is benign. Inherited CNVs may have different and unrecognized breakpoints and mechanisms such as mosaicism, incomplete penetrance and variable expression, which can result in inherited CNVs having significantly different impacts among individuals [115]. For example, both deletions and duplications of 1q21.1 are associated with varying levels of ID, microcephaly, dysmorphic features and congenital anomalies, despite the 1q21.1 CNV being inherited from both mildly affected and unaffected parents [38,39].
Clinicians and clinical laboratories often rely on available databases and medical literature that examine the phenotype and frequency at which the CNV has been identified. Yet ambiguity remains about the possible pathogenic effects of a number of CNVs. The 15q11 region is prone to unequal recombination events (mediated by low copy number repeats) leading to deletion, duplication and triplication of the genomic region. Duplications and triplications of 15q11 have been associated with autism [117-120], drug-resistant epileptic seizures [120-123] and schizophrenia [123-125]. Despite a number of studies reporting the pathogenic nature of the 15q11 duplication, researchers have also refuted that increased dose of 15q11 alone is sufficient for a disease phenotype [44,116]. Whether a parent of origin effect or other epigenetic factors contribute to a disease phenotype has yet to be discerned.
Prenatal CNV analysis
Studies evaluating the efficacy of microarray-based CNV analysis have mainly been performed in pediatric populations, although CNV analysis has been performed in spontaneous miscarriages [109], in fetuses terminated for MCA [126,127] and for prenatal diagnosis [128-132]. Indications for pursuing prenatal CNV analysis are similar to the indications for pediatric CNV analysis, and they include an increased risk for chromosomal abnormalities [128,129] and a family history of an intragenic CNV [133]. Unique to prenatal testing, CNV analysis is also performed as a result of advanced maternal age, an abnormal fetal ultrasound or parental anxiety [134].
Initial concerns over the use of prenatal CNV analysis have faded with the experience of clinicians and the laboratory. Similar to postnatal microarray studies, the use of prenatal microarray analysis has resulted in a significant increase in the identification of genomic alterations. In a recent meta-analysis by Hilleman et al., prenatal aCGH appears to increase the detection rate of fetal chromosomal imbalances by 3.6% over traditional karytoype analysis [135], and, similar to postnatal MCA studies, prenatal CNV analysis will identify a significantly increased rate of chromosomal abnormalities when a structural fetal anomaly is present [135,136].
Currently, CNV analysis is not recommended to replace traditional prenatal cytogenetic testing [137], although it is often used as a first-tier option for invasive prenatal testing [138,139]. Prenatal diagnosis allows potential parents to make informed decisions about reproduction. Prenatal CNV analysis extends parental autonomy to decisions over microdeletion or duplication syndromes as well as chromosome abnormalities. Whether microarray analysis is utilized as first-tier testing or in limited circumstances such as fetal anomalies, genetic counseling is a necessary component of prenatal CNV testing [140-142].
Genetic counseling
CNV analysis has provided an ability to identify disease-causing alterations in an unprecedented number of diseases and phenotypes. Despite the promise of CNV analysis, testing may reveal a variant of unknown significance, CNVs with incomplete penetrance or variable expressivity, or unanticipated findings such as misattributed paternity. As a result, an emphasis has been placed on the genetic counseling of patients and families undergoing testing.
As noted above, CNV analysis may identify results that are relatively ambiguous, which may make the interpretation of results relatively complicated. This difficulty is intensified in the prenatal setting, as the decision to continue a pregnancy is often made after the results of prenatal CNV analysis are available. Prenatal aCGH analysis may increase the number of variants of unknown significance by 1% to 2% compared with prenatal karyotype analysis alone [135,138]. The potential of indeterminate results has led many to question the utility of CNV analysis in prenatal diagnosis [143] and has raised concerns on the possible emotional harm on the expectant parents [144]. As a result, informed consent and genetic counseling are paramount prior to undergoing prenatal CNV analysis.
Although delivery of counseling will differ among clinicians, genetic counseling should inform patients and families undergoing CNV analysis of the potential benefit of testing as well as the potential risks of testing, such as variants of unknown significance [128,141]. Genetic counselors, geneticists and medical geneticists are familiar with discussing such issues, which are not unique to CNV analysis. Genetic counseling remains integral to providing clinical CNV analysis whether it is performed in the postnatal or prenatal setting.
Ethical concerns
The unbiased genomic nature of microarray-based CNV analysis is both a benefit and a concern for the clinician. CNV analysis may also discover information that was not intended, such as CNVs that predispose for adult-onset disorders, regions associated with neoplasia and misattributed paternity. In evaluating patients for developmental delay and/or congenital anomalies, individuals have been noted to have CNVs in cancer predisposition syndromes such as familial adenomatous polyposis [145], Peutz-Jeghers syndrome and Li-Fraumeni syndrome [146].
There is significant debate over disclosing incidental findings in genetic research, although the moral obligation to disclose information about pre-symptomatic conditions or neoplasia syndromes is distinct from disclosing misattributed paternity [147,148]. Genetic counseling for CNV analysis should include the possibility of unintended or incidental results. Prior to undergoing testing it is important to discuss both policies about disclosure of unintended results and how results will be relayed with the patient. Rarely, CNV analysis may reveal results that have legal consequences. SNP-arrays will also identify consanguinity as a result of LOH. Depending on the age of the parent and degree of relation, consanguinity may indicate sexual abuse [140,149,150], and laboratories should have institutional policies concerning the possible legal implications of such testing.
The ethical issues surrounding CNV analysis and HTS technologies are not dissimilar from those of genetic testing in general. The importance of informed consent and minimizing the risk of privacy violations are emphasized due to the vast amount of genetic information generated by CNV analysis. Informed consent for CNV analysis may be difficult due to the nature of the testing as opposed to informed consent for the testing of one single gene. Although it is important to include specific information such as the possibility of identifying pre-symptomatic conditions, informed consent should address both the goal and general methodology of the testing and not every possible result [151].
There has been a recent shift in both clinical testing and research testing to share genetic results and phenotypic data. In part this is due to the need of large data sets to differentiate between benign and pathogenic CNVs. The potential harm of loss of privacy or confidentiality may be increased due to the sharing of data, although the actual risk of privacy violations is hard to estimate [152]. It is also important for the clinician to make the patient aware of the laboratory's policy on data sharing. Potential mistrust of the clinician may occur if the patient is unaware of the potential risk of loss of privacy and discovers that the clinical laboratory shares genetic and phenotypic data [151].
Towards the future: high-throughput sequencing technologies
Although microarray technology is the current mainstay of the clinical cytogenetic laboratory, HTS technologies are a powerful tool for the analysis of genetic variation as well as mutation detection in patient cohorts. The strength of HTS is its potential ability to detect all forms of variation, including single nucleotide variations), small (<50 bp) insertions/deletions, CNVs and copy neutral structural variations on a single platform. HTS technologies hold great promise as the future method of choice for detection of all forms of genomic variation, including CNVs.
Structural variation may be underrepresented regardless of whether microarray analysis or HTS technology is utilized. Currently no single platform or strategy identifies all forms of genomic variation and the choice of method for identifying CNVs may ultimately limit results [153]. The understanding of the benefits and limitations of each technology becomes paramount as use in clinical practice is expanding. We suggest a testing algorithm to identify genomic variation in clinical samples based on available technologies for mutation detection and the clinical presentation of the patient (Figure 1).
Figure 1.
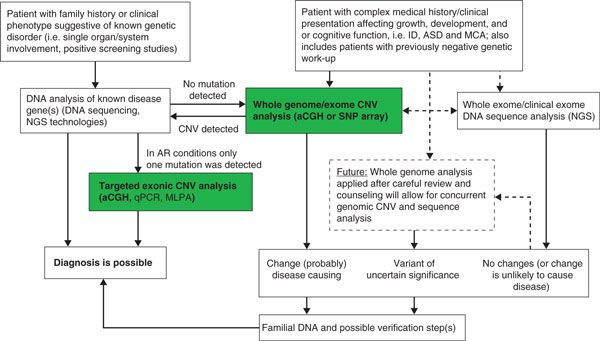
Clinical copy number variation analysis algorithm. Whole genome or exome copy number variation (CNV) analysis is an accepted first-line screening tool for the evaluation of patients with complex clinical presentations, and also intellectual disability (ID), autism spectrum disorder (ASD) or multiple congenital anomalies (MCA). If negative, whole exome/clinical exome analysis is now clinically available for subsequent analysis. CNVs detected by array-based comparative genomic hybridization (aCGH) or SNP-based microarray (SNP-array) may lead to a diagnosis or point to a coexisting mutation in the affected gene(s) in autosomal recessive (AR) traits. Targeted exonic CNV analysis can similarly be applied to identify deletions in genes with a heterozygous mutation detected by DNA sequencing. The emergence of whole genome DNA sequence analysis on a clinical basis will allow for integrated whole exome CNV/whole exome sequence analysis in the near future. ARMLPA, multiplex ligation-dependent probe amplification; NGS, next-generation high-throughput DNA sequencing; qPCR, quantitative PCR.
Conclusion
CNV analysis is a powerful tool for gene discovery, evaluating pathogenic effects of genomic alterations and establishing a diagnosis for patients with a number of phenotypes. The goal of clinical CNV analysis is designed to identify structural alterations that would establish a genetic diagnosis in an individual or family. A genetic diagnosis may aid in the clinical management of an individual and allows for accurate genetic counseling, including providing recurrence risks and prenatal testing options, and a genetic diagnosis may relieve the family's anxiety surrounding the etiology of disease.
Despite the noted benefit of CNV analysis, it is often difficult to determine the pathogenic effects of a CNV. This ambiguity may create an added burden of uncertainty, which is often shared between the laboratory, the clinician and the family. Appropriate genetic counseling helps inform the family of possible outcomes, including a normal result, variants of unknown significance and the possibility of incidental findings. As the ability to detect CNVs continues to increase so must the ability to discern the pathogenic effects of CNVs. A current framework exists to investigate the possible effects of CNVs in the human genome. Continued collaboration between researchers, clinicians and families is imperative to both maximize the benefit of CNV analysis as well as minimize the risk to patients undergoing testing.
Abbreviations
aCGH: array-based comparative genomic hybridization; ASD: autism spectrum disorder; BAC: bacterial artificial chromosome; bp: base pair; CNV: copy number variation; FISH: fluorescence in situ hybridization; HTS: high-throughput sequencing; ID: intellectual disability; kb: kilobase; LOH: loss of heterozygosity; MCA: multiple congenital anomalies; PCR: polymerase chain reaction; SNP: single nucleotide polymorphism; SNP-array: SNP-based microarray.
Competing interests
The authors declare that they have no competing interests.
Box 1. Web resources
The URLs for websites referred to herein are as follows:
Database of Genomic Variants http://projects.tcag.ca/variation/
NCBI Database of genomic structural variation (dbVar) http://www.ncbi.nlm.nih.gov/dbvar/
DECIPHER http://decipher.sanger.ac.uk
ECARUCA http://umcecaruca01.extern.umcn.nl:8080/ecaruca/ecaruca.jsp
International Standards for Cytogenomic Arrays Database https://www.iscaconsortium.org
Contributor Information
Curtis R Coughlin, II, Email: Curtis.Coughlin@childrenscolorado.org.
Gunter H Scharer, Email: gscharer@mcw.edu.
Tamim H Shaikh, Email: Tamim.Shaikh@ucdenver.edu.
References
- Bejjani BA, Shaffer LG. Clinical utility of contemporary molecular cytogenetics. Annu Rev Genomics Hum Genet. 2008;9:71–86. doi: 10.1146/annurev.genom.9.081307.164207. [DOI] [PubMed] [Google Scholar]
- Nishioka K, Hayashi S, Farrer MJ, Singleton AB, Yoshino H, Imai H, Kitami T, Sato K, Kuroda R, Tomiyama H, Mizoguchi K, Murata M, Toda T, Imoto I, Inazawa J, Mizuno Y, Hattori N. Clinical heterogeneity of alpha-synuclein gene duplication in Parkinson's disease. Ann Neurol. 2006;59:298–309. doi: 10.1002/ana.20753. [DOI] [PubMed] [Google Scholar]
- Rovelet-Lecrux A, Hannequin D, Raux G, Le Meur N, Laquerrière A, Vital A, Dumanchin C, Feuillette S, Brice A, Vercelletto M, Dubas F, Frebourg T, Campion D. APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat Genet. 2006;38:24–26. doi: 10.1038/ng1718. [DOI] [PubMed] [Google Scholar]
- Farrer M, Kachergus J, Forno L, Lincoln S, Wang D-S, Hulihan M, Maraganore D, Gwinn-Hardy K, Wszolek Z, Dickson D, Langston JW. Comparison of kindreds with parkinsonism and alpha-synuclein genomic multiplications. Ann Neurol. 2004;55:174–179. doi: 10.1002/ana.10846. [DOI] [PubMed] [Google Scholar]
- Gonzalez E, Kulkarni H, Bolivar H, Mangano A, Sanchez R, Catano G, Nibbs RJ, Freedman BI, Quinones MP, Bamshad MJ, Murthy KK, Rovin BH, Bradley W, Clark RA, Anderson SA, O'Connell RJ, Agan BK, Ahuja SS, Bologna R, Sen L, Dolan MJ, Ahuja SK. The influence of CCL3L1 gene-containing segmental duplications on HIV-1/AIDS susceptibility. Science. 2005;307:1434–1440. doi: 10.1126/science.1101160. [DOI] [PubMed] [Google Scholar]
- Cook EH Jr, Scherer SW. Copy-number variations associated with neuropsychiatric conditions. Nature. 2008;455:919–923. doi: 10.1038/nature07458. [DOI] [PubMed] [Google Scholar]
- Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, Yamrom B, Yoon S, Krasnitz A, Kendall J, Leotta A, Pai D, Zhang R, Lee Y-H, Hicks J, Spence SJ, Lee AT, Puura K, Lehtimäki T, Ledbetter D, Gregersen PK, Bregman J, Sutcliffe JS, Jobanputra V, Chung W, Warburton D, King M-C, Skuse D, Geschwind DH, Gilliam TC. et al. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaikh TH, Gai X, Perin JC, Glessner JT, Xie H, Murphy K, O'Hara R, Casalunovo T, Conlin LK, D'Arcy M, Frackelton EC, Geiger EA, Haldeman-Englert C, Imielinski M, Kim CE, Medne L, Annaiah K, Bradfield JP, Dabaghyan E, Eckert A, Onyiah CC, Ostapenko S, Otieno FG, Santa E, Shaner JL, Skraban R, Smith RM, Elia J, Goldmuntz E, Spinner NB. et al. High-resolution mapping and analysis of copy number variations in the human genome: a data resource for clinical and research applications. Genome Res. 2009;19:1682–1690. doi: 10.1101/gr.083501.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iafrate AJ, Feuk L, Rivera MN, Listewnik ML, Donahoe PK, Qi Y, Scherer SW, Lee C. Detection of large-scale variation in the human genome. Nat Genet. 2004;36:949–951. doi: 10.1038/ng1416. [DOI] [PubMed] [Google Scholar]
- Sebat J, Lakshmi B, Troge J, Alexander J, Young J, Lundin P, Månér S, Massa H, Walker M, Chi M, Navin N, Lucito R, Healy J, Hicks J, Ye K, Reiner A, Gilliam TC, Trask B, Patterson N, Zetterberg A, Wigler M. Large-scale copy number polymorphism in the human genome. Science. 2004;305:525–528. doi: 10.1126/science.1098918. [DOI] [PubMed] [Google Scholar]
- Conrad DF, Andrews TD, Carter NP, Hurles ME, Pritchard JK. A high-resolution survey of deletion polymorphism in the human genome. Nat Genet. 2006;38:75–81. doi: 10.1038/ng1697. [DOI] [PubMed] [Google Scholar]
- Feuk L, Carson AR, Scherer SW. Structural variation in the human genome. Nat Rev Genet. 2006;7:85–97. doi: 10.1038/nrg1767. [DOI] [PubMed] [Google Scholar]
- Kidd JM, Cooper GM, Donahue WF, Hayden HS, Sampas N, Graves T, Hansen N, Teague B, Alkan C, Antonacci F, Haugen E, Zerr T, Yamada NA, Tsang P, Newman TL, Tüzün E, Cheng Z, Ebling HM, Tusneem N, David R, Gillett W, Phelps KA, Weaver M, Saranga D, Brand A, Tao W, Gustafson E, McKernan K, Chen L, Malig M. et al. Mapping and sequencing of structural variation from eight human genomes. Nature. 2008;453:56–64. doi: 10.1038/nature06862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarroll SA, Hadnott TN, Perry GH, Sabeti PC, Zody MC, Barrett JC, Dallaire S, Gabriel SB, Lee C, Daly MJ, Altshuler DM. Common deletion polymorphisms in the human genome. Nat Genet. 2006;38:86–92. doi: 10.1038/ng1696. [DOI] [PubMed] [Google Scholar]
- Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, Fiegler H, Shapero MH, Carson AR, Chen W, Cho EK, Dallaire S, Freeman JL, González JR, Gratacòs M, Huang J, Kalaitzopoulos D, Komura D, MacDonald JR, Marshall CR, Mei R, Montgomery L, Nishimura K, Okamura K, Shen F, Somerville MJ, Tchinda J, Valsesia A, Woodwark C, Yang F. et al. Global variation in copy number in the human genome. Nature. 2006;444:444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp AJ, Locke DP, McGrath SD, Cheng Z, Bailey JA, Vallente RU, Pertz LM, Clark RA, Schwartz S, Segraves R, Oseroff VV, Albertson DG, Pinkel D, Eichler EE. Segmental duplications and copy-number variation in the human genome. Am J Hum Genet. 2005;77:78–88. doi: 10.1086/431652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills RE, Walter K, Stewart C, Handsaker RE, Chen K, Alkan C, Abyzov A, Yoon SC, Ye K, Cheetham RK, Chinwalla A, Conrad DF, Fu Y, Grubert F, Hajirasouliha I, Hormozdiari F, Iakoucheva LM, Iqbal Z, Kang S, Kidd JM, Konkel MK, Korn J, Khurana E, Kural D, Lam HYK, Leng J, Li R, Li Y, Lin C-Y, Luo R. et al. Mapping copy number variation by population-scale genome sequencing. Nature. 2011;470:59–65. doi: 10.1038/nature09708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad DF, Pinto D, Redon R, Feuk L, Gokcumen O, Zhang Y, Aerts J, Andrews TD, Barnes C, Campbell P, Fitzgerald T, Hu M, Ihm CH, Kristiansson K, Macarthur DG, Macdonald JR, Onyiah I, Pang AWC, Robson S, Stirrups K, Valsesia A, Walter K, Wei J, Tyler-Smith C, Carter NP, Lee C, Scherer SW, Hurles ME. Origins and functional impact of copy number variation in the human genome. Nature. 2010;464:704–712. doi: 10.1038/nature08516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman JL, Perry GH, Feuk L, Redon R, McCarroll SA, Altshuler DM, Aburatani H, Jones KW, Tyler-Smith C, Hurles ME, Carter NP, Scherer SW, Lee C. Copy number variation: new insights in genome diversity. Genome Res. 2006;16:949–961. doi: 10.1101/gr.3677206. [DOI] [PubMed] [Google Scholar]
- The 1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boone PM, Bacino CA, Shaw CA, Eng PA, Hixson PM, Pursley AN, Kang S-HL, Yang Y, Wiszniewska J, Nowakowska BA, del Gaudio D, Xia Z, Simpson-Patel G, Immken LL, Gibson JB, Tsai AC-H, Bowers JA, Reimschisel TE, Schaaf CP, Potocki L, Scaglia F, Gambin T, Sykulski M, Bartnik M, Derwinska K, Wisniowiecka-Kowalnik B, Lalani SR, Probst FJ, Bi W, Beaudet AL. et al. Detection of clinically relevant exonic copy-number changes by array CGH. Hum Mutat. 2010;31:1326–1342. doi: 10.1002/humu.21360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaikh TH, Kurahashi H, Saitta SC, O'Hare AM, Hu P, Roe BA, Driscoll DA, McDonald-McGinn DM, Zackai EH, Budarf ML, Emanuel BS. Chromosome 22-specific low copy repeats and the 22q11.2 deletion syndrome: genomic organization and deletion endpoint analysis. Hum Mol Genet. 2000;9:489–501. doi: 10.1093/hmg/9.4.489. [DOI] [PubMed] [Google Scholar]
- Inoue K, Lupski JR. Molecular mechanisms for genomic disorders. Annu Rev Genomics Hum Genet. 2002;3:199–242. doi: 10.1146/annurev.genom.3.032802.120023. [DOI] [PubMed] [Google Scholar]
- Shaikh TH, O'Connor RJ, Pierpont ME, McGrath J, Hacker AM, Nimmakayalu M, Geiger E, Emanuel BS, Saitta SC. Low copy repeats mediate distal chromosome 22q11.2 deletions: sequence analysis predicts breakpoint mechanisms. Genome Res. 2007;17:482–491. doi: 10.1101/gr.5986507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Binsbergen E. Origins and breakpoint analyses of copy number variations: up close and personal. Cytogenet Genome Res. 2011;135:271–276. doi: 10.1159/000330267. [DOI] [PubMed] [Google Scholar]
- Carvalho CMB, Zhang F, Liu P, Patel A, Sahoo T, Bacino CA, Shaw C, Peacock S, Pursley A, Tavyev YJ, Ramocki MB, Nawara M, Obersztyn E, Vianna-Morgante AM, Stankiewicz P, Zoghbi HY, Cheung SW, Lupski JR. Complex rearrangements in patients with duplications of MECP2 can occur by fork stalling and template switching. Hum Mol Genet. 2009;18:2188–2203. doi: 10.1093/hmg/ddp151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JA, Carvalho CMB, Lupski JR. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell. 2007;131:1235–1247. doi: 10.1016/j.cell.2007.11.037. [DOI] [PubMed] [Google Scholar]
- Korbel JO, Urban AE, Affourtit JP, Godwin B, Grubert F, Simons JF, Kim PM, Palejev D, Carriero NJ, Du L, Taillon BE, Chen Z, Tanzer A, Saunders ACE, Chi J, Yang F, Carter NP, Hurles ME, Weissman SM, Harkins TT, Gerstein MB, Egholm M, Snyder M. Paired-end mapping reveals extensive structural variation in the human genome. Science. 2007;318:420–426. doi: 10.1126/science.1149504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, Carter NP, Church DM, Crolla JA, Eichler EE, Epstein CJ, Faucett WA, Feuk L, Friedman JM, Hamosh A, Jackson L, Kaminsky EB, Kok K, Krantz ID, Kuhn RM, Lee C, Ostell JM, Rosenberg C, Scherer SW, Spinner NB, Stavropoulos DJ, Tepperberg JH, Thorland EC, Vermeesch JR, Waggoner DJ, Watson MS. et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010;86:749–764. doi: 10.1016/j.ajhg.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledbetter DH, Riccardi VM, Airhart SD, Strobel RJ, Keenan BS, Crawford JD. Deletions of chromosome 15 as a cause of the Prader-Willi syndrome. N Engl J Med. 1981;304:325–329. doi: 10.1056/NEJM198102053040604. [DOI] [PubMed] [Google Scholar]
- Smith AC, McGavran L, Robinson J, Waldstein G, Macfarlane J, Zonona J, Reiss J, Lahr M, Allen L, Magenis E. Interstitial deletion of (17)(p11.2p11.2) in nine patients. Am J Med Genet. 1986;24:393–414. doi: 10.1002/ajmg.1320240303. [DOI] [PubMed] [Google Scholar]
- Nickerson E, Greenberg F, Keating MT, McCaskill C, Shaffer LG. Deletions of the elastin gene at 7q11.23 occur in approximately 90% of patients with Williams syndrome. Am J Hum Genet. 1995;56:1156–1161. [PMC free article] [PubMed] [Google Scholar]
- Roa BB, Garcia CA, Lupski JR. Charcot-Marie-Tooth disease type 1A: molecular mechanisms of gene dosage and point mutation underlying a common inherited peripheral neuropathy. Int J Neurol. 1991. pp. 97–107. [PubMed]
- Douglas J, Tatton-Brown K, Coleman K, Guerrero S, Berg J, Cole TRP, Fitzpatrick D, Gillerot Y, Hughes HE, Pilz D, Raymond FL, Temple IK, Irrthum A, Schouten JP, Rahman N. Partial NSD1 deletions cause 5% of Sotos syndrome and are readily identifiable by multiplex ligation dependent probe amplification. J Med Genet. 2005;42:e56. doi: 10.1136/jmg.2005.031930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahan P, Li Y, Izumi M, Graubert TA. The impact of copy number variation on local gene expression in mouse hematopoietic stem and progenitor cells. Nat Genet. 2009;41:430–437. doi: 10.1038/ng.350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henrichsen CN, Chaignat E, Reymond A. Copy number variants, diseases and gene expression. Hum Mol Genet. 2009;18:R1–8. doi: 10.1093/hmg/ddp011. [DOI] [PubMed] [Google Scholar]
- Rivera-Brugués N, Albrecht B, Wieczorek D, Schmidt H, Keller T, Göhring I, Ekici AB, Tzschach A, Garshasbi M, Franke K, Klopp N, Wichmann H-E, Meitinger T, Strom TM, Hempel M. Cohen syndrome diagnosis using whole genome arrays. J Med Genet. 2011;48:136–140. doi: 10.1136/jmg.2010.082206. [DOI] [PubMed] [Google Scholar]
- Brunetti-Pierri N, Berg JS, Scaglia F, Belmont J, Bacino CA, Sahoo T, Lalani SR, Graham B, Lee B, Shinawi M, Shen J, Kang S-HL, Pursley A, Lotze T, Kennedy G, Lansky-Shafer S, Weaver C, Roeder ER, Grebe TA, Arnold GL, Hutchison T, Reimschisel T, Amato S, Geragthy MT, Innis JW, Obersztyn E, Nowakowska B, Rosengren SS, Bader PI, Grange DK. et al. Recurrent reciprocal 1q21.1 deletions and duplications associated with microcephaly or macrocephaly and developmental and behavioral abnormalities. Nat Genet. 2008;40:1466–1471. doi: 10.1038/ng.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mefford HC, Sharp AJ, Baker C, Itsara A, Jiang Z, Buysse K, Huang S, Maloney VK, Crolla JA, Baralle D, Collins A, Mercer C, Norga K, de Ravel T, Devriendt K, Bongers EMHF, de Leeuw N, Reardon W, Gimelli S, Bena F, Hennekam RC, Male A, Gaunt L, Clayton-Smith J, Simonic I, Park SM, Mehta SG, Nik-Zainal S, Woods CG, Firth HV. et al. Recurrent rearrangements of chromosome 1q21.1 and variable pediatric phenotypes. N Engl J Med. 2008;359:1685–1699. doi: 10.1056/NEJMoa0805384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christiansen J, Dyck JD, Elyas BG, Lilley M, Bamforth JS, Hicks M, Sprysak KA, Tomaszewski R, Haase SM, Vicen-Wyhony LM, Somerville MJ. Chromosome 1q21.1 contiguous gene deletion is associated with congenital heart disease. Circ Res. 2004;94:1429–1435. doi: 10.1161/01.RES.0000130528.72330.5c. [DOI] [PubMed] [Google Scholar]
- Sharp AJ, Hansen S, Selzer RR, Cheng Z, Regan R, Hurst JA, Stewart H, Price SM, Blair E, Hennekam RC, Fitzpatrick CA, Segraves R, Richmond TA, Guiver C, Albertson DG, Pinkel D, Eis PS, Schwartz S, Knight SJL, Eichler EE. Discovery of previously unidentified genomic disorders from the duplication architecture of the human genome. Nat Genet. 2006;38:1038–1042. doi: 10.1038/ng1862. [DOI] [PubMed] [Google Scholar]
- Sharp AJ, Mefford HC, Li K, Baker C, Skinner C, Stevenson RE, Schroer RJ, Novara F, De Gregori M, Ciccone R, Broomer A, Casuga I, Wang Y, Xiao C, Barbacioru C, Gimelli G, Bernardina BD, Torniero C, Giorda R, Regan R, Murday V, Mansour S, Fichera M, Castiglia L, Failla P, Ventura M, Jiang Z, Cooper GM, Knight SJL, Romano C. et al. A recurrent 15q13.3 microdeletion syndrome associated with mental retardation and seizures. Nat Genet. 2008;40:322–328. doi: 10.1038/ng.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DT, Shen Y, Weiss LA, Korn J, Anselm I, Bridgemohan C, Cox GF, Dickinson H, Gentile J, Harris DJ, Hegde V, Hundley R, Khwaja O, Kothare S, Luedke C, Nasir R, Poduri A, Prasad K, Raffalli P, Reinhard A, Smith SE, Sobeih MM, Soul JS, Stoler J, Takeoka M, Tan W-H, Thakuria J, Wolff R, Yusupov R, Gusella JF. et al. Microdeletion/duplication at 15q13.2q13.3 among individuals with features of autism and other neuropsychiatric disorders. J Med Genet. 2009;46:242–248. doi: 10.1136/jmg.2008.059907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Bon BWM, Mefford HC, Menten B, Koolen DA, Sharp AJ, Nillesen WM, Innis JW, de Ravel TJL, Mercer CL, Fichera M, Stewart H, Connell LE, Ounap K, Lachlan K, Castle B, Van der Aa N, van Ravenswaaij C, Nobrega MA, Serra-Juhé C, Simonic I, de Leeuw N, Pfundt R, Bongers EM, Baker C, Finnemore P, Huang S, Maloney VK, Crolla JA, van Kalmthout M, Elia M. et al. Further delineation of the 15q13 microdeletion and duplication syndromes: a clinical spectrum varying from non-pathogenic to a severe outcome. J Med Genet. 2009;46:511–523. doi: 10.1136/jmg.2008.063412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helbig I, Mefford HC, Sharp AJ, Guipponi M, Fichera M, Franke A, Muhle H, de Kovel C, Baker C, von Spiczak S, Kron KL, Steinich I, Kleefuss-Lie AA, Leu C, Gaus V, Schmitz B, Klein KM, Reif PS, Rosenow F, Weber Y, Lerche H, Zimprich F, Urak L, Fuchs K, Feucht M, Genton P, Thomas P, Visscher F, de Haan G-J, Møller RS. et al. 15q13.3 microdeletions increase risk of idiopathic generalized epilepsy. Nat Genet. 2009;41:160–162. doi: 10.1038/ng.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramamoorthy A, Flockhart DA, Hosono N, Kubo M, Nakamura Y, Skaar TC. Differential quantification of CYP2D6 gene copy number by four different quantitative real-time PCR assays. Pharmacogenet Genomics. 2010;20:451–454. doi: 10.1097/FPC.0b013e32833a1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'haene B, Vandesompele J, Hellemans J. Accurate and objective copy number profiling using real-time quantitative PCR. Methods. 2010;50:262–270. doi: 10.1016/j.ymeth.2009.12.007. [DOI] [PubMed] [Google Scholar]
- Weaver S, Dube S, Mir A, Qin J, Sun G, Ramakrishnan R, Jones RC, Livak KJ. Taking qPCR to a higher level: Analysis of CNV reveals the power of high throughput qPCR to enhance quantitative resolution. Methods. 2010;50:271–276. doi: 10.1016/j.ymeth.2010.01.003. [DOI] [PubMed] [Google Scholar]
- Armour JA, Sismani C, Patsalis PC, Cross G. Measurement of locus copy number by hybridisation with amplifiable probes. Nucleic Acids Res. 2000;28:605–609. doi: 10.1093/nar/28.2.605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Dunnen JT, White SJ. MLPA and MAPH: sensitive detection of deletions and duplications. Curr Protoc Hum Genet. 2006;Chapter 7:Unit 7.14. doi: 10.1002/0471142905.hg0714s51. [DOI] [PubMed] [Google Scholar]
- Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 2002;30:e57. doi: 10.1093/nar/gnf056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorstman JAS, Jalali GR, Rappaport EF, Hacker AM, Scott C, Emanuel BS. MLPA: a rapid, reliable, and sensitive method for detection and analysis of abnormalities of 22q. Hum Mutat. 2006;27:814–821. doi: 10.1002/humu.20330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medvedev P, Stanciu M, Brudno M. Computational methods for discovering structural variation with next-generation sequencing. Nat Methods. 2009;6:S13–20. doi: 10.1038/nmeth.1374. [DOI] [PubMed] [Google Scholar]
- Yoon S, Xuan Z, Makarov V, Ye K, Sebat J. Sensitive and accurate detection of copy number variants using read depth of coverage. Genome Res. 2009;19:1586–1592. doi: 10.1101/gr.092981.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaikh TH. Oligonucleotide arrays for high-resolution analysis of copy number alteration in mental retardation/multiple congenital anomalies. Genet Med. 2007;9:617–625. doi: 10.1097/GIM.0b013e318148bb81. [DOI] [PubMed] [Google Scholar]
- Ballif BC, Rorem EA, Sundin K, Lincicum M, Gaskin S, Coppinger J, Kashork CD, Shaffer LG, Bejjani BA. Detection of low-level mosaicism by array CGH in routine diagnostic specimens. Am J Med Genet A. 2006;140:2757–2767. doi: 10.1002/ajmg.a.31539. [DOI] [PubMed] [Google Scholar]
- Cheung SW, Shaw CA, Scott DA, Patel A, Sahoo T, Bacino CA, Pursley A, Li J, Erickson R, Gropman AL, Miller DT, Seashore MR, Summers AM, Stankiewicz P, Chinault AC, Lupski JR, Beaudet AL, Sutton VR. Microarray-based CGH detects chromosomal mosaicism not revealed by conventional cytogenetics. Am J Med Genet A. 2007;143A:1679–1686. doi: 10.1002/ajmg.a.31740. [DOI] [PubMed] [Google Scholar]
- Scott SA, Cohen N, Brandt T, Toruner G, Desnick RJ, Edelmann L. Detection of low-level mosaicism and placental mosaicism by oligonucleotide array comparative genomic hybridization. Genet Med. 2010;12:85–92. doi: 10.1097/GIM.0b013e3181cc75d0. [DOI] [PubMed] [Google Scholar]
- Conlin LK, Thiel BD, Bonnemann CG, Medne L, Ernst LM, Zackai EH, Deardorff MA, Krantz ID, Hakonarson H, Spinner NB. Mechanisms of mosaicism, chimerism and uniparental disomy identified by single nucleotide polymorphism array analysis. Hum Mol Genet. 2010;19:1263–1275. doi: 10.1093/hmg/ddq003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González JR, Rodríguez-Santiago B, Cáceres A, Pique-Regi R, Rothman N, Chanock SJ, Armengol L, Pérez-Jurado LA. A fast and accurate method to detect allelic genomic imbalances underlying mosaic rearrangements using SNP array data. BMC Bioinformatics. 2011;12:166. doi: 10.1186/1471-2105-12-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson J, Morton NE, Collins A. Extended tracts of homozygosity in outbred human populations. Hum Mol Genet. 2006;15:789–795. doi: 10.1093/hmg/ddi493. [DOI] [PubMed] [Google Scholar]
- Li L-H, Ho S-F, Chen C-H, Wei C-Y, Wong W-C, Li L-Y, Hung S-I, Chung W-H, Pan W-H, Lee M-TM, Tsai F-J, Chang C-F, Wu J-Y, Chen Y-T. Long contiguous stretches of homozygosity in the human genome. Hum Mutat. 2006;27:1115–1121. doi: 10.1002/humu.20399. [DOI] [PubMed] [Google Scholar]
- Miyazawa H, Kato M, Awata T, Kohda M, Iwasa H, Koyama N, Tanaka T, Kyo S, Okazaki Y, Hagiwara K. Homozygosity haplotype allows a genomewide search for the autosomal segments shared among patients. Am J Hum Genet. 2007;80:1090–1102. doi: 10.1086/518176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Orr A, Guernsey DL, Robitaille J, Asselin G, Samuels ME, Dubé M-P. Application of homozygosity haplotype analysis to genetic mapping with high-density SNP genotype data. PLoS ONE. 2009;4:e5280. doi: 10.1371/journal.pone.0005280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrandt F, Heeringa SF, Rüschendorf F, Attanasio M, Nürnberg G, Becker C, Seelow D, Huebner N, Chernin G, Vlangos CN, Zhou W, O'Toole JF, Hoskins BE, Wolf MTF, Hinkes BG, Chaib H, Ashraf S, Schoeb DS, Ovunc B, Allen SJ, Vega-Warner V, Wise E, Harville HM, Lyons RH, Washburn J, Macdonald J, Nürnberg P, Otto EA. A systematic approach to mapping recessive disease genes in individuals from outbred populations. PLoS Genet. 2009;5:e1000353. doi: 10.1371/journal.pgen.1000353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locke DP, Sharp AJ, McCarroll SA, McGrath SD, Newman TL, Cheng Z, Schwartz S, Albertson DG, Pinkel D, Altshuler DM, Eichler EE. Linkage disequilibrium and heritability of copy-number polymorphisms within duplicated regions of the human genome. Am J Hum Genet. 2006;79:275–290. doi: 10.1086/505653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudmant PH, Kitzman JO, Antonacci F, Alkan C, Malig M, Tsalenko A, Sampas N, Bruhn L, Shendure J, Eichler EE. Diversity of human copy number variation and multicopy genes. Science. 2010;330:641–646. doi: 10.1126/science.1197005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankiewicz P, Beaudet AL. Use of array CGH in the evaluation of dysmorphology, malformations, developmental delay, and idiopathic mental retardation. Curr Opin Genet Dev. 2007;17:182–192. doi: 10.1016/j.gde.2007.04.009. [DOI] [PubMed] [Google Scholar]
- Lakich D, Kazazian HH Jr, Antonarakis SE, Gitschier J. Inversions disrupting the factor VIII gene are a common cause of severe haemophilia A. Nat Genet. 1993;5:236–241. doi: 10.1038/ng1193-236. [DOI] [PubMed] [Google Scholar]
- Castaldo G, D'Argenio V, Nardiello P, Zarrilli F, Sanna V, Rocino A, Coppola A, Di Minno G, Salvatore F. Haemophilia A: molecular insights. Clin Chem Lab Med. 2007;45:450–461. doi: 10.1515/CCLM.2007.093. [DOI] [PubMed] [Google Scholar]
- Osborne LR, Li M, Pober B, Chitayat D, Bodurtha J, Mandel A, Costa T, Grebe T, Cox S, Tsui LC, Scherer SW. A 1.5 million-base pair inversion polymorphism in families with Williams-Beuren syndrome. Nat Genet. 2001;29:321–325. doi: 10.1038/ng753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimelli G, Pujana MA, Patricelli MG, Russo S, Giardino D, Larizza L, Cheung J, Armengol L, Schinzel A, Estivill X, Zuffardi O. Genomic inversions of human chromosome 15q11-q13 in mothers of Angelman syndrome patients with class II (BP2/3) deletions. Hum Mol Genet. 2003;12:849–858. doi: 10.1093/hmg/ddg101. [DOI] [PubMed] [Google Scholar]
- Hochstenbach R, van Binsbergen E, Engelen J, Nieuwint A, Polstra A, Poddighe P, Ruivenkamp C, Sikkema-Raddatz B, Smeets D, Poot M. Array analysis and karyotyping: workflow consequences based on a retrospective study of 36,325 patients with idiopathic developmental delay in the Netherlands. Eur J Med Genet. 2009;52:161–169. doi: 10.1016/j.ejmg.2009.03.015. [DOI] [PubMed] [Google Scholar]
- Jacobs PA, Matsuura JS, Mayer M, Newlands IM. A cytogenetic survey of an institution for the mentally retarded: I. Chromosome abnormalities. Clin Genet. 1978;13:37–60. doi: 10.1111/j.1399-0004.1978.tb04127.x. [DOI] [PubMed] [Google Scholar]
- Ligon AH, Beaudet AL, Shaffer LG. Simultaneous, multilocus FISH analysis for detection of microdeletions in the diagnostic evaluation of developmental delay and mental retardation. Am J Hum Genet. 1997;61:51–59. doi: 10.1086/513904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries BBA, Pfundt R, Leisink M, Koolen DA, Vissers LELM, Janssen IM, Reijmersdal S van, Nillesen WM, Huys EHLPG, Leeuw N de, Smeets D, Sistermans EA, Feuth T, van Ravenswaaij-Arts CMA, van Kessel AG, Schoenmakers EFPM, Brunner HG, Veltman JA. Diagnostic genome profiling in mental retardation. Am J Hum Genet. 2005;77:606–616. doi: 10.1086/491719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman JM, Baross A, Delaney AD, Ally A, Arbour L, Armstrong L, Asano J, Bailey DK, Barber S, Birch P, Brown-John M, Cao M, Chan S, Charest DL, Farnoud N, Fernandes N, Flibotte S, Go A, Gibson WT, Holt RA, Jones SJM, Kennedy GC, Krzywinski M, Langlois S, Li HI, McGillivray BC, Nayar T, Pugh TJ, Rajcan-Separovic E. et al. Oligonucleotide microarray analysis of genomic imbalance in children with mental retardation. Am J Hum Genet. 2006;79:500–513. doi: 10.1086/507471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koolen DA, Pfundt R, de Leeuw N, Hehir-Kwa JY, Nillesen WM, Neefs I, Scheltinga I, Sistermans E, Smeets D, Brunner HG, van Kessel AG, Veltman JA, de Vries BBA. Genomic microarrays in mental retardation: a practical workflow for diagnostic applications. Hum Mutat. 2009;30:283–292. doi: 10.1002/humu.20883. [DOI] [PubMed] [Google Scholar]
- Wagenstaller J, Spranger S, Lorenz-Depiereux B, Kazmierczak B, Nathrath M, Wahl D, Heye B, Glaser D, Liebscher V, Meitinger T, Strom TM. Copy-number variations measured by single-nucleotide-polymorphism oligonucleotide arrays in patients with mental retardation. Am J Hum Genet. 2007;81:768–779. doi: 10.1086/521274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw-Smith C, Redon R, Rickman L, Rio M, Willatt L, Fiegler H, Firth H, Sanlaville D, Winter R, Colleaux L, Bobrow M, Carter NP. Microarray based comparative genomic hybridisation (array-CGH) detects submicroscopic chromosomal deletions and duplications in patients with learning disability/mental retardation and dysmorphic features. J Med Genet. 2004;41:241–248. doi: 10.1136/jmg.2003.017731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning M, Hudgins L. Array-based technology and recommendations for utilization in medical genetics practice for detection of chromosomal abnormalities. Genet Med. 2010;12:742–745. doi: 10.1097/GIM.0b013e3181f8baad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Dies KA, Holm IA, Bridgemohan C, Sobeih MM, Caronna EB, Miller KJ, Frazier JA, Silverstein I, Picker J, Weissman L, Raffalli P, Jeste S, Demmer LA, Peters HK, Brewster SJ, Kowalczyk SJ, Rosen-Sheidley B, McGowan C, Duda AW 3rd, Lincoln SA, Lowe KR, Schonwald A, Robbins M, Hisama F, Wolff R, Becker R, Nasir R, Urion DK, Milunsky JM. et al. Clinical genetic testing for patients with autism spectrum disorders. Pediatrics. 2010;125:e727–735. doi: 10.1542/peds.2009-1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X-Y, Phung MT, Shaw CA, Pham K, Neil SE, Patel A, Sahoo T, Bacino CA, Stankiewicz P, Kang S-HL, Lalani S, Chinault AC, Lupski JR, Cheung SW, Beaudet AL. Genomic imbalances in neonates with birth defects: high detection rates by using chromosomal microarray analysis. Pediatrics. 2008;122:1310–1318. doi: 10.1542/peds.2008-0297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming JE, Geiger E, James AC, Ciprero KL, Nimmakayalu M, Zhang Y, Huang A, Vaddi M, Rappaport E, Zackai EH, Shaikh TH. Rapid detection of submicroscopic chromosomal rearrangements in children with multiple congenital anomalies using high density oligonucleotide arrays. Hum Mutat. 2006;27:467–473. doi: 10.1002/humu.20322. [DOI] [PubMed] [Google Scholar]
- Edelmann L, Hirschhorn K. Clinical utility of array CGH for the detection of chromosomal imbalances associated with mental retardation and multiple congenital anomalies. Ann N Y Acad Sci. 2009;1151:157–166. doi: 10.1111/j.1749-6632.2008.03610.x. [DOI] [PubMed] [Google Scholar]
- Saam J, Gudgeon J, Aston E, Brothman AR. How physicians use array comparative genomic hybridization results to guide patient management in children with developmental delay. Genet Med. 2008;10:181–186. doi: 10.1097/GIM.0b013e3181634eca. [DOI] [PubMed] [Google Scholar]
- Dobyns WB, Curry CJ, Hoyme HE, Turlington L, Ledbetter DH. Clinical and molecular diagnosis of Miller-Dieker syndrome. Am J Hum Genet. 1991;48:584–594. [PMC free article] [PubMed] [Google Scholar]
- Krepischi-Santos ACV, Vianna-Morgante AM, Jehee FS, Passos-Bueno MR, Knijnenburg J, Szuhai K, Sloos W, Mazzeu JF, Kok F, Cheroki C, Otto PA, Mingroni-Netto RC, Varela M, Koiffmann C, Kim CA, Bertola DR, Pearson PL, Rosenberg C. Whole-genome array-CGH screening in undiagnosed syndromic patients: old syndromes revisited and new alterations. Cytogenet Genome Res. 2006;115:254–261. doi: 10.1159/000095922. [DOI] [PubMed] [Google Scholar]
- Potocki L, Chen KS, Park SS, Osterholm DE, Withers MA, Kimonis V, Summers AM, Meschino WS, Anyane-Yeboa K, Kashork CD, Shaffer LG, Lupski JR. Molecular mechanism for duplication 17p11.2 - the homologous recombination reciprocal of the Smith-Magenis microdeletion. Nat Genet. 2000;24:84–87. doi: 10.1038/71743. [DOI] [PubMed] [Google Scholar]
- Potocki L, Bi W, Treadwell-Deering D, Carvalho CMB, Eifert A, Friedman EM, Glaze D, Krull K, Lee JA, Lewis RA, Mendoza-Londono R, Robbins-Furman P, Shaw C, Shi X, Weissenberger G, Withers M, Yatsenko SA, Zackai EH, Stankiewicz P, Lupski JR. Characterization of Potocki-Lupski syndrome (dup(17)(p11.2p11.2)) and delineation of a dosage-sensitive critical interval that can convey an autism phenotype. Am J Hum Genet. 2007;80:633–649. doi: 10.1086/512864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaffer LG, Theisen A, Bejjani BA, Ballif BC, Aylsworth AS, Lim C, McDonald M, Ellison JW, Kostiner D, Saitta S, Shaikh T. The discovery of microdeletion syndromes in the post-genomic era: review of the methodology and characterization of a new 1q41q42 microdeletion syndrome. Genet Med. 2007;9:607–616. doi: 10.1097/GIM.0b013e3181484b49. [DOI] [PubMed] [Google Scholar]
- Mazzeu JF, Krepischi-Santos AC, Rosenberg C, Szuhai K, Knijnenburg J, Weiss JMM, Kerkis I, Mustacchi Z, Colin G, Mombach R, Pavanello R de CM, Otto PA, Vianna-Morgante AM. Chromosome abnormalities in two patients with features of autosomal dominant Robinow syndrome. Am J Med Genet A. 2007;143A:1790–1795. doi: 10.1002/ajmg.a.31661. [DOI] [PubMed] [Google Scholar]
- Rice GM, Qi Z, Selzer R, Richmond T, Thompson K, Pauli RM, Yu J. Microdissection-based high-resolution genomic array analysis of two patients with cytogenetically identical interstitial deletions of chromosome 1q but distinct clinical phenotypes. Am J Med Genet A. 2006;140:1637–1643. doi: 10.1002/ajmg.a.31349. [DOI] [PubMed] [Google Scholar]
- Mazzeu JF, Vianna-Morgante AM, Krepischi ACV, Oudakker A, Rosenberg C, Szuhai K, McGill J, Maccraughan J, van Bokhoven H, Brunner HG. Deletions encompassing 1q41q42.1 and clinical features of autosomal dominant Robinow syndrome. Clin Genet. 2010;77:404–407. doi: 10.1111/j.1399-0004.2009.01355.x. [DOI] [PubMed] [Google Scholar]
- Filges I, Röthlisberger B, Boesch N, Weber P, Wenzel F, Huber AR, Heinimann K, Miny P. Interstitial deletion 1q42 in a patient with agenesis of corpus callosum: Phenotype-genotype comparison to the 1q41q42 microdeletion suggests a contiguous 1q4 syndrome. Am J Med Genet A. 2010;152A:987–993. doi: 10.1002/ajmg.a.33330. [DOI] [PubMed] [Google Scholar]
- Kantarci S, Ackerman KG, Russell MK, Longoni M, Sougnez C, Noonan KM, Hatchwell E, Zhang X, Pieretti Vanmarcke R, Anyane-Yeboa K, Dickman P, Wilson J, Donahoe PK, Pober BR. Characterization of the chromosome 1q41q42.12 region, and the candidate gene DISP1, in patients with CDH. Am J Med Genet A. 2010. pp. 2493–2504. [DOI] [PMC free article] [PubMed]
- Rosenfeld JA, Lacassie Y, El-Khechen D, Escobar LF, Reggin J, Heuer C, Chen E, Jenkins LS, Collins AT, Zinner S, Babcock M, Morrow B, Schultz RA, Torchia BS, Ballif BC, Tsuchiya KD, Shaffer LG. New cases and refinement of the critical region in the 1q41q42 microdeletion syndrome. Eur J Med Genet. 2011;54:42–49. doi: 10.1016/j.ejmg.2010.10.002. [DOI] [PubMed] [Google Scholar]
- Vissers LELM, Veltman JA, van Kessel AG, Brunner HG. Identification of disease genes by whole genome CGH arrays. Hum Mol Genet. 2005. pp. R215–223. [DOI] [PubMed]
- Wong L-JC, Dimmock D, Geraghty MT, Quan R, Lichter-Konecki U, Wang J, Brundage EK, Scaglia F, Chinault AC. Utility of oligonucleotide array-based comparative genomic hybridization for detection of target gene deletions. Clin Chem. 2008;54:1141–1148. doi: 10.1373/clinchem.2008.103721. [DOI] [PubMed] [Google Scholar]
- Tayeh MK, Chin ELH, Miller VR, Bean LJH, Coffee B, Hegde M. Targeted comparative genomic hybridization array for the detection of single- and multiexon gene deletions and duplications. Genet Med. 2009;11:232–240. doi: 10.1097/GIM.0b013e318195e191. [DOI] [PubMed] [Google Scholar]
- Lalani SR, Stockton DW, Bacino C, Molinari LM, Glass NL, Fernbach SD, Towbin JA, Craigen WJ, Graham JM Jr, Hefner MA, Lin AE, McBride KL, Davenport SL, Belmont JW. Toward a genetic etiology of CHARGE syndrome: I. A systematic scan for submicroscopic deletions. Am J Med Genet A. 2003;118A:260–266. doi: 10.1002/ajmg.a.20002. [DOI] [PubMed] [Google Scholar]
- Vissers LELM, van Ravenswaaij CMA, Admiraal R, Hurst JA, de Vries BBA, Janssen IM, van der Vliet WA, Huys EHLPG, de Jong PJ, Hamel BCJ, Schoenmakers EFPM, Brunner HG, Veltman JA, van Kessel AG. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat Genet. 2004;36:955–957. doi: 10.1038/ng1407. [DOI] [PubMed] [Google Scholar]
- Diskin SJ, Hou C, Glessner JT, Attiyeh EF, Laudenslager M, Bosse K, Cole K, Mossé YP, Wood A, Lynch JE, Pecor K, Diamond M, Winter C, Wang K, Kim C, Geiger EA, McGrady PW, Blakemore AIF, London WB, Shaikh TH, Bradfield J, Grant SFA, Li H, Devoto M, Rappaport ER, Hakonarson H, Maris JM. Copy number variation at 1q21.1 associated with neuroblastoma. Nature. 2009;459:987–991. doi: 10.1038/nature08035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teshiba R, Masumoto K, Esumi G, Nagata K, Kinoshita Y, Tajiri T, Taguchi T, Yamamoto K. Identification of TCTE3 as a gene responsible for congenital diaphragmatic hernia using a high-resolution single-nucleotide polymorphism array. Pediatr Surg Int. 2011;27:193–198. doi: 10.1007/s00383-010-2778-z. [DOI] [PubMed] [Google Scholar]
- Khan MA, Rafiq MA, Noor A, Ali N, Ali G, Vincent JB, Ansar M. A novel deletion mutation in the TUSC3 gene in a consanguineous Pakistani family with autosomal recessive nonsyndromic intellectual disability. BMC Med Genet. 2011;12:56. doi: 10.1186/1471-2350-12-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank B, Bermejo JL, Hemminki K, Sutter C, Wappenschmidt B, Meindl A, Kiechle-Bahat M, Bugert P, Schmutzler RK, Bartram CR, Burwinkel B. Copy number variant in the candidate tumor suppressor gene MTUS1 and familial breast cancer risk. Carcinogenesis. 2007;28:1442–1445. doi: 10.1093/carcin/bgm033. [DOI] [PubMed] [Google Scholar]
- Bejjani BA, Shaffer LG. Application of array-based comparative genomic hybridization to clinical diagnostics. J Mol Diagn. 2006;8:528–533. doi: 10.2353/jmoldx.2006.060029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bejjani BA, Saleki R, Ballif BC, Rorem EA, Sundin K, Theisen A, Kashork CD, Shaffer LG. Use of targeted array-based CGH for the clinical diagnosis of chromosomal imbalance: is less more? Am J Med Genet A. 2005;134:259–267. doi: 10.1002/ajmg.a.30621. [DOI] [PubMed] [Google Scholar]
- Schaeffer AJ, Chung J, Heretis K, Wong A, Ledbetter DH, Lese-Martin C. Comparative genomic hybridization-array analysis enhances the detection of aneuploidies and submicroscopic imbalances in spontaneous miscarriages. Am J Hum Genet. 2004;74:1168–1174. doi: 10.1086/421250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung SW, Shaw CA, Yu W, Li J, Ou Z, Patel A, Yatsenko SA, Cooper ML, Furman P, Stankiewicz P, Stankiewicz P, Lupski JR, Chinault AC, Beaudet AL. Development and validation of a CGH microarray for clinical cytogenetic diagnosis. Genet Med. 2005;7:422–432. doi: 10.1097/01.GIM.0000170992.63691.32. [DOI] [PubMed] [Google Scholar]
- Shaffer LG, Bejjani BA. Medical applications of array CGH and the transformation of clinical cytogenetics. Cytogenet Genome Res. 2006;115:303–309. doi: 10.1159/000095928. [DOI] [PubMed] [Google Scholar]
- Shaffer LG, Bejjani BA, Torchia B, Kirkpatrick S, Coppinger J, Ballif BC. The identification of microdeletion syndromes and other chromosome abnormalities: cytogenetic methods of the past, new technologies for the future. Am J Med Genet C Semin Med Genet. 2007;145C:335–345. doi: 10.1002/ajmg.c.30152. [DOI] [PubMed] [Google Scholar]
- Tsuchiya KD, Shaffer LG, Aradhya S, Gastier-Foster JM, Patel A, Rudd MK, Biggerstaff JS, Sanger WG, Schwartz S, Tepperberg JH, Thorland EC, Torchia BA, Brothman AR. Variability in interpreting and reporting copy number changes detected by array-based technology in clinical laboratories. Genet Med. 2009;11:866–873. doi: 10.1097/GIM.0b013e3181c0c3b0. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Revenga L, Mila M, Rosenberg C, Lamb A, Lee C. Structural variation in the human genome: the impact of copy number variants on clinical diagnosis. Genet Med. 2007;9:600–606. doi: 10.1097/GIM.0b013e318149e1e3. [DOI] [PubMed] [Google Scholar]
- Lee C, Iafrate AJ, Brothman AR. Copy number variations and clinical cytogenetic diagnosis of constitutional disorders. Nat Genet. 2007;39:S48–54. doi: 10.1038/ng2092. [DOI] [PubMed] [Google Scholar]
- Stankiewicz P, Pursley AN, Cheung SW. Challenges in clinical interpretation of microduplications detected by array CGH analysis. Am J Med Genet A. 2010;152A:1089–1100. doi: 10.1002/ajmg.a.33216. [DOI] [PubMed] [Google Scholar]
- Cook EH Jr, Lindgren V, Leventhal BL, Courchesne R, Lincoln A, Shulman C, Lord C, Courchesne E. Autism or atypical autism in maternally but not paternally derived proximal 15q duplication. Am J Hum Genet. 1997;60:928–934. [PMC free article] [PubMed] [Google Scholar]
- Hogart A, Leung KN, Wang NJ, Wu DJ, Driscoll J, Vallero RO, Schanen NC, LaSalle JM. Chromosome 15q11-13 duplication syndrome brain reveals epigenetic alterations in gene expression not predicted from copy number. J Med Genet. 2009;46:86–93. doi: 10.1136/jmg.2008.061580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai G, Edelmann L, Goldsmith JE, Cohen N, Nakamine A, Reichert JG, Hoffman EJ, Zurawiecki DM, Silverman JM, Hollander E, Soorya L, Anagnostou E, Betancur C, Buxbaum JD. Multiplex ligation-dependent probe amplification for genetic screening in autism spectrum disorders: efficient identification of known microduplications and identification of a novel microduplication in ASMT. BMC Med Genomics. 2008;1:50. doi: 10.1186/1755-8794-1-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis NR, Veltman MWM, Thompson R, Craig E, Bolton PF, Thomas NS. Clinical findings in 33 subjects with large supernumerary marker(15) chromosomes and 3 subjects with triplication of 15q11-q13. Am J Med Genet A. 2006;140:434–441. doi: 10.1002/ajmg.a.31091. [DOI] [PubMed] [Google Scholar]
- Kitsiou-Tzeli S, Tzetis M, Sofocleous C, Vrettou C, Xaidara A, Giannikou K, Pampanos A, Mavrou A, Kanavakis E. De novo interstitial duplication of the 15q11.2-q14 PWS/AS region of maternal origin: Clinical description, array CGH analysis, and review of the literature. Am J Med Genet A. 2010;152A:1925–1932. doi: 10.1002/ajmg.a.33447. [DOI] [PubMed] [Google Scholar]
- Orrico A, Zollino M, Galli L, Buoni S, Marangi G, Sorrentino V. Late-onset Lennox-Gastaut syndrome in a patient with 15q11.2-q13.1 duplication. Am J Med Genet A. 2009;149A:1033–1035. doi: 10.1002/ajmg.a.32785. [DOI] [PubMed] [Google Scholar]
- Michelson M, Eden A, Vinkler C, Leshinsky-Silver E, Kremer U, Lerman-Sagie T, Lev D. Familial partial trisomy 15q11-13 presenting as intractable epilepsy in the child and schizophrenia in the mother. Eur J Paediatr Neurol. 2011;15:230–233. doi: 10.1016/j.ejpn.2010.11.001. [DOI] [PubMed] [Google Scholar]
- Stewart LR, Hall AL, Kang S-HL, Shaw CA, Beaudet AL. High frequency of known copy number abnormalities and maternal duplication 15q11-q13 in patients with combined schizophrenia and epilepsy. BMC Med Genet. 2011;12:154. doi: 10.1186/1471-2350-12-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingason A, Kirov G, Giegling I, Hansen T, Isles AR, Jakobsen KD, Kristinsson KT, le Roux L, Gustafsson O, Craddock N, Möller H-J, McQuillin A, Muglia P, Cichon S, Rietschel M, Ophoff RA, Djurovic S, Andreassen OA, Pietiläinen OPH, Peltonen L, Dempster E, Collier DA, St Clair D, Rasmussen HB, Glenthøj BY, Kiemeney LA, Franke B, Tosato S, Bonetto C, Saemundsen E. et al. Maternally derived microduplications at 15q11-q13: implication of imprinted genes in psychotic illness. Am J Psychiatry. 2011;168:408–417. doi: 10.1176/appi.ajp.2010.09111660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Caignec C, Boceno M, Saugier-Veber P, Jacquemont S, Joubert M, David A, Frebourg T, Rival JM. Detection of genomic imbalances by array based comparative genomic hybridisation in fetuses with multiple malformations. J Med Genet. 2005;42:121–128. doi: 10.1136/jmg.2004.025478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valduga M, Philippe C, Bach Segura P, Thiebaugeorges O, Miton A, Beri M, Bonnet C, Nemos C, Foliguet B, Jonveaux P. A retrospective study by oligonucleotide array-CGH analysis in 50 fetuses with multiple malformations. Prenat Diagn. 2010;30:333–341. doi: 10.1002/pd.2460. [DOI] [PubMed] [Google Scholar]
- Van den Veyver IB, Patel A, Shaw CA, Pursley AN, Kang S-HL, Simovich MJ, Ward PA, Darilek S, Johnson A, Neill SE, Bi W, White LD, Eng CM, Lupski JR, Cheung SW, Beaudet AL. Clinical use of array comparative genomic hybridization (aCGH) for prenatal diagnosis in 300 cases. Prenat Diagn. 2009;29:29–39. doi: 10.1002/pd.2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahoo T, Cheung SW, Ward P, Darilek S, Patel A, del Gaudio D, Kang SHL, Lalani SR, Li J, McAdoo S, Burke A, Shaw CA, Stankiewicz P, Chinault AC, Van den Veyver IB, Roa BB, Beaudet AL, Eng CM. Prenatal diagnosis of chromosomal abnormalities using array-based comparative genomic hybridization. Genet Med. 2006;8:719–727. doi: 10.1097/01.gim.0000245576.47154.63. [DOI] [PubMed] [Google Scholar]
- Shaffer LG, Coppinger J, Alliman S, Torchia BA, Theisen A, Ballif BC, Bejjani BA. Comparison of microarray-based detection rates for cytogenetic abnormalities in prenatal and neonatal specimens. Prenat Diagn. 2008;28:789–795. doi: 10.1002/pd.2053. [DOI] [PubMed] [Google Scholar]
- Kleeman L, Bianchi DW, Shaffer LG, Rorem E, Cowan J, Craigo SD, Tighiouart H, Wilkins-Haug LE. Use of array comparative genomic hybridization for prenatal diagnosis of fetuses with sonographic anomalies and normal metaphase karyotype. Prenat Diagn. 2009;29:1213–1217. doi: 10.1002/pd.2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evangelidou P, Sismani C, Ioannides M, Christodoulou C, Koumbaris G, Kallikas I, Georgiou I, Velissariou V, Patsalis PC. Clinical application of whole-genome array CGH during prenatal diagnosis: Study of 25 selected pregnancies with abnormal ultrasound findings or apparently balanced structural aberrations. Mol Cytogenet. 2010;3:24. doi: 10.1186/1755-8166-3-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JA, Cheung SW, Ward PA, Inoue K, Lupski JR. Prenatal diagnosis of PLP1 copy number by array comparative genomic hybridization. Prenat Diagn. 2005;25:1188–1191. doi: 10.1002/pd.1308. [DOI] [PubMed] [Google Scholar]
- Shaffer LG, Dabell MP, Rosenfeld JA, Neill NJ, Ballif BC, Coppinger J, Diwan NR, Chong K, Shohat M, Chitayat D. Referral patterns for microarray testing in prenatal diagnosis. Prenat Diagn. 2012;32:344–350. doi: 10.1002/pd.3856. [DOI] [PubMed] [Google Scholar]
- Hillman SC, Pretlove S, Coomarasamy A, McMullan DJ, Davison EV, Maher ER, Kilby MD. Additional information from array comparative genomic hybridization technology over conventional karyotyping in prenatal diagnosis: a systematic review and meta-analysis. Ultrasound Obstet Gynecol. 2011;37:6–14. doi: 10.1002/uog.7754. [DOI] [PubMed] [Google Scholar]
- Tyreman M, Abbott KM, Willatt LR, Nash R, Lees C, Whittaker J, Simonic I. High resolution array analysis: diagnosing pregnancies with abnormal ultrasound findings. J Med Genet. 2009;46:531–541. doi: 10.1136/jmg.2008.065482. [DOI] [PubMed] [Google Scholar]
- ACOG Committee Opinion No. 446: array comparative genomic hybridization in prenatal diagnosis. Obstet Gynecol. 2009;114:1161–1163. doi: 10.1097/AOG.0b013e3181c33cad. [DOI] [PubMed] [Google Scholar]
- Armengol L, Nevado J, Serra-Juhé C, Plaja A, Mediano C, García-Santiago FA, García-Aragonés M, Villa O, Mansilla E, Preciado C, Fernández L, Ángeles Mori M, García-Pérez L, Lapunzina PD, Pérez-Jurado LA. Clinical utility of chromosomal microarray analysis in invasive prenatal diagnosis. Hum Genet. 2012;131:513–523. doi: 10.1007/s00439-011-1095-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaffer LG, Dabell MP, Fisher AJ, Coppinger J, Bandholz AM, Ellison JW, Ravnan JB, Torchia BS, Ballif BC, Rosenfeld JA. Experience with microarray-based comparative genomic hybridization for prenatal diagnosis in over 5000 pregnancies. Prenat Diagn. 2012. pp. 1–10. [DOI] [PMC free article] [PubMed]
- Schaaf CP, Wiszniewska J, Beaudet AL. Copy number and SNP arrays in clinical diagnostics. Annu Rev Genomics Hum Genet. 2011;12:25–51. doi: 10.1146/annurev-genom-092010-110715. [DOI] [PubMed] [Google Scholar]
- Darilek S, Ward P, Pursley A, Plunkett K, Furman P, Magoulas P, Patel A, Cheung SW, Eng CM. Pre- and postnatal genetic testing by array-comparative genomic hybridization: genetic counseling perspectives. Genet Med. 2008;10:13–18. doi: 10.1097/GIM.0b013e31815f1ddb. [DOI] [PubMed] [Google Scholar]
- Savage MS, Mourad MJ, Wapner RJ. Evolving applications of microarray analysis in prenatal diagnosis. Curr Opin Obstet Gynecol. 2011;23:103–108. doi: 10.1097/GCO.0b013e32834457c7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuster E. Microarray genetic screening: a prenatal roadblock for life? Lancet. 2007;369:526–529. doi: 10.1016/S0140-6736(07)60239-6. [DOI] [PubMed] [Google Scholar]
- McGillivray G, Rosenfeld JA, McKinlay Gardner RJ, Gillam LH. Genetic counselling and ethical issues with chromosome microarray analysis in prenatal testing. Prenat Diagn. 2012;32:389–395. doi: 10.1002/pd.3849. [DOI] [PubMed] [Google Scholar]
- Adams SA, Coppinger J, Saitta SC, Stroud T, Kandamurugu M, Fan Z, Ballif BC, Shaffer LG, Bejjani BA. Impact of genotype-first diagnosis: the detection of microdeletion and microduplication syndromes with cancer predisposition by aCGH. Genet Med. 2009;11:314–322. doi: 10.1097/GIM.0b013e3181a028a5. [DOI] [PubMed] [Google Scholar]
- Adam MP, Justice AN, Schelley S, Kwan A, Hudgins L, Martin CL. Clinical utility of array comparative genomic hybridization: uncovering tumor susceptibility in individuals with developmental delay. J Pediatr. 2009;154:143–146. doi: 10.1016/j.jpeds.2008.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho MK. Understanding incidental findings in the context of genetics and genomics. J Law Med Ethics. 2008;36:280–285, 212. doi: 10.1111/j.1748-720X.2008.00270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabor HK, Cho MK. Ethical implications of array comparative genomic hybridization in complex phenotypes: points to consider in research. Genet Med. 2007;9:626–631. doi: 10.1097/GIM.0b013e3181485688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaudet AL. Ethical issues raised by common copy number variants and single nucleotide polymorphisms of certain and uncertain significance in general medical practice. Genome Med. 2010;2:42. doi: 10.1186/gm163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaaf CP, Scott DA, Wiszniewska J, Beaudet AL. Identification of incestuous parental relationships by SNP-based DNA microarrays. Lancet. 2011;377:555–556. doi: 10.1016/S0140-6736(11)60201-8. [DOI] [PubMed] [Google Scholar]
- Tabor HK, Berkman BE, Hull SC, Bamshad MJ. Genomics really gets personal: how exome and whole genome sequencing challenge the ethical framework of human genetics research. Am J Med Genet A. 2011;155A:2916–2924. doi: 10.1002/ajmg.a.34357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caulfield T, McGuire AL, Cho M, Buchanan JA, Burgess MM, Danilczyk U, Diaz CM, Fryer-Edwards K, Green SK, Hodosh MA, Juengst ET, Kaye J, Kedes L, Knoppers BM, Lemmens T, Meslin EM, Murphy J, Nussbaum RL, Otlowski M, Pullman D, Ray PN, Sugarman J, Timmons M. Research ethics recommendations for whole-genome research: consensus statement. PLoS Biol. 2008;6:e73. doi: 10.1371/journal.pbio.0060073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang AW, MacDonald JR, Pinto D, Wei J, Rafiq MA, Conrad DF, Park H, Hurles ME, Lee C, Venter JC, Kirkness EF, Levy S, Feuk L, Scherer SW. Towards a comprehensive structural variation map of an individual human genome. Genome Biol. 2010;11:R52. doi: 10.1186/gb-2010-11-5-r52. [DOI] [PMC free article] [PubMed] [Google Scholar]