

Abstract
Common variable immunodeficiency (CVID) describes a heterogeneous subset of hypogammaglobulinemias of unknown etiology. Typically, patients present with recurrent bacterial infections of the respiratory and gastrointestinal tract. A significant proportion of CVID patients develops additional autoimmune, inflammatory or lymphoproliferative complications. CVID is the most frequent symptomatic primary immunodeficiency encountered in adults. Informative monogenetic defects have been found in single patients and families but in most cases the pathogenesis is still elusive. Numerous immunological studies have demonstrated phenotypic and functional abnormalities of T cells, B cells and antigen-presenting cells. A hallmark is the impaired memory B-cell formation that has been taken advantage of for classifying CVID patients. Clinical multi-center studies have demonstrated a correlation between immunological markers and clinical presentation. Long-term outcome is significantly influenced by delay of diagnosis and treatment and the presence of chronic inflammatory complications. While immunoglobulin replacement therapy plus antibiotics can control infections in most cases, patients with non-infectious inflammatory complications such as granulomatous inflammation, interstitial lung disease, inflammatory bowel disease, lymphoproliferation and developing malignancies still represent a therapeutic challenge. In this review we provide a systematic overview of the immunological, clinical, diagnostic and therapeutic aspects of CVID and highlight recent developments in these fields.
Definition of common variable immunodeficiency
The diagnosis 'common variable immunodeficiency' (CVID) describes patients presenting with hypogammaglobulinemia of unknown origin and variable immunological and clinical phenotypes. The most common symptoms are severe, recurrent and sometimes chronic bacterial infections mainly of the respiratory and gastrointestinal tracts.
Based on the 1999 criteria issued by the American and European societies for immunodeficiency [1], the diagnosis of CVID can be made if the following criteria are fulfilled: a male or female patient who exhibits a marked decrease of IgG (at least two standard deviations below the mean for age) and of at least one of the IgM or IgA isotypes; onset of immunodeficiency at greater than 2 years of age; absence of isohemagglutinins and/or poor response to vaccines; and other defined causes of hypogammaglobulinemia have been excluded. Most important is the exclusion of other primary immunodeficiencies and secondary causes of hypogammaglobulinemia (Table 1).
Table 1.
Primary and secondary causes of hypogammaglobulinemia to be distinguished from common variable immunodeficiency
| Cause | Diagnosis (examples) |
|---|---|
| Primary immunodeficiencies | |
| Agammaglobulinemias | X-chromosomal agammaglobulinemia (BTK), rare AR forms |
| Class switch recombination deficiencies | X-chromosomal form: CD40L |
| Autosomal recessive forms: CD40, AID, UNG | |
| X-chromosome lymphoproliferative | SAP, XIAP deficiency |
| syndrome | |
| Combined immunodeficiency | Hypomorphic variants of severe combined immunodeficiencies |
| Other forms of combined immunodeficiency (ORAI, STIM, DOCK8 a.o.) | |
| Other defined primary immunodeficiencies | WHIM (warts, hypogammaglobulinemia, infections, myelokathexis) syndrome DiGeorge syndrome |
| Chromosomal instability syndromes | Ataxia telangiectasia (ATM) |
| Nijmegen breakage syndrome (NBS1) | |
| ICF (immunodeficiency, chromosomal instability, facial abnormalities) syndrome (DNMT3B) | |
| Secondary cause | |
| Malignancy | Chronic lymphatic leukemia |
| Immunodeficiency and thymoma (Good syndrome) | |
| Malignant lymphoma | |
| Protein loss | Loss of immunoglobulins (for example, renal or gastrointestinal protein loss, severe burns, lymphangiectasis) |
| Hyperkatabolism of immunoglobulins (for example, myotonic dystrophy types 1 and 2) | |
| Drug induced | Anticonvulsants (carbamazepine, valproic acid, phenytoine) |
| Sulfasalazine | |
| Gold salts | |
| Glucocorticoids | |
| Azathioprine | |
| D-Penicillamine | |
| Antimalarial agents (very rare) | |
| Methotrexate (very rare) | |
| Alkylating agents (cyclophosphamid, chlorambucil) | |
| Anti-CD20 (rituximab) | |
| Imatinib | |
| Infectious | Congenital cytomegalovirus, rubella or Toxoplasma gondii infection, neonatal HIV infection |
| Ebstein-Barr virus infection |
AID, activation-induced cytidine deaminase; a.o., and others; AR, autosomal recessive; ATM, ataxia telangiectasia mutated; BTK, Bruton agammaglobulinemia tyrosine kinase; DNMT3B, DNA (cytosine-5-)-methyltransferase 3 beta; DOCK8, dedicator of cytokinesis 8; NBS1, Nijmegen breakage syndrome 1; ORAI, ORAI calcium release-activated calcium modulator 1; SAP, SLAM associated protein/SH2 domain protein 1A; STIM, stromal interaction molecule 1; UNG, uracil-DNA glycosylase; XIAP, X-linked inhibitor of apoptosis.
It is important to note that only a small percentage of patients taking any of the drugs mentioned in Table 1 will develop a secondary hypogammaglobulinemia, suggesting an individual predisposition. While some of the drug reactions are due to toxic effects, others may be induced by an allergic reaction.
The listed infections usually do not cause hypogammaglobulinemia; therefore, an underlying predisposition is also likely in these patients. Only mutations in SH2D1A (encoding SAP) causing X-chromosomal lymphoproliferative syndrome are confirmed to be associated with Epstein Barr virus-driven hypogammaglobulinemia.
Epidemiology
CVID encompasses the largest group of symptomatic primary immunodeficiencies, with an estimated incidence between 1:10,000 and 1:50,000 [1,2]. There are regional differences in incidence, with CVID being a rare diagnosis among Asians and Afro-Americans [3,4]. There is no gender predisposition and the age of onset is usually in the second to third decade of life, although a smaller group of patients already manifests CVID in childhood [3,4], and, in general, CVID may occur at any age [5].
Genetics of common variable immunodeficiency
In contrast to most other primary immunodeficiencies, more than 90% of documented CVID patients are lacking a definite molecular genetic diagnosis or other causal explanation for their disease. Only 10 to 20% of CVID patients have a positive family history, while most cases occur sporadically [3,4]. Four out of five 'CVID families' show autosomal dominant inheritance. In some larger pedigrees, individuals with selective IgA deficiency (sIgAD), CVID and intermediate forms can be observed side by side [6,7]. This finding and cases of progression from sIgAD towards CVID [8] indicate a possible common genetic predisposition. Autosomal recessive CVID is rarely seen in Europe and North America but is more frequent in regions and ethnic groups with higher rates of consanguinity [4,9].
Genetic linkage analysis of large collections of familial CVID/sIgAD patients [10-12] or singular large pedigrees with multiple CVID/sIgAD cases [6] revealed possible genetic loci on chromosome 4q [6], chromosome 6 [10,12] and chromosome 16q [11]. These early genome-wide microsatellite-marker studies found the strongest association with the HLA region [10,12]; they were recently confirmed by a genome-wide single nucleotide polymorphism (SNP) genotyping array approach in several hundred CVID patients [13]. This study also revealed several structural chromosomal abnormalities unique to CVID and many novel candidate genes significantly associated with CVID or its clinical complications [13].
In a minority of patients with CVID, distinct molecular genetic defects have been identified. These genes associated with a CVID phenotype are ICOS (inducible costimulator) [14], TACI (transmembrane activator and calcium-modulating cyclophilin ligand interactor) [15,16], CD19 [17], BAFF-R [18], CD81 [19], CD20 [20], CD21 [21] and LRBA (lipopolysaccharide responsive beige-like anchor protein) [22]. These defects are very rare, however, only occurring in single cases or single families and adding up to less than 3% of CVID patients. The exceptions are TACI mutations, which are seen in up to 10% of CVID cases but occur also in 1% of the healthy population, and thus must be regarded as disease modifiers rather than disease-causing gene defects.
The currently known monogenetic defects associated with CVID are summarized in Table 2. These mono-genetic defects affect only very few patients, but do provide important prototypic disease models by indicating weak points in terminal B-cell differentiation.
Table 2.
Monogenetic defects associated with or causing common variable immunodeficiency
| Defect (gene, chromosome) | Frequency | Onset | Ig serum levels | Clinical symptoms |
|---|---|---|---|---|
| ICOS deficiency (ICOS, 2q33) | 5 families, 11 patients | Late onset, early onset | IgG, A, M low | URI, LRI, GI, SP, LP, AI, SG |
| CD19 deficiency (CD19, 16p11.2) | 6 families, 9 patients | Early onset | IgG and IgA low, IgM variable | URI, LRI, GI, SP, LP, AI, SG, GN |
| BAFFR deficiency (TNFRSF13C, 22q13.1-q13.31) | 1 family, 2 patients | Late onset | IgG and IgM low, IgA normal | URI, LRI |
| CD81 deficiency (CD81, 11p15.5) | 1 family, 1 patient | Early onset | IgG low, IgA and IgM normal | URI, LRI, GN |
| CD20 deficiency (CD20, 11q12) | 1 family, 1 patient | Early onset | IgG low, IgA and IgM normal | URI |
| CD21 deficiency (CR2, 1q32) | 1 family, 1 patient | Late onset | IgG and IgA low, IgM normal | URI, GI, SP |
| TACI (TNFRSF13B, 17p11.2) | About 8 to 10% of CVID patients | Late onset, early onset | IgG and IgA low, IgM variable | URI, LRI, GI, SP, AI, LP, SG |
| LRBA deficiency (LRBA, 4q31.3) | 4 families, 5 patients | Early onset (<15 years) | IgG and IgA low, IgM variable | URI, LRI, AI, GI, SG |
AI, autoimmunity (autoimmune hemolytic anemia, immune thrombocytopenic purpura, neutropenia); GI, gastrointestinal tract involvement (gastritis, celiac like disease, Crohn's like disease); GN, glomerulonephritis; LP, lymphoproliferation; LRI, lower respiratory tract infection (bronchitis, pneumonia); SG, sarkoid-like granulomatous disease; SP, splenomegaly; URI, upper respiratory tract infection (sinusitis, rhinitis, otitis media, pharyngitis).
Immunopathology of common variable immunodeficiency
The immune system of CVID patients has been investigated by many studies, describing both phenotypic and functional abnormalities in the adaptive and, more recently, also in the innate immune system. However, the plethora of these defects, their unequal distribution within different CVID cohorts and the lack of a real comprehensive and combined analysis of all of them have so far precluded a definitive mapping of all immunopathogenic pathways leading to CVID.
Based on the most common defects found in T cells and B cells of the adaptive immune system, several classification systems have been introduced [23-26].
Disturbances of T cells
For many years abnormalities of CD4+ and CD8+ T cell numbers or function have been known and described in subgroups of CVID patients. In a significant proportion of CVID patients a reduction of total CD4+ T-cell counts and the naive CD4+CD45RA+ subset has been observed [23,27,28]. Regulatory CD25+FoxP3+-CD4+ T cells are also diminished [29-32] in a subgroup of CVID patients who present clinically with increased autoimmunity, granulomas, splenomegaly and an expansion of CD21low B cells [32].
The T-cell compartment of some CVID patients expresses surface marker patterns indicative of chronic activation; in contrast to CD4+ T cells, the CD8+ T cells of these patients may numerically expand, explaining the frequently inverted CD4/CD8 T-cell ratio seen in CVID. These disturbances of the CD8+ T-cell pool can be associated with disturbed cytokine secretion [33], lower memory B-cell numbers and severe clinical courses [34], chronic or recurrent cytomegalovirus infections [35] and polyclonal expansions of 'large granular lymphocytes' in combination with splenomegaly [36].
Giovanetti and colleagues [23] defined clinically relevant subgroups of CVID patients based on the reduction of naive CD4+ T cells. Their group I patients exhibited a severe reduction of naive CD4+ T cells, signs of massive T-cell activation, association with splenomegaly and a more severe course of the disease [23].
The French DEFI study group delineated a CVID subgroup with clinically relevant T-cell insufficiency and coined the term 'late onset combined immunodeficiency' (LOCID) for these patients [37]. Inclusion criteria were CD4+ T cells below 200/μl or evidence of opportunistic infections, which occurred in 3.5% and 5.4%, respectively, of their studied cohort. LOCID patients often had a consanguine background, and suffered more often from sarkoid-like granulomas, gastrointestinal complaints, splenomegaly and lymphoma; in general, LOCID patients were more sick and required more intensive therapy [37].
Disturbances of B cells
The total number of peripheral B cells is slightly reduced in about 40 to 50% of CVID patients [26]. In some patients elevated numbers of B cells are reported, often associated with polyclonal lymphoid organ infiltration and autoimmunity [5]. In only about 10% of CVID patients are B cells dramatically reduced or absent [26]. Disease progression tends to be more rapid and severe in these patients [3,38] and the X-linked form of agammaglobulinemia and Good's syndrome (B-cell aplasia associated with thymoma) have to be excluded (Table 1).
On a cellular level, hypogammaglobulinemia and defective specific antibody responses are matched by impaired terminal B-cell differentiation into memory B cells and plasma cells. The depletion of plasma cells in lymphoid organs (gut-associated lymphatic tissue, lymph nodes, bone marrow) has been demonstrated in several studies [39-41]. Furthermore, class-switched memory B cells are reduced in 80 to 90% of CVID patients [24-26]. Since this finding is not specific for CVID, it is not suitable as a diagnostic criterion but has been used to classify CVID patients into clinically and immunologically more homogeneous subgroups [24-26]. The 'Freiburg' classification distinguishes three groups of CVID patients [25] based on the percentage of switched memory B cells and the expansion of activated so-called CD21low B cells. The 'Paris' classification distinguishes three CVID subgroups [24] based on the reduction of total versus switched memory B cells. For both classification schemes several studies demonstrated that classification of CVID patients based on B-cell phenotypes is useful for identifying clinical subtypes, adapting therapeutic regimens (vaccination), assessing risks of certain complications and performing pathogenic research [42-46].
In 2008 a European multi-center trial combined both classification systems and proposed the EUROclass classification [26]. B cells were phenotyped for CD19, IgD/IgM, CD27, CD21 and CD38 expression; patients with more than 1% circulating B cells (B+, >90% of all patients) were distinguished from those with less than 1% (B-, <10% of all patients). The B+ group was further split into patients with normal or reduced percentages of switched memory B cells (smB+, >2% of total B cells; smB-, <2% of total B cells). Further subgroups were established depending on the expansion of CD21low B cells or transitional B cells. The EUROclass trial confirmed the clinical association of reduced switched memory B cells and expanded CD21low B cells with splenomegaly and granulomatous disease and revealed for the first time an expansion of transitional B cells in patients with lymphadenopathy [26].
The disturbed memory B-cell formation points towards an impaired germinal center reaction in secondary lymphoid organs of most CVID patients. This assumption is further supported by decreased rates of somatic hypermutations in CD27+ B cells of CVID patients [47,48], a phenomenon that inversely correlates with an increased risk of chronic lung damage [47]. Histopathological studies of secondary lymphoid organs in CVID that would allow a closer look at possible pathomechanisms in situ are still rare. Taubenheim and colleagues [41] showed in three patients an intact development of the centroblast/centrocyte stage, including the sequential expression of BCL-6 and Blimp-1, but the subsequent development into plasmablasts and plasma cells was disturbed.
B-cell activation is triggered by stimulation of the B-cell receptor, CD40, cytokine receptors and pattern recognition receptors such as Toll-like receptors (TLRs). Currently, defects of B-cell receptor activation [44] as well as the TLRs [49-51] have been identified in subgroups of patients. The underlying cause remains unknown for both defects.
Disturbances of antigen presenting cells and innate immunity receptors
Professional antigen presenting cells, such as dendritic cells (DCs), interact with naive T cells in the T-cell areas of secondary lymphoid organs. As part of the germinal center reaction they cooperate with cognate T and B cells to promote their further differentiation. Outside of germinal centers plasmacytoid DCs may initiate immunoglobulin class switching and terminal B-cell differentiation independent of T-cell help but via signals through TLRs and the cytokines BAFF (B-cell activating factor) and APRIL (a proliferation inducing ligand). These two pathways are closely linked together particularly at the level of TLR9 and the BAFF/APRIL receptor TACI [52,53]. When DCs from CVID patients were differentiated in cell culture experiments their maturation was impaired, resulting in diminished interleukin-12 production and impaired up-regulation of co-stimulatory molecules. This might limit the ability of CVID DCs to contact and successfully interact with T cells [54,55]. In addition, TLR9 expression and response of plasmacytoid DCs and B cells to CpG stimulation is reduced [49]. Further investigations in CVID patients revealed an additional dysfunction of TLR7 and TLR8 signaling [50,51]. The recently described relationship between TACI and the TLR9 signaling pathway [52] strengthens the assumption that these disturbances of the TLR system in CVID patients are of pathophysiological relevance even though no genetic mutations in the TLR pathway have been established so far.
Clinical presentation of common variable immunodeficiency
Infections
Over 90% of CVID patients suffer from an increased susceptibility to bacterial pathogens affecting mucous membranes of the upper and lower airways and, to a lesser extent, of the gastrointestinal tract [3,4,38]. Table 3 summarizes frequencies of specific infections and pathogens encountered in two consecutive studies on the Mount Sinai Hospital CVID cohort in New York [3,38] and the French DEFI cohort study [4]. In the DEFI cohort study, approximately two-thirds of the 252 patients presented with sinusitis or bronchitis and 50% had at least one bout of pneumonia during their life [4]. About one-third of patients had developed bronchiectasis as a result of chronic and recurrent infections. Frequently detected pathogens were Streptococcus pneumoniae, Haemophilus influenzae, Staphylococcus aureus and Moraxella catharralis. Recurrent and chronic diarrhea was present in approximately 40% of patients and in about half of them pathogens like Giardia lamblia followed by Salmonella and Campylobacter jejuni were identified. Acute and chronic gastritis caused by Helicobacter pylori is frequently diagnosed in CVID patients [3]. Up to 10% of CVID patients are described as suffering from increased rates of Herpes zoster infections. In contrast, typical opportunistic infections are quite unusual and evoke the possibility of an underlying combined immunodeficiency [37]. A rare but typical complication of hypogammaglobulinemia is oligoarthritis due to Mycoplasma species (Figure 1). Clinically, the condition presents as reactive arthritis with synovial culture techniques being often negative; therefore, the microbiological diagnosis has to include multipathogen PCR in synovial fluid. In the differential diagnosis of hypogammaglobulinemic oligoarthritis, serological investigations are not helpful since most patients do not exhibit an adequate antibody response against the respective pathogens or the test results are influenced by intravenous immunoglobulin substitution therapy. In patients with suspected reactive arthritis, therefore, we recommend to initially determine IgG and IgA serum concentrations before proceeding to extensive and potentially meaningless antibacterial antibody responses.
Table 3.
Infectious complications in common variable immunodeficiency
| Mount Sinai 1999 [3](n = 248) | Mount Sinai 2011 [38](n = 473) | DEFI 2008 [4](n = 252) | |
|---|---|---|---|
| Recurrent (respiratory tract) infections | 98% (243/248) | 94% (445/473) | 91% (230/252) |
| Pneumoniaa | 76.6% (190/248) | 40% (187/473) | 58% (147/252) |
| Meningitis | <1% (2/248) | NS | 7.9% (20/252) |
| Viral hepatitis | 6.5% (16/248) | 3.4% (16/473) | 1.2% (3/252) |
| Giardia enteritis | 3.2% (8/248) | 2.3% (11/473) | 13.9% (35/252) |
| (Recurrent) Herpes zoster | 3.6% (9/248) | 2.5% (12/473) | 11% (27/252) |
| Pneumocystis jiroveci infection | 2.8% (7/248) | 1.3% (6/473) | <1% (2/252) |
| Salmonella infection | 1.2% (3/248) | <1% (4/473) | 7.5% (19/252) |
| Mycoplasma infection | 2.8% (7/248) | <1% (4/473) | NS |
| Candidiasis | 1.2% (3/248) | <1% (3/473) | <1% (2/252) |
| Mycobacterial disease | <1% (1/248) | <1% (3/473) | <1% (2/252) |
| Papilloma viral infection | NS | <1% (3/473) | <1% (13/252) |
aPredominantly bacterial, lobar or bronchopneumonia, X-ray proven. NS, not specified.
Figure 1.
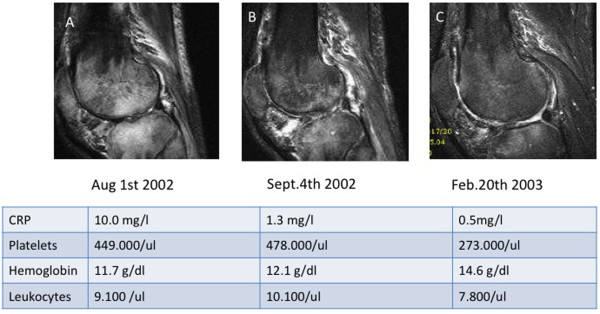
Oligoarthritis due to Mycoplasma salivarius as an early manifestation of common variable immunodeficiency. A male aged 36 years was healthy until he developed recurrent upper respiratory tract infections and a first bout of pneumonia 18 months prior to these images being taken. Five months later he presented with refractory right-sided gonarthritis to an orthopedic surgeon. Despite multiple sterile knee taps, arthroscopy and a Baker cyst resection, joint inflammation continued and extended to the right shoulder and the right ankle. He was referred to the Division of Rheumatology and Clinical Immunology at Freiburg University Hospital for further diagnosis and treatment of 'multifocal osteomyelitis and oligoarthritis of unknown origin'. On admission he presented with three tender and swollen joints (right knee, shoulder, ankle), moderately elevated C-reactive protein (CRP) levels (5 to 29 mg/dl) and severe hypogammaglobulinemia: IgG 1.7 g/L, IgA <0.6 g/L, IgM <0.3 g/L. Diagnosis of CVID was established and the patient was started on monthly intravenous immunoglobulin infusions (500 mg/kg) plus various ineffective antibiotic regimens (initially cefuroxime plus neomycin, then clarithromycin and metronidazol). A diagnostic puncture of the right shoulder eventually revealed Mycoplasma salivarius by multiplex PCR diagnostics. From that point on the patient was put on doxicycline (200 mg/daily orally) and the inflammatory process rapidly improved. Doxicycline was stopped after 4 weeks, whereas monthly intravenous immunoglobulin was continued. As of today, the patient has been back to work for 7 years and is clinically doing well. Magnetic resonance imaging follow-up (T2, TIRM sequences of the right knee) and laboratory parameters at three time points (A, B, C) nicely show the improvement of the severe arthritis and osteomyelitis of the right knee.
Granulomatous lesions
Approximately 10 to 20% of CVID patients develop granulomatous interstitial lung disease. Microbial testing of these lesions often reveals no specific pathogen; the reported detection of human herpes virus-8 in a US CVID cohort [56] could not be confirmed in larger European patient groups (unpublished data), indicating that the underlying cause remains unknown and probably is multifactorial. Patients with granulomatous interstitial lung disease have a significantly poorer prognosis than other CVID patients [38,57]. The granulomatous disease to some extent resembles sarcoidosis; in addition to lung and lymph nodes, also the liver, skin, spleen, bone marrow, gastrointestinal tract, brain and kidney (in decreasing frequency) may be affected [58].
Gastrointestinal symptoms
Diarrhea is un-bloody if associated with a sprue-like disease and bloody when resulting from chronic inflammatory bowel disease. The sprue-like villous atrophy seen in CVID is often not gluten-sensitive and resembles more autoimmune enteropathy. The involvement of the colon in CVID is reminiscent of Crohn's disease and ulcerative colitis, but can be distinguished histologically [59]. The nodular lymphoid hyperplasia that may occur both in the duodenum and ileum may be asymptomatic or associated with unformed stools [3].
Liver disease and abnormal liver function tests are found in 10% of CVID patients [3,60]. The most common liver disease in CVID represents nodular regenerative hyperplasia of the liver tissue [60,61] or seronegative, granulomatous hepatitis; autoimmune hepatitis is not a typical entity found in these patients. Usually, liver function in CVID patients is still preserved but portal hypertension may develop [60,61]. Liver disease heralds a poorer prognosis [38]. In any case of a suspected hepatopathy in CVID, seronegative hepatitis B and C as well as cytomegalovirus or Epstein Barr virus hepatitis must be ruled out by searching for hepatitis antigen or viral RNA, respectively.
Autoimmunity
Autoimmunity is present in about 30% of CVID patients [3,26,38,62,63]. Table 4 summarizes frequencies of autoimmune disease encountered in two consecutive studies on the Mount Sinai Hospital CVID cohort in New York [3,38] and the French DEFI cohort study [62].
Table 4.
Common autoimmune manifestations in common variable immunodeficiency
| Mount Sinai 1999 [3](n = 248) | Mount Sinai 2011 [38](n = 473) | DEFI 2010 [62](n = 311) | |
|---|---|---|---|
| AIHA | 4.8% (12/248) | 7% (33/473) | 5.4% (17/311) |
| ITP | 6% (15/248) | 14.2% (67/473) | 13.2% (41/311) |
| Neutropenia | <1% (2/248) | <1% (<5/473) | 3.2% (10/311) |
| Rheumatoid arthritis | 3.6% (9/248) | 3.2% (15/473) | 2.6% (8/311) |
| Vitiligo | NS | <1% (<5/473) | 3.9% (12/311) |
| Sicca syndrome, Sjögren's syndrome | <1% (2/248) | <1% (<5/473) | 4.2% (13/311) |
| Autoimmune thyroiditis, diabetes mellitus, | <1% (2/248) | <1% (<5/473 | 3.9% (12/311) |
| multiple sclerosis | |||
| Alopecia | 1.6% (4/248) | 1.1% (5/473) | NS |
| Pernicious anemia | 1.2% (3/248) | <1% (<5/473) | NS |
| Systemic lupus erythematosus | <1% (2/248) | <1% (<5/473) | <1% (1/311) |
AIHA, autoimmune hemolytic anemia; ITP, immune thrombocytopenic purpura; NS, not specified.
Particularly common are autoimmune thrombocytopenia (10 to 12%) and autoimmune hemolytic anemia (5 to 7%), showing a significant correlation with splenomegaly [26,64]. Cytopenias can manifest before, simultaneously with or after the diagnosis of immunodeficiency. Immunologically, autoimmune cytopenias are associated with low numbers of class-switched memory B cells, low numbers of regulatory T cells, expanded CD21low B cells [25,65], and nodular T-cell infiltrates of the bone marrow [40]. Autoimmune thyroid disease, vitiligo, pernicious anemia, psoriasis, rheumatoid arthritis and systemic lupus erythematosus are observed in CVID cohorts at decreasing frequency [3,5,26] (Table 4).
Lymphoproliferation and malignancies
Benign lymphoproliferation is found in 40 to 50% of CVID patients, often as splenomegaly, and in approximately 10 to 20% as local or diffuse lymphadenopathy [5,26]. Histologically, several subsets can be distinguished, with follicular hyperplasia and granulomatous inflammation being the most common ones [66]. In conjunction with lymphoproliferation, CVID patients carry an increased risk of developing lymphoma [3,5]. Most lymphomas are of the B-cell non-Hodgkin lymphoma type [3]. In addition to lymphomas, stomach cancers represent an important malignant manifestation in CVID [3,5,63,67]. The increased risk of cancer in CVID may result from impaired immunity to potentially carcinogenic pathogens (for example, Helicobacter pylori, Epstein-Barr virus) or impaired tumor cell surveillance. In this context it is notable that patients of a CVID subgroup exhibit increased radiosensitivity, known to be a risk factor for increased tumor incidence [68].
Diagnosis and follow-up of common variable immunodeficiency
The diagnosis of CVID can only be made after the exclusion of a variety of other causes of hypogammaglobulinemia (Table 1). Its rarity and high clinical variability lead to a significant delay in diagnosis between four [69] and nine years [63] after onset of symptoms. Clinically, the leading symptom in most patients is the classical susceptibility to bacterial airway infections as described above. As mentioned before, opportunistic infections are always suggestive of LOCID. Some CVID cases manifest initially with autoimmune cytopenias; thus, CVID needs to be excluded in patients with immune thrombocytopenic purpura, autoimmune hemolytic anemia and autoimmune neutropenia.
An inexpensive, quantitative determination of serum immunoglobulins is the first and most important step in the diagnosis of CVID. Required for the diagnosis of CVID is the diminution of at least two isotypes (IgG and IgA or IgM). IgG is typically below 5 g/L (normal range 7 to 16 g/L) and IgA is markedly reduced or not detectable in most patients. IgM is also below the normal range in up to 80% of patients.
While drug-induced hypogammaglobulinemia (for example, with rituximab; Table 1) may be revealed by the patient's history, proteinuria is detected by Uristix® and intestinal protein loss may be suspected from the medical history in combination with a decreased serum albumin concentration. Lymphomas tend to be the most difficult differential diagnosis in secondary hypogammaglobulinemia. They require histological examination of lymph node and bone marrow biopsies, notably in CVID patients with ongoing lymphoproliferation. The differential diagnosis of other genetically defined immunodeficiencies is rare and should be done in a specialized center.
Besides quantitative determination of serum immunoglobulins, the basic laboratory tests should include a differential blood count, liver and kidney function parameters and C-reactive protein determination. The routine examinations are complemented by the determination of specific antibodies against protein antigens (tetanus, diphtheria and hepatitis B virus and hepatitis A virus in vaccinated patients) and antibodies against pneumococcal capsular polysaccharides. These studies are particularly meaningful if the patient has been vaccinated for diagnostic purposes prior to the start of immunoglobulin substitution.
The next stage of diagnosis is flow cytometric analysis of lymphocyte subpopulations, including total T, B and natural killer cells, to distinguish late manifesting X-linked agammaglobulinemia (B cells <0.1%) and combined immunodeficiencies (CD4 cells <200/μl). The classification of CVID patients with the separation of B-cell subpopulations is reserved for specialized immunodeficiency centers. A bone marrow biopsy should be performed in patients with low B-cell numbers (<1%) [40] and if lymphoma or myelodysplasia is suspected. In addition, several diagnostic procedures at first visit and during follow-ups are indicated for the control of possible secondary complications (summarized in Table 5).
Table 5.
Initial and follow-up diagnostics in common variable immunodeficiency
| System | Type of diagnostic procedure | Intervals |
|---|---|---|
| Hematopoietic system | Blood formula and differential blood counts | (3-)6 months; more often in case of known autoimmune cytopenia |
| Coombs test in any case of newly developing anemia | On demand | |
| Bone marrow biopsy in case of suspicion of lymphoma or myelodysplasia | On demand at diagnosis or during follow-up | |
| Immune system | IgG, IgA, IgM (aiming at trough levels >7g/L) | |
| In intravenous immunoglobulin-substituted patients | Monthly trough levels | |
| In subcutaneous immunoglobulin-substituted patients | 1-3 months | |
| Immunofixation in serum and urine, ß2-microglobulin | At diagnosis | |
| CD3, CD4, CD8, CD19, CD56 lymphocyte subsets | Initially and repeatedly in case of suspected combined immunodeficiency | |
| Vaccination responses to tetanus, diphtheria, hepatitis, pneumococcal polysaccharides | At diagnosis | |
| Classification (detailed T/B-cell evaluation) | At diagnosis | |
| Microbiology (direct culture of pathogens, PCR, antigen ELISA) | Purulent sputum: determine colonizing pathogens and resistance pattern to antibiotics | On demand |
| In case of chronic bronchiectasis, control of colonising pathogens (Pseudomonas spp., Haemophilius influenzae, Streptococcus pneumoniae, Staphylococcus aureus, Candida spp. and others) with sensitivity to antibiotics | 6-12 months and on demand | |
| Lungs | Spirometry, CO diffusion test, blood gases | 12 months |
| Chest X-ray | (12-)24 months | |
| HR-CT in case of proven GILD | 24 months and on demand | |
| Bronchoscopy + BAL in case of suspicion of GILD | At diagnosis; on demand | |
| Lymphoproliferation | Abdomen sonography CT-Abdomen/MRT | 12 months |
| Lymph node biopsy | On demand; in case of suspected lymphoma | |
| Gastrointestinal tract | Oesophogastroscopy | In case of clinical symptoms and every |
| 24 months in case of increased risk for developing intestinal malignancy | ||
| Colonoscopy | On demand | |
| Central nervous system | MRT, liquor analysis in case of neurological symptoms (exclusion of enteroviral infection) | On demand |
BAL, bronchoalveolar lavage; CT, computed tomography; GILD, granulomatous interstitial lung disease; HR-CT, high-resolution computed tomography; MRT, magnetic resonance computed tomography.
Therapy, natural course and prognosis
Current therapy of CVID can be categorized as follows: regular and sufficient substitution with immunoglobulins (IgG trough levels >7.0 g/L); targeted antibiotic treatment of (breakthrough) infections; adequate treatment of complications; and in selected patients with severe hematological changes (chronic transfusion need, leukopenia, thrombocytopenia), secondary malignancies and suspected combined immunodeficiency, allogeneic peripheral stem cell transplantation is being considered in experienced centers [70].
The immunoglobulin replacement therapy is the mainstay of therapy; 90% of CVID patients are on either intravenous (IVIg) or subcutaneous (SCIg) treatment [71-74]. Intramuscular administration is no longer recommended because this route does not ensure effective serum levels but is associated with a higher rate of side effects. The current standard dosage when administered intravenously is 400 to 600 mg/kg every 3 to 4 weeks. For subcutaneous administration, this corresponds to 100 to 150 mg/kg per week. The goal is the control of infections, which is reached at different individual IgG trough levels [63]. As a target value, IgG trough levels of more than 7 g/L are desirable before the next infusion. Patients with existing chronic lung disease (for example, bronchiectasis) or inflammatory bowel disease often require higher doses of IgG and may not reach the desired trough level. In chronic sinusitis additional careful local therapy (saline lavage, expectorant and decongestant therapy) is mandatory.
First line therapy in autoimmune cytopenias and lympho proliferation are steroids. In case of failure, immunosuppressive drugs, rituximab or splenectomy have been reported as options. The inflammatory and granulomatous lesions of the lungs, liver and intestine respond poorly to the immunoglobulin replacement therapy alone and therefore often require corticosteroids, eventually in combination with immunosuppressants (cyclosporin A, azathioprin and others). Prospective trials on the effectiveness of immunosuppressive drugs in CVID are still lacking.
The life expectancy of CVID patients has considerably improved over the past 30 years [5,63], from initially 12 years to currently over 50 years [3]. Reduced survival was significantly associated with age at diagnosis, lower baseline IgG, higher IgM and fewer peripheral B cells. The risk of death was 11 times higher for patients with non-infectious complications such as lymphoma, chronic hepatitis, structural lung disease and chronic gastrointestinal disease [38].
Thus, the development of better surrogate diagnostic markers for the presence and activity of these secondary complications as well as new therapeutic approaches are a major challenge for the coming years in the care of CVID patients.
Conclusion
CVID represents the most common primary immunodeficiency. Besides an increased susceptibility to infections it frequently presents with signs of autoimmunity, notably autoimmune cytopenias and rheumatic diseases. Its early diagnosis and treatment are important for a favorable outcome. While in most patients susceptibility to infections can be sufficiently covered by immunoglobulin replacement therapy and antibiotics, other manifestations, such as autoimmunity, granulomatous disease, interstitial lung disease, chronic diarrhea, lympho proliferation and developing malignancies, need special attention and treatment, which is best offered in close collaboration between primary care doctors and specialized immunodeficiency centers.
Note
This article is part of the series on Adult immunodeficiency, edited by Hans-Hartmut Peter. Other articles in this series can be found at http://arthritis-research.com/series/immunodeficiency
Abbreviations
CVID: common variable immunodeficiency; DC: dendritic cell; LOCID: late onset combined immunodeficiency; sIgAD: selective IgA deficiency; TLR: Toll-like receptor.
Competing interests
KW received a grant from Baxter. HHP is member of the scientific advisory board of Pfizer's Prevenar development program in Germany.
Contributor Information
Ulrich Salzer, Email: ulrich.salzer@uniklinik-freiburg.de.
Klaus Warnatz, Email: klaus.warnatz@uniklinik-freiburg.de.
Hans Hartmut Peter, Email: hans-hartmut.peter@uniklinik-freiburg.de.
Acknowledgements
The research was funded by grants from the German Research Foundation through the SFB620 projects C1 (KW, HHP) and C7 (US), the 7th European Union framework program grant numer HEALTH-F2-2008-201549 (KW, US), and the Federal Ministry of Education and Research (BMBF 01 EO 0803; KW, HHP, US).
References
- Conley ME. Diagnostic guidelines - An International Consensus document. Clin Immunol. 1999;93:189. doi: 10.1006/clim.1999.4798. [DOI] [PubMed] [Google Scholar]
- Bonilla FA, Bernstein IL, Khan DA, Ballas ZK, Chinen J, Frank MM, Kobrynski LJ, Levinson AI, Mazer B, Nelson RP Jr, Orange JS, Routes JM, Shearer WT, Sorensen RU. American Academy of Allergy; Asthma and Immunology; American College of Allergy; Asthma and Immunology; Joint Council of Allergy; Asthma and Immunology. Practice parameter for the diagnosis and management of primary immunodeficiency. Ann Allergy Asthma Immunol. 2005;94(5 Suppl 1):S1–63. doi: 10.1016/s1081-1206(10)61142-8. [DOI] [PubMed] [Google Scholar]
- Cunningham-Rundles C, Bodian C. Common variable immunodeficiency: clinical and immunological features of 248 patients. Clin Immunol. 1999;92:34–48. doi: 10.1006/clim.1999.4725. [DOI] [PubMed] [Google Scholar]
- Oksenhendler E, Gérard L, Fieschi C, Malphettes M, Mouillot G, Jaussaud R, Viallard JF, Gardembas M, Galicier L, Schleinitz N, Suarez F, Soulas-Sprauel P, Hachulla E, Jaccard A, Gardeur A, Théodorou I, Rabian C, Debré P. DEFI Study Group. Infections in 252 patients with common variable immunodeficiency. Clin Infect Dis. 2008;46:1547–1554. doi: 10.1086/587669. [DOI] [PubMed] [Google Scholar]
- Chapel H, Lucas M, Lee M, Bjorkander J, Webster D, Grimbacher B, Fieschi C, Thon V, Abedi MR, Hammarstrom L. Common variable immunodeficiency disorders: division into distinct clinical phenotypes. Blood. 2008;112:277–286. doi: 10.1182/blood-2007-11-124545. [DOI] [PubMed] [Google Scholar]
- Finck A, Van der Meer JW, Schaffer AA, Pfannstiel J, Fieschi C, Plebani A, Webster AD, Hammarstrom L, Grimbacher B. Linkage of autosomaldominant common variable immunodeficiency to chromosome 4q. Eur J Hum Genet. 2006;14:867–875. doi: 10.1038/sj.ejhg.5201634. [DOI] [PubMed] [Google Scholar]
- Vorechovsky I, Zetterquist H, Paganelli R, Koskinen S, Webster AD, Bjorkander J, Smith CI, Hammarstrom L. Family and linkage study of selective IgA deficiency and common variable immunodeficiency. Clin Immunol Immunopathol. 1995;77:185–192. doi: 10.1006/clin.1995.1142. [DOI] [PubMed] [Google Scholar]
- Espanol T, Catala M, Hernandez M, Caragol I, Bertran JM. Development of a common variable immunodeficiency in IgA-deficient patients. Clin Immunol Immunopathol. 1996;80:333–335. doi: 10.1006/clin.1996.0132. [DOI] [PubMed] [Google Scholar]
- Aghamohammadi A, Farhoudi A, Moin M, Rezaei N, Kouhi A, Pourpak Z, Yaseri N, Movahedi M, Gharagozlou M, Zandieh F, Yazadni F, Arshi S, Mohammadzadeh I, Ghazi BM, Mahmoudi M, Tahaei S, Isaeian A. Clinical and immunological features of 65 Iranian patients with common variable immunodeficiency. Clin Diagn Lab Immunol. 2005;12:825–832. doi: 10.1128/CDLI.12.7.825-832.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kralovicova J, Hammarstrom L, Plebani A, Webster AD, Vorechovsky I. Finescale mapping at IGAD1 and genome-wide genetic linkage analysis implicate HLA-DQ/DR as a major susceptibility locus in selective IgA deficiency and common variable immunodeficiency. J Immunol. 2003;170:2765–2775. doi: 10.4049/jimmunol.170.5.2765. [DOI] [PubMed] [Google Scholar]
- Schaffer AA, Pfannstiel J, Webster AD, Plebani A, Hammarstrom L, Grimbacher B. Analysis of families with common variable immunodeficiency (CVID) and IgA deficiency suggests linkage of CVID to chromosome 16q. Hum Genet. 2006;118:725–729. doi: 10.1007/s00439-005-0101-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder HW Jr, Zhu ZB, March RE, Campbell RD, Berney SM, Nedospasov SA, Turetskaya RL, Atkinson TP, Go RC, Cooper MD, Volanakis JE. Susceptibility locus for IgA deficiency and common variable immunodeficiency in the HLA-DR3, -B8, -A1 haplotypes. Mol Med. 1998;4:72–86. [PMC free article] [PubMed] [Google Scholar]
- Orange JS, Glessner JT, Resnick E, Sullivan KE, Lucas M, Ferry B, Kim CE, Hou C, Wang F, Chiavacci R, Kugathasan S, Sleasman JW, Baldassano R, Perez EE, Chapel H, Cunningham-Rundles C, Hakonarson H. Genome-wide association identifies diverse causes of common variable immunodeficiency. J Allergy Clin Immunol. 2011;127:1360–1367 e1366. doi: 10.1016/j.jaci.2011.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimbacher B, Hutloff A, Schlesier M, Glocker E, Warnatz K, Dräger R, Eibel H, Fischer B, Schäffer AA, Mages HW, Kroczek RA, Peter HH. Homozygous loss of ICOS is associated with adult-onset common variable immunodeficiency. Nat Immunol. 2003;4:261–268. doi: 10.1038/ni902. [DOI] [PubMed] [Google Scholar]
- Castigli E, Wilson SA, Garibyan L, Rachid R, Bonilla F, Schneider L, Geha RS. TACI is mutant in common variable immunodeficiency and IgA deficiency. Nat Genet. 2005;37:829–834. doi: 10.1038/ng1601. [DOI] [PubMed] [Google Scholar]
- Salzer U, Chapel HM, Webster AD, Pan-Hammarström Q, Schmitt-Graeff A, Schlesier M, Peter HH, Rockstroh JK, Schneider P, Schäffer AA, Hammarström L, Grimbacher B. Mutations in TNFRSF13B encoding TACI are associated with common variable immunodeficiency in humans. Nat Genet. 2005;37:820–828. doi: 10.1038/ng1600. [DOI] [PubMed] [Google Scholar]
- van Zelm MC, Reisli I, van der Burg M, Castaño D, van Noesel CJ, van Tol MJ, Woellner C, Grimbacher B, Patiño PJ, van Dongen JJ, Franco JL. An antibody-deficiency syndrome due to mutations in the CD19 gene. N Engl J Med. 2006;354:1901–1912. doi: 10.1056/NEJMoa051568. [DOI] [PubMed] [Google Scholar]
- Warnatz K, Salzer U, Rizzi M, Fischer B, Gutenberger S, Böhm J, Kienzler AK, Pan-Hammarström Q, Hammarström L, Rakhmanov M, Schlesier M, Grimbacher B, Peter HH, Eibel H. B-cell activating factor receptor deficiency is associated with an adult-onset antibody deficiency syndrome in humans. Proc Natl Acad Sci USA. 2009;106:13945–13950. doi: 10.1073/pnas.0903543106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Zelm MC, Smet J, Adams B, Mascart F, Schandené L, Janssen F, Ferster A, Kuo CC, Levy S, van Dongen JJ, van der Burg M. CD81 gene defect in humans disrupts CD19 complex formation and leads to antibody deficiency. J Clin Invest. 2010;120:1265–1274. doi: 10.1172/JCI39748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuijpers TW, Bende RJ, Baars PA, Grummels A, Derks IA, Dolman KM, Beaumont T, Tedder TF, van Noesel CJ, Eldering E, van Lier RA. CD20 deficiency in humans results in impaired T cell-independent antibody responses. J Clin Invest. 2010;120:214–222. doi: 10.1172/JCI40231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiel J, Kimmig L, Salzer U, Grudzien M, Lebrecht D, Hagena T, Draeger R, Völxen N, Bergbreiter A, Jennings S, Gutenberger S, Aichem A, Illges H, Hannan JP, Kienzler AK, Rizzi M, Eibel H, Peter HH, Warnatz K, Grimbacher B, Rump JA, Schlesier M. Genetic CD21 deficiency is associated with hypogammaglobulinemia. J Allergy Clin Immunol. 2012;129:801–810. doi: 10.1016/j.jaci.2011.09.027. [DOI] [PubMed] [Google Scholar]
- Lopez-Herrera G, Tampella G, Pan-Hammarström Q, Herholz P, Trujillo-Vargas CM, Phadwal K, Simon AK, Moutschen M, Etzioni A, Mory A, Srugo I, Melamed D, Hultenby K, Liu C, Baronio M, Vitali M, Philippet P, Dideberg V, Aghamohammadi A, Rezaei N, Enright V, Du L, Salzer U, Eibel H, Pfeifer D, Veelken H, Stauss H, Lougaris V, Plebani A, Gertz EM. et al. Deleterious mutations in LRBA are associated with a syndrome of immune deficiency and autoimmunity. Am J Hum Genet. 2012;90:986–1001. doi: 10.1016/j.ajhg.2012.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giovannetti A, Pierdominici M, Mazzetta F, Marziali M, Renzi C, Mileo AM, De Felice M, Mora B, Esposito A, Carello R, Pizzuti A, Paggi MG, Paganelli R, Malorni W, Aiuti F. Unravelling the complexity of T cell abnormalities in common variable immunodeficiency. J Immunol. 2007;178:3932–3943. doi: 10.4049/jimmunol.178.6.3932. [DOI] [PubMed] [Google Scholar]
- Piqueras B, Lavenu-Bombled C, Galicier L, Bergeron-van der Cruyssen F, Mouthon L, Chevret S, Debre P, Schmitt C, Oksenhendler E. Common variable immunodeficiency patient classification based on impaired B cell memory differentiation correlates with clinical aspects. J Clin Immunol. 2003;23:385–400. doi: 10.1023/A:1025373601374. [DOI] [PubMed] [Google Scholar]
- Warnatz K, Denz A, Drager R, Braun M, Groth C, Wolff-Vorbeck G, Eibel H, Schlesier M, Peter HH. Severe deficiency of switched memory B cells (CD27(+)IgM(-)IgD(-)) in subgroups of patients with common variable immunodeficiency: a new approach to classify a heterogeneous disease. Blood. 2002;99:1544–1551. doi: 10.1182/blood.V99.5.1544. [DOI] [PubMed] [Google Scholar]
- Wehr C, Kivioja T, Schmitt C, Ferry B, Witte T, Eren E, Vlkova M, Hernandez M, Detkova D, Bos PR, Poerksen G, von Bernuth H, Baumann U, Goldacker S, Gutenberger S, Schlesier M, Bergeron-van der Cruyssen F, Le Garff M, Debré P, Jacobs R, Jones J, Bateman E, Litzman J, van Hagen PM, Plebani A, Schmidt RE, Thon V, Quinti I, Espanol T, Webster AD. et al. The EUROclass trial: defining subgroups in common variable immunodeficiency. Blood. 2008;111:77–85. doi: 10.1182/blood-2007-06-091744. [DOI] [PubMed] [Google Scholar]
- De Vera MJ, Al-Harthi L, Gewurz AT. Assessing thymopoiesis in patients with common variable immunodeficiency as measured by T-cell receptor excision circles. Ann Allergy Asthma Immunol. 2004;93:478–484. doi: 10.1016/S1081-1206(10)61416-0. [DOI] [PubMed] [Google Scholar]
- Isgro A, Marziali M, Mezzaroma I, Luzi G, Mazzone AM, Guazzi V, Andolfi G, Cassani B, Aiuti A, Aiuti F. Bone marrow clonogenic capability, cytokine production, and thymic output in patients with common variable immunodeficiency. J Immunol. 2005;174:5074–5081. doi: 10.4049/jimmunol.174.8.5074. [DOI] [PubMed] [Google Scholar]
- Arumugakani G, Wood PM, Carter CR. Frequency of Treg cells is reduced in CVID patients with autoimmunity and splenomegaly and is associated with expanded CD21lo B lymphocytes. J Clin Immunol. 2010;30:292–300. doi: 10.1007/s10875-009-9351-3. [DOI] [PubMed] [Google Scholar]
- Horn J, Manguiat A, Berglund LJ, Knerr V, Tahami F, Grimbacher B, Fulcher DA. Decrease in phenotypic regulatory T cells in subsets of patients with common variable immunodeficiency. Clin Exp Immunol. 2009;156:446–454. doi: 10.1111/j.1365-2249.2009.03913.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melo KM, Carvalho KI, Bruno FR, Ndhlovu LC, Ballan WM, Nixon DF, Kallas EG, Costa-Carvalho BT. A decreased frequency of regulatory T cells in patients with common variable immunodeficiency. PLoS One. 2009;4:e6269. doi: 10.1371/journal.pone.0006269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu GP, Chiang D, Song SJ, Hoyte EG, Huang J, Vanishsarn C, Nadeau KC. Regulatory T cell dysfunction in subjects with common variable immunodeficiency complicated by autoimmune disease. Clin Immunol. 2009;131:240–253. doi: 10.1016/j.clim.2008.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm AM, Sivertsen EA, Tunheim SH, Haug T, Bjerkeli V, Yndestad A, Aukrust P, Froland SS. Gene expression analysis of peripheral T cells in a subgroup of common variable immunodeficiency shows predominance of CCR7(-) effector-memory T cells. Clin Exp Immunol. 2004;138:278–289. doi: 10.1111/j.1365-2249.2004.02630.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viallard JF, Blanco P, Andre M, Etienne G, Liferman F, Neau D, Vidal E, Moreau JF, Pellegrin JL. CD8+HLA-DR+ T lymphocytes are increased in common variable immunodeficiency patients with impaired memory B-cell differentiation. Clin Immunol. 2006;119:51–58. doi: 10.1016/j.clim.2005.11.011. [DOI] [PubMed] [Google Scholar]
- Raeiszadeh M, Kopycinski J, Paston SJ, Diss T, Lowdell M, Hardy GA, Hislop AD, Workman S, Dodi A, Emery V, Webster AD. The T cell response to persistent herpes virus infections in common variable immunodeficiency. Clin Exp Immunol. 2006;146:234–242. doi: 10.1111/j.1365-2249.2006.03209.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm AM, Tjonnfjord G, Yndestad A, Beiske K, Muller F, Aukrust P, Froland SS. Polyclonal expansion of large granular lymphocytes in common variable immunodeficiency - association with neutropenia. Clin Exp Immunol. 2006;144:418–424. doi: 10.1111/j.1365-2249.2006.03086.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malphettes M, Gérard L, Carmagnat M, Mouillot G, Vince N, Boutboul D, Bérezné A, Nove-Josserand R, Lemoing V, Tetu L, Viallard JF, Bonnotte B, Pavic M, Haroche J, Larroche C, Brouet JC, Fermand JP, Rabian C, Fieschi C, Oksenhendler E. DEFI Study Group. Late-onset combined immune deficiency: a subset of common variable immunodeficiency with severe T cell defect. Clin Infect Dis. 2009;49:1329–1338. doi: 10.1086/606059. [DOI] [PubMed] [Google Scholar]
- Resnick ES, Moshier EL, Godbold JH, Cunningham-Rundles C. Morbidity and mortality in common variable immune deficiency over 4 decades. Blood. 2011;119:1650–1657. doi: 10.1182/blood-2011-09-377945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbst EW, Armbruster M, Rump JA, Buscher HP, Peter HH. Intestinal B cell defects in common variable immunodeficiency. Clin Exp Immunol. 1994;95:215–221. doi: 10.1111/j.1365-2249.1994.tb06513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochtrop ML, Goldacker S, May AM, Rizzi M, Draeger R, Hauschke D, Stehfest C, Warnatz K, Goebel H, Technau-Ihling K, Werner M, Salzer U, Eibel H, Schlesier M, Peter HH. T and B lymphocyte abnormalities in bone marrow biopsies of common variable immunodeficiency. Blood. 2011;118:309–318. doi: 10.1182/blood-2010-11-321695. [DOI] [PubMed] [Google Scholar]
- Taubenheim N, von Hornung M, Durandy A, Warnatz K, Corcoran L, Peter HH, Eibel H. Defined blocks in terminal plasma cell differentiation of common variable immunodeficiency patients. J Immunol. 2005;175:5498–5503. doi: 10.4049/jimmunol.175.8.5498. [DOI] [PubMed] [Google Scholar]
- Berglund LJ, Wong SW, Fulcher DA. B-cell maturation defects in common variable immunodeficiency and association with clinical features. Pathology. 2008;40:288–294. doi: 10.1080/00313020801911470. [DOI] [PubMed] [Google Scholar]
- Detková D, de Gracia J, Lopes-da-Silva S, Vendrell M, Alvarez A, Guarner L, Vidaller A, Rodrigo MJ, Caragol I, Espanol T, Hernández M. Common variable immunodeficiency: association between memory B cells and lung diseases. Chest. 2007;131:1883–1889. doi: 10.1378/chest.06-2994. [DOI] [PubMed] [Google Scholar]
- Foerster C, Voelxen N, Rakhmanov M, Keller B, Gutenberger S, Goldacker S, Thiel J, Feske S, Peter HH, Warnatz K. B cell receptor-mediated calcium signaling is impaired in B lymphocytes of type Ia patients with common variable immunodeficiency. J Immunol. 2010;184:7305–7313. doi: 10.4049/jimmunol.1000434. [DOI] [PubMed] [Google Scholar]
- Goldacker S, Draeger R, Warnatz K, Huzly D, Salzer U, Thiel J, Eibel H, Schlesier M, Peter HH. Active vaccination in patients with common variable immunodeficiency (CVID) Clin Immunol. 2007;124:294–303. doi: 10.1016/j.clim.2007.04.011. [DOI] [PubMed] [Google Scholar]
- Ko J, Radigan L, Cunningham-Rundles C. Immune competence and switched memory B cells in common variable immunodeficiency. Clin Immunol. 2005;116:37–41. doi: 10.1016/j.clim.2005.03.019. [DOI] [PubMed] [Google Scholar]
- Andersen P, Permin H, Andersen V, Schejbel L, Garred P, Svejgaard A, Barington T. Deficiency of somatic hypermutation of the antibody light chain is associated with increased frequency of severe respiratory tract infection in common variable immunodeficiency. Blood. 2005;105:511–517. doi: 10.1182/blood-2003-12-4359. [DOI] [PubMed] [Google Scholar]
- Bonhomme D, Hammarstrom L, Webster D, Chapel H, Hermine O, Le Deist F, Lepage E, Romeo PH, Levy Y. Impaired antibody affinity maturation process characterizes a subset of patients with common variable immunodeficiency. J Immunol. 2000;165:4725–4730. doi: 10.4049/jimmunol.165.8.4725. [DOI] [PubMed] [Google Scholar]
- Cunningham-Rundles C, Radigan L, Knight AK, Zhang L, Bauer L, Nakazawa A. TLR9 activation is defective in common variable immune deficiency. J Immunol. 2006;176:1978–1987. doi: 10.4049/jimmunol.176.3.1978. [DOI] [PubMed] [Google Scholar]
- Yu JE, Knight AK, Radigan L, Marron TU, Zhang L, Sanchez-Ramon S, Cunningham-Rundles C. Toll-like receptor 7 and 9 defects in common variable immunodeficiency. J Allergy Clin Immunol. 2009;124:349–356. doi: 10.1016/j.jaci.2009.05.019. 356. e1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu JE, Zhang L, Radigan L, Sanchez-Ramon S, Cunningham-Rundles C. TLR-mediated B cell defects and IFN-alpha in common variable immunodeficiency. J Clin Immunol. 2012;32:50–60. doi: 10.1007/s10875-011-9602-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He B, Santamaria R, Xu W, Cols M, Chen K, Puga I, Shan M, Xiong H, Bussel JB, Chiu A, Puel A, Reichenbach J, Marodi L, Döffinger R, Vasconcelos J, Issekutz A, Krause J, Davies G, Li X, Grimbacher B, Plebani A, Meffre E, Picard C, Cunningham-Rundles C, Casanova JL, Cerutti A. The transmembrane activator TACI triggers immunoglobulin class switching by activating B cells through the adaptor MyD88. Nat Immunol. 2010;11:836–845. doi: 10.1038/ni.1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treml LS, Carlesso G, Hoek KL, Stadanlick JE, Kambayashi T, Bram RJ, Cancro MP, Khan WN. TLR stimulation modifies BLyS receptor expression in follicular and marginal zone B cells. J Immunol. 2007;178:7531–7539. doi: 10.4049/jimmunol.178.12.7531. [DOI] [PubMed] [Google Scholar]
- Bayry J, Lacroix-Desmazes S, Kazatchkine MD, Galicier L, Lepelletier Y, Webster D, Lévy Y, Eibl MM, Oksenhendler E, Hermine O, Kaveri SV. Common variable immunodeficiency is associated with defective functions of dendritic cells. Blood. 2004;104:2441–2443. doi: 10.1182/blood-2004-04-1325. [DOI] [PubMed] [Google Scholar]
- Cunningham-Rundles C, Radigan L. Deficient IL-12 and dendritic cell function in common variable immune deficiency. Clin Immunol. 2005;115:147–153. doi: 10.1016/j.clim.2004.12.007. [DOI] [PubMed] [Google Scholar]
- Wheat WH, Cool CD, Morimoto Y, Rai PR, Kirkpatrick CH, Lindenbaum BA, Bates CA, Ellison MC, Serls AE, Brown KK, Routes JM. Possible role of human herpesvirus 8 in the lymphoproliferative disorders in common variable immunodeficiency. J Exp Med. 2005;202:479–484. doi: 10.1084/jem.20050381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates CA, Ellison MC, Lynch DA, Cool CD, Brown KK, Routes JM. Granulomatous-lymphocytic lung disease shortens survival in common variable immunodeficiency. J Allergy Clin Immunol. 2004;114:415–421. doi: 10.1016/j.jaci.2004.05.057. [DOI] [PubMed] [Google Scholar]
- Ardeniz O, Cunningham-Rundles C. Granulomatous disease in common variable immunodeficiency. Clin Immunol. 2009;133:198–207. doi: 10.1016/j.clim.2009.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels JA, Lederman HM, Maitra A, Montgomery EA. Gastrointestinal tract pathology in patients with common variable immunodeficiency (CVID): a clinicopathologic study and review. Am J Surg Pathol. 2007;31:1800–1812. doi: 10.1097/PAS.0b013e3180cab60c. [DOI] [PubMed] [Google Scholar]
- Ward C, Lucas M, Piris J, Collier J, Chapel H. Abnormal liver function in common variable immunodeficiency disorders due to nodular regenerative hyperplasia. Clin Exp Immunol. 2008;153:331–337. doi: 10.1111/j.1365-2249.2008.03711.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malamut G, Ziol M, Suarez F, Beaugrand M, Viallard JF, Lascaux AS, Verkarre V, Bechade D, Poynard T, Hermine O, Cellier C. Nodular regenerative hyperplasia: the main liver disease in patients with primary hypogammaglobulinemia and hepatic abnormalities. J Hepatol. 2008;48:74–82. doi: 10.1016/j.jhep.2007.08.011. [DOI] [PubMed] [Google Scholar]
- Boileau J, Mouillot G, Gerard L, Carmagnat M, Rabian C, Oksenhendler E, Pasquali JL, Korganow AS. Autoimmunity in common variable immunodeficiency: correlation with lymphocyte phenotype in the French DEFI study. J Autoimmun. 2011;36:25–32. doi: 10.1016/j.jaut.2010.10.002. [DOI] [PubMed] [Google Scholar]
- Quinti I, Soresina A, Spadaro G, Martino S, Donnanno S, Agostini C, Claudio P, Franco D, Maria Pesce A, Borghese F, Guerra A, Rondelli R, Plebani A. Italian Primary Immunodeficiency Network. Long-term follow-up and outcome of a large cohort of patients with common variable immunodeficiency. J Clin Immunol. 2007;27:308–316. doi: 10.1007/s10875-007-9075-1. [DOI] [PubMed] [Google Scholar]
- Sève P, Bourdillon L, Sarrot-Reynauld F, Ruivard M, Jaussaud R, Bouhour D, Bonotte B, Gardembas M, Poindron V, Thiercelin MF, Broussolle C, Oksenhendler E. DEF-I Study Group. Autoimmune hemolytic anemia and common variable immunodeficiency: a case-control study of 18 patients. Medicine (Baltimore) 2008;87:177–184. doi: 10.1097/MD.0b013e31817a90ba. [DOI] [PubMed] [Google Scholar]
- Mouillot G, Carmagnat M, Gérard L, Garnier JL, Fieschi C, Vince N, Karlin L, Viallard JF, Jaussaud R, Boileau J, Donadieu J, Gardembas M, Schleinitz N, Suarez F, Hachulla E, Delavigne K, Morisset M, Jacquot S, Just N, Galicier L, Charron D, Debré P, Oksenhendler E, Rabian C. DEFI Study Group. B-cell and T-cell phenotypes in CVID patients correlate with the clinical phenotype of the disease. J Clin Immunol. 2010;30:746–755. doi: 10.1007/s10875-010-9424-3. [DOI] [PubMed] [Google Scholar]
- Sander CA, Medeiros LJ, Weiss LM, Yano T, Sneller MC, Jaffe ES. Lymphoproliferative lesions in patients with common variable immunodeficiency syndrome. Am J Surg Pathol. 1992;16:1170–1182. doi: 10.1097/00000478-199212000-00004. [DOI] [PubMed] [Google Scholar]
- Mellemkjaer L, Hammarstrom L, Andersen V, Yuen J, Heilmann C, Barington T, Bjorkander J, Olsen JH. Cancer risk among patients with IgA deficiency or common variable immunodeficiency and their relatives: a combined Danish and Swedish study. Clin Exp Immunol. 2002;130:495–500. doi: 10.1046/j.1365-2249.2002.02004.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorechovsky I, Scott D, Haeney MR, Webster DA. Chromosomal radiosensitivity in common variable immune deficiency. Mutat Res. 1993;290:255–264. doi: 10.1016/0027-5107(93)90166-D. [DOI] [PubMed] [Google Scholar]
- Seymour B, Miles J, Haeney M. Primary antibody deficiency and diagnostic delay. J Clin Pathol. 2005;58:546–547. doi: 10.1136/jcp.2004.016204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzi M, Neumann C, Fielding AK, Marks R, Goldacker S, Thaventhiran J, Tarzi MD, Schlesier M, Salzer U, Eibel H, Warnatz K, Finke J, Grimbacher B, Peter HH. Outcome of allogeneic stem cell transplantation in adults with common variable immunodeficiency. J Allergy Clin Immunol. 2011;128:1371–1374.e2. doi: 10.1016/j.jaci.2011.07.055. [DOI] [PubMed] [Google Scholar]
- Ballow M. Immunoglobulin therapy: methods of delivery. J Allergy Clin Immunol. 2008;122:1038–1039. doi: 10.1016/j.jaci.2008.08.012. [DOI] [PubMed] [Google Scholar]
- Gardulf A. Immunoglobulin treatment for primary antibody deficiencies: advantages of the subcutaneous route. BioDrugs. 2007;21:105–116. doi: 10.2165/00063030-200721020-00005. [DOI] [PubMed] [Google Scholar]
- Gardulf A, Andersen V, Bjorkander J, Ericson D, Froland SS, Gustafson R, Hammarstrom L, Jacobsen MB, Jonsson E, Moller G. et al. Subcutaneous immunoglobulin replacement in patients with primary antibody deficiencies: safety and costs. Lancet. 1995;345:365–369. doi: 10.1016/S0140-6736(95)90346-1. [DOI] [PubMed] [Google Scholar]
- Orange JS, Hossny EM, Weiler CR, Ballow M, Berger M, Bonilla FA, Buckley R, Chinen J, El-Gamal Y, Mazer BD, Nelson RP Jr, Patel DD, Secord E, Sorensen RU, Wasserman RL, Cunningham-Rundles C. Primary Immunodeficiency Committee of the American Academy of Allergy; Asthma and Immunology. Use of intravenous immunoglobulin in human disease: a review of evidence by members of the Primary Immunodeficiency Committee of the American Academy of Allergy, Asthma and Immunology. J Allergy Clin Immunol. 2006;117(4 Suppl):S525–553. doi: 10.1016/j.jaci.2006.01.015. [DOI] [PubMed] [Google Scholar]