

Abstract
Recent genome-wide association studies have implicated the tumor necrosis factor receptor-associated factor 3-interacting protein 2 (TRAF3IP2) gene and its product, nuclear factor-kappa-B activator 1 (Act1), in the development of psoriatic arthritis (PsA). The high level of sequence homology of the TRAF3IP2 (Act1) gene across the animal kingdom and the presence of the Act1 protein in multiple cell types strongly suggest that the protein is of importance in normal cellular function. Act1 is an adaptor protein for the interleukin-17 (IL-17) receptor, and recent observations have highlighted the significance of IL-17 signaling and localized inflammation in autoimmune diseases. This review summarizes data from recent genome-wide association studies as well as immunological and molecular investigations of Act1. Together, these studies provide new insight into the role of IL-17 signaling in PsA. It is well established that IL-17 activation of tumor necrosis factor receptor-associated factor 6 (TRAF6) signaling pathways normally leads to nuclear factor-kappa-B-mediated inflammation. However, the dominant PsA-associated TRAF3IP2 (Act1) gene single-nucleotide polymorphism (rs33980500) results in decreased binding of Act1 to TRAF6. This key mutation in Act1 could lead to a greater association of the IL-17 receptor with TRAF2/TRAF5 and this in turn suggests an alternative function for IL-17 in PsA. The recent observations described and discussed in this review raise the clinically significant possibility of redefining the immunological role of IL-17 in PsA and provide a basis for defining future studies to elucidate the molecular and cellular functions of Act1.
Introduction
In 2010, a new association between the tumor necrosis factor receptor-associated factor 3-interacting protein 2 (TRAF3IP2) gene and the immune-mediated diseases psoriatic arthritis (PsA) and psoriatic vulgaris (PsV) was identified by a genome-wide association study (GWAS) [1]. The TRAF3IP2 gene encodes nuclear factor-kappa-B (NF-κB) activator 1 (Act1), an interleukin-17 receptor (IL-17R) adaptor protein. TRAF3IP2/Act1 is also known as CIKS; however, for the purposes of clarity in this review, the gene/protein will be referred to exclusively as Act1. The Act1 gene shows high evolutionary conservation throughout the animal kingdom [1,2], and the protein has been found in a range of cell types both structural (for example, epithelial) and immunological, most notably B cells, T helper (Th) cells [3,4], and innate immune cells such as neutrophils [5]. The sequence conservation of the gene, the presence of the Act1 protein in multiple cell types, and its function as an adaptor protein strongly suggest that Act1 plays an important role in immune cell signaling.
Here, the impact of the PsA-associated variants of Act1 on the molecular interactions of the protein and their known and potential effects on downstream signaling and gene expression are reviewed. Normally, the binding of interleukin-17 (IL-17) to IL-17R leads to Act1 engagement and activation of the tumor necrosis factor receptor-associated factor (TRAF) 6 protein and subsequent NF-κB-mediated release of pro-inflammatory cytokines. Recent studies suggest that PsA-associated modifications of the Act1 protein may result in a greater association of IL-17R activation with an alternative pathway of TRAF2/TRAF5-mediated signaling [1,6] and subsequent changes in the expression of chemokines such as CXCL1 [6]. Collectively, these observations suggest an alternative immunological role of IL-17 in PsA and provide a basis for defining future molecular and cellular studies to establish the normal and disease-associated roles of Act1.
Structure of Act1, an IL-17 receptor adaptor protein encoded by the TRAF3IP2 gene
Act1 is an IL-17R adaptor protein that consists of 574 amino acids (Figure 1). The structure and function of the protein have been characterized by multiple groups [7,8]. Act1 has four domains: a C-terminal SEFIR (similar expression to fibroblast growth factor genes/IL-17R) domain, two TRAF-binding domains, and a helix-loop-helix domain. Several TRAF proteins, including TRAF 2, 3, 5, and 6, have been shown to bind to Act1 and hence Act1 serves to connect the IL-17R to TRAFs and their downstream signaling pathways. Act1-deficient cells fail to respond to IL-17 [3,9], confirming that Act1 plays a key role in the downstream signaling of the IL-17R.
Figure 1.
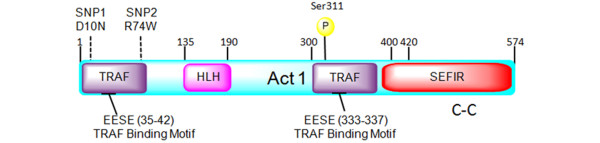
Structure of the Act1 adaptor protein. Act1 contains two TRAF-binding motifs (residues EESE (35 to 42) and EESE (333 to 337)), a helix-loop-helix (HLH) domain (residues 135 to 190), a SEFIR domain (residues 394 to 574), and a coil-coiled (C-C) domain (residues 470 to 500). The SNPs (SNP1 (D10N or Asp10Asn) = rs33980500 and SNP2 (R74W or Arg74Trp) = rs13190932) and the experimentally determined phosphorylation locations are shown above the Act1 domain structure. Act1, nuclear factor-kappa-B activator 1; SEFIR, similar expression to fibroblast growth factor genes/interleukin-17 receptor; SNP, single-nucleotide polymorphism; TRAF, tumor necrosis factor receptor-associated factor.
Single-nucleotide polymorphisms in the TRAF3IP2 gene
The human genome sequencing project, completed in 2003 [10], supported subsequent large-scale GWASs [11]. Differences in single-nucleotide polymorphism (SNP) frequencies between patients and controls have led to the identification of new genetic loci being associated with diseases. Large numbers of patient samples are required for GWASs in order to distinguish common SNPs in a population from SNPs that can be associated with a disease. Once an SNP association has been made, further studies are required to identify the specific gene(s) affected and how the identified genes might contribute to the disease [12]. GWASs have had a major impact on our understanding of a range of autoimmune diseases, including Crohn's disease, psoriasis, inflammatory bowel disease, celiac disease, and multiple sclerosis [13-16]. Here, we have reviewed current knowledge regarding the PsA-associated SNPs in the TRAF3IP2 (Act1) gene and their potential influence on the function of the Act1 protein to give novel insights into potential alterations of IL-17 signaling in PsA.
The PsA GWAS was composed of 609 German patients with PsA and 990 controls which where replicated in six European cohorts. In total, 5,488 individuals were involved in the GWAS [1]. Previously, associations at two well-known susceptibility loci (HLA-C and IL-12B) were replicated, and four new associations at TRAF3IP2 were identified: two intronic variants (rs13196377 and rs13210247) and two coding variants (rs33980500 and rs13190932). This GWAS identified a significant association between the region coding for the TRAF3IP2 (Act1) gene with both PsA and PsV as compared with controls (Figure 2) [1]. The TRAF3IP2 association with psoriasis has been replicated in another GWAS, which involved 2,622 patients with psoriasis and 5,667 controls [16]. Both of the studies suggest that the pathogenesis of psoriasis and PsA may be caused by TRAF3IP2 gene mutations that result in a deregulation of the immune system.
Figure 2.
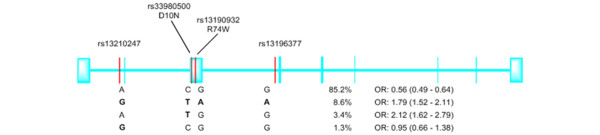
The structure (exon/intron) of the TRAF3IP2/Act1 gene and the location of single-nucleotide polymorphisms from the genome-wide association study. The risk alleles are in bold. For each haplotype, odds ratios (ORs) (and 95% confidence intervals) compare the occurrence of the haplotype versus all three other alleles. The single-nucleotide polymorphisms (rs33980500 and rs13190932) occur within the N-terminal region of TRAF3IP2 (Figure 1). Reproduced with permission from Huffmeier and colleagues [1]. Act1, nuclear factor-kappa-B activator 1; TRAF3IP2, tumor necrosis factor receptor-associated factor 3-interacting protein 2.
From their location alone, it appears that exonic SNPs may directly affect the function of the protein as they occur close to the N-terminal TRAF-binding motif of the Act1 protein. A mammalian-two-hybrid approach was used to measure the effects of PsA-associated SNPs on the binding between Act1 and TRAF6 [1]. Whereas the SNP rs33980500 (Table 1) [1,17] encodes aspartic acid instead of asparagine at position 10 (D10N), and this decreases Act1's ability to bind TRAF6, the rs13190932 SNP (Table 1), which encodes arginine at position 74 in the place of tryptophan (R74W), does not affect TRAF6 binding [1]. As TRAF6 may play a key role in the downstream signaling pathways of the IL-17R, the association of the D10N mutation with PsA suggests that altered Act1-mediated IL-17R signaling may play a role in PsA. More recently, it was confirmed that the rs33980500 (p.D10N) mutation affects the binding of Act1 to TRAF6, and the importance of this mutation was revealed by the observation that other recently discovered SNPs in the TRAF3IP2 gene do not influence this interaction [18].
Table 1.
TRAF3IP2 (Act1) gene chromosome 6q21 single-nucleotide polymorphisms associated with psoriatic arthritis
| Single-nucleotide polymorphism | Locus | Allele | Risk allele | C6q21 position | Exonic/Intronic | Reference(s) |
|---|---|---|---|---|---|---|
| rs33980500 (p.D10N) | TRAF3IP2 | C | T | 111913262 | Exonic | [1,17] |
| rs13196377 | TRAF3IP2 | G | A | 112008531 | Intronic | [1] |
| rs13190932 (p.R74W) | TRAF3IP2 | G | A | 112019763 | Exonic | [1] |
| rs13210247 | TRAF3IP2 | A | G | 112029413 | Intronic | [1,17] |
Act1, nuclear factor-kappa-B activator 1; TRAF3IP2, tumor necrosis factor receptor-associated factor 3-interacting protein 2.
Act1 binding to the IL-17 receptor complex
The IL-17 cytokine family is composed of six separate cytokines (IL-17A to IL-17F). Each of these cytokines has a similar protein structure that includes four highly conserved cysteine residues. In vertebrates, the IL-17R family contains five (IL-17RA to IL-17RE) different receptor subunits [19], all of which are membrane-spanning proteins. The IL-17R subunits differ in their affinities for IL-17 cytokine family members. There are also multiple cytokine/receptor interactions within the IL-17 and IL-17R family. For example, it is known that IL-17A exists in homo-dimeric and hetero-dimeric IL-17A/IL-17F forms which bind to a dimeric receptor complex composed of two IL-17RA subunits or a heterogeneous receptor of IL -17RA and IL-17RC subunits [20,21]. The similarity in IL-17A and IL-17F function is not surprising as they share high sequence homology [22].
Bioinformatic approaches have shown that, similar to Act1, the IL-17R subunits contain a SEFIR domain (Figure 3) [23]. It has been demonstrated experimentally, by using various cell lines and mouse models, that Act1 binds to IL-17RA and IL-17RC through their SEFIR domains [24]. Furthermore, the IL-17R SEFIR domain has sequence and structural similarities to the Toll/IL-1R (TIR) domain [23], which is well characterized as providing specific binding sites for many adaptor proteins, including myeloid differentiation primary response gene 88 (MyD88) and MyD88 associate ligand (Mal), which are crucial for Toll-like receptor (TLR) cell signaling [25].
Figure 3.
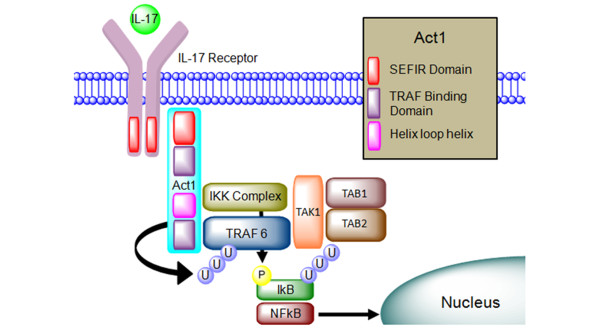
The IL-17R/Act1/TRAF protein complex. The extracellular IL-17R binds the IL-17 cytokines. Act1 proteins bind to the intra-cellular SEFIR motif, which is present on both of the receptors. The Act1 adaptor proteins contain TRAF motifs, which bind different TRAF proteins. Downstream progression leads to activation of pro-inflammatory genes. Act1, nuclear factor-kappa-B activator 1; IKK, IkB kinase; IL-17, interleukin-17; IL-17R, interleukin-17 receptor; NF-κB, nuclear factor-kappa-B; SEFIR, similar expression to fibroblast growth factor genes/interleukin-17 receptor; TAB, transforming growth factor-beta-activated kinase 1 (TAK1)-binding protein; TAK1, transforming growth factor-beta-activated kinase 1; TRAF, tumor necrosis factor receptor-associated factor.
IL-17 receptor and TLR4 signaling
Many proteins have been shown to be required for both IL-17R and TLR4 signaling. The relatively well-understood functions of these proteins in TLR4 signaling pathways give potential insight into how the 'equivalent' proteins may function in IL-17R signaling (Figure 4). TLR4 is an innate immune receptor that is activated by lipopolysaccharides, resulting in an inflammatory response. Once TLR4 is activated, the adaptor proteins MyD88 and Mal bind to the receptor complex through a TIR-TIR interaction [26]. Prior to the interaction of Mal with the TLR, Mal is phosphorylated on tyrosine 86, 106, and 159 by Bruton's tyrosine kinase (Btk) [27,28]. MyD88 binds to the TLR4/Mal protein complex and then itself binds to the interleukin receptor-associated kinase 4 (IRAK4). IRAK4 catalyzes the phosphorylation of itself and IRAK1. IRAK1 subsequently phosphorylates pellino-1 to influence the protein's E3 ubiquitin ligase activity [29]. The phosphorylation and E3 ubiquitin ligase activity of pellino-1 are critical for the formation of a TRAF6/transforming growth factor-beta-kinase 1 (TRAF6-TAK1) complex [30]. Once activated, TRAF6, an E3 ubiquitin ligase, binds to the E2 ubiquitination enzyme complex (UBC13/UEV1A). From the binding of TRAF6 to the E2 complex (UBC13/UEV1A) onwards, the TLR4 and IL-17R signaling pathways are comparable. Thus, the proteins downstream of TRAF6 which lead to the activation of NF-κB, a master transcription factor responsible for the regulation of pro-inflammatory gene expression, have been identified in both the IL-17R [31,32] and TLR [33] signaling pathways.
Figure 4.
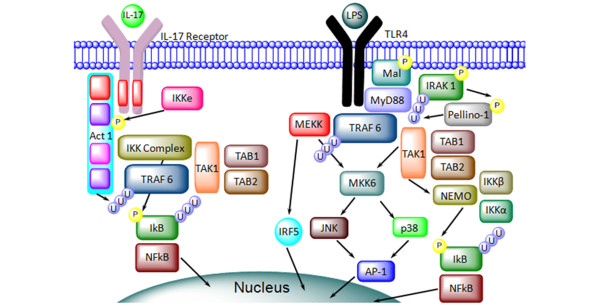
Comparison of the dual roles of TRAF6 in the TLR and IL-17R signaling pathways. Downstream signaling interactions and post translational modifications of proteins involved in the TLR/IL-17R signaling pathways. TRAF6 is a key signaling protein in inflammation. See 'IL-17 receptor and TLR4 signaling' section of text for details. Act1, nuclear factor-kappa-B activator 1; AP-1, activator protein 1; IKK, IkB kinase; IL-17, interleukin-17; IL-17R, interleukin-17 receptor; IRAK4, interleukin receptor associated kinase 4; IRF5, interferon regulatory factor 5; JNK, c-Jun N-terminal kinases; LPS, lipopolysaccharide; Mal, MyD88 associate ligand; MEKK, MAP kinase/ERK kinase kinase; MyD88, myeloid differentiation primary response gene; NEMO, nuclear factor-kappa-B essential modulator protein; NF-κB, nuclear factor-kappa-B; TAB, transforming growth factor-beta-activated kinase 1 (TAK1)-binding protein; TAK1, transforming growth factor-beta-activated kinase 1; TLR, Toll-like receptor; TRAF6, tumor necrosis factor receptor-associated factor 6.
The UBC-TRAF6 complex catalyzes the formation of Lys63-linked polyubiquitin chains on TRAF6 itself and on the IKK-γ protein (which is also known as the NF-κB essential modulator protein, or NEMO) [25]. The ubiquitination of TRAF6 in this complex promotes association with the TAK complex [34], which is composed of TAK1 and the TAK1-binding proteins (TAB2 and TAB3). TAB2 and TAB3 bind to the polyubiquitination chains on IRAK1, which increases the stability of the overall complex [29]. This complex in turn interacts with a number of downstream proteins, including IRF5, MEKK 2/3, and the IKK complex. The IKK complex is composed of two kinases (IKKα and IKKβ) and a regulatory subunit IKKγ (NEMO), and its activation results in the phosphorylation of specific serine residues in IkB proteins. Phosphorylated IkB proteins are subsequently polyubiquitinated and degraded by the 26S proteasome, allowing NF-κB to move into the nucleus, where it becomes active [35].
Although less is known definitively about IL-17R signaling, it seems likely that Act1 plays roles similar to those of the TLR4 adaptor and structural proteins. Thus, Act1 binds to the IL-17R SEFIR domain directly, and this is analogous to Mal's capacity to bind to the TLR4 TIR domain. In the IL-17R and TLR4 signaling pathways, Act1 and pellino-1 (respectively) are phosphorylated by IKKε, and this results in the formation of ubiquitindependent complexes. The IKKε-mediated phosphorylation of Act1 and pellino-1 is likely to be required for the formation of these signaling complexes [6,7,36,37]. The phosphorylation of Act1 regulates its binding to TRAF 2, 5, and 6 [6], and it has been shown that, when Act1 binds to TRAF6, its novel E3 ubiquitin ligase activity [8] results in the ubiquitination of TRAF6 in a non-degradative way [37]. As in TLR4 signaling, TRAF6 ubiquitination is required for the release of NF-κB through the TAK1 and IKK complexes.
In the IL-17R signaling pathway, Act1 binds directly to the IKK complex. This has been confirmed in vitro and by co-immunoprecipitation from cells, including non-transfected cells [38]. The Act1 protein is thought to mainly interact with NEMO/IKKγ in the IKK complex; however, other observations suggest that it may also interact with IKKα and IKKβ [38]. The co-immunoprecipitation experiments used mutated and wild-type Act1 proteins to show that the amino acids 274 to 374 (on Act1) are required for the binding of Act1 to each of the proteins within the IKK complex (NEMO, IKKα, and IKKβ) [38]. The phosphorylation of IKKα and IKKβ at two serine residues in their T-Loop and the ubiquitination and occasionally the phosphorylation of NEMO/IKKγ have been shown to be required for the activation of the IKK complex and the subsequent release of transcription factor NF-κB [39]. The key role of Act1 has been confirmed by downregulation of the Act1 expression, which results in a decrease in the release of the NF-κB into the nucleus [40].
In conclusion, it is apparent that relatively few Act1 protein interactions and altered post-translational modifications have been characterized and so our overall view of the IL-17R signaling pathway is more simplistic when compared with that of the TLR4 signaling pathway. This is not surprising as to date the TLR pathway has received much greater scrutiny, and, of course, it remains possible that future investigations will reveal greater complexities in IL-17R signaling pathways and, more specifically, Act1's role in these pathways.
Function of IL-17 in psoriatic arthritis
There are multiple cell types that express IL-17 in both the innate and adaptive immune systems. Innate immune cells such as macrophages, mast cells, and neutrophils express higher levels of IL-17 in spondyloarthritis, a family of inflammatory rheumatic diseases which includes PsA [5,41]. The main adaptive immune cell associated with the expression of IL-17 is a T-cell subset known as Th17. Increased levels of Th17 cells have been observed in blood, synovial tissue/fluid, and skin samples [42,43] of patients with PsA. Production of the Th-cell subset Th17 is driven by a complex set of different cytokines: IL-1/21/6 and transforming growth factor-beta [20]. Th17 cells produce and release IL-17 (A-F), which binds to and activates the IL-17Rs. The main function of IL-17 is to coordinate local tissue inflammation by upregulating pro-inflammatory and neutrophil-mobilizing chemokines and cytokines. These include IL-6, granulocyte-macro-phage colony-stimulating factor, tumor necrosis factor (TNF), IL-1, CXCL1, CXCL2, CCL7, CCL2, and CCL20 [6]. IL-17Rs are present and functional on epithelial cells in inflamed tissue [21,39].
The upregulation of Th17 cells or increased IL-17 production is generally thought to be pro-inflammatory, particularly in the context of the pathogenesis in auto-immune diseases [44]. This could be associated with the main functions of IL-17 (A/F) in inflammation or alternatively with the functions of IL-17C-IL-17RE. A model for the binding of IL-17C to the IL-17RE receptor was designed to show how this signaling pathway could regulate the functions of Th17 cells in inflamed tissues [45]. Act1 has been associated with the IL-17RE receptor and other receptors such as CD40/BAFF (CD40/B cell-activating factor of TNF family) receptors expressed by B cells. So any alteration in protein interactions caused by the presence of PsA-associated SNPs could potentially modify the functions of many different innate and adaptive immune cells in inflamed tissues [4,45]. Specific to PsA, IL-17 could have a regulatory role on fibroblast-like synoviocytes, osteoblasts, and chondrocytes, impacting both synovial inflammation and joint destruction [43,46,47]. While the pro-inflammatory effects of IL-17 are considerably weaker than those of TNF or IL-1, it has been shown that IL-17 can act synergistically with both proteins to increase significantly the expression of pro-inflammatory genes [37,48,49]. The synergy between IL-17 and TNF suggests that Act1 has functions that are independent of TRAF6-mediated activation of NF-κB.
Role of the TRAF2/5 signaling pathway
Multiple research groups have shown that Act1 interacts with other TRAF proteins (for example, TRAF5 and TRAF2) [6,37,50]. The TRAF2 and TRAF5 proteins are known to play multiple signaling roles in the IL-17R and TNF receptor signaling pathways [51]. TNF inhibitors such as infliximab are widely used as a treatment for PsA [52,53]. In IL-17R and TNF signaling, both proteins seem to have redundant functions, as the downstream effects observed when cells are treated with IL-17/TNF were repressed only if both of the proteins were inhibited [54,55]. The TRAF2/5 pathway is associated with the downstream activation of MAPK, p38, Erk, and mRNA stability of CXCL1 and CXCL2 [6]. CXCL1 is a chemokine expressed by epithelial cells, macrophages, and neutrophils. The main function of CXCL1 is to attract neutrophils, which is a feature found in the skin and synovial tissue in PsA [56]. The inhibition of IKKε, which modifies Act1 to increase TRAF2/5 binding, causes a decrease in the production of pro-inflammatory cytokines/chemokines associated with the synergy between IL-17 and TNF [6]. Pro-inflammatory chemokine mRNA instability is promoted by the protein RNA-splicing-regulatory factor SF2 (alternative splicing factor, or ASF), which is associated with the IL-17 TRAF2/5 signaling pathway (Figure 5). ASF promotes chemokine instability in a sequence-specific way, and the inhibition of ASF prolongs CXCL1 mRNA half-life [50]. For target mRNA, this could be a regulatory mechanism used by cells to prevent overexpression of specific chemokine proteins [50]. ASF is very abundant within cells, but the majority of the protein is located within the nucleus. The TRAF2/5-dependent binding of ASF within the cytoplasm interrupts the selectively enhanced decay of target mRNA. Thus, increased ASF inhibition should cause a larger pro-inflammatory response of epithelial cells to IL-17 [44,45].
Figure 5.
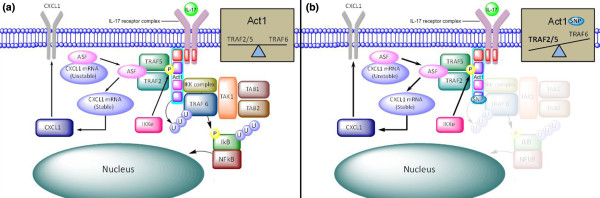
TRAF6 and TRAF2/5 signaling pathways. (a) The endogenous TRAF6 and TRAF2/5 signaling pathways and post-translational modifications. (b) The presence of the psoriatic arthritis-associated single-nucleotide polymorphism (SNP) (rs33980500) could affect the proteins downstream of Act1. The decrease in TRAF6 binding could result in an increase in the TRAF2/5 signaling pathway and this in turn may lead to increased CXCL1 mRNA and protein expression. Act1, nuclear factor-kappa-B activator 1; ASF, alternative splicing factor; IKK, IkB kinase; IL-17, interleukin-17; IL-17R, interleukin-17 receptor; NF-κB, nuclear factor-kappa-B; TAB, transforming growth factor-beta-activated kinase 1 (TAK1)-binding protein; TAK1, transforming growth factor-beta-activated kinase 1; TRAF6, tumor necrosis factor receptor-associated factor 6.
Like TRAF6, the TRAF2/5 proteins are activated by the E3 ligase activity of Act1 [57]. However, TAK proteins do not interact with TRAF5, and this suggests that TRAF5 does not activate NF-κB by the formation of the TRAF-TAK complex [50]. Nonetheless, it is important to note that within TNF signaling the TRAF2/5 proteins have been associated with the release/activation of NF-κB [55]. In the TNF signaling pathway, TRAF5/6 interacts with a wide variety of proteins (NF-κB-inducing kinase (NIK), MAP kinase/ERK kinase kinase 1 (MEKK1), transforming growth factor, atypical protein kinase C, or factor β-activated kinase), and this interaction phosphorylates the IKK complex, leading to the downstream release of NF-κB.
TRAF3IP2 SNP (rs33980500) and the TRAF2/5 IL-17 signaling pathway
IL-17 has multiple functions in an immune response. Some of these functions are mediated through TRAF2 and TRAF5 proteins that are independent of the TRAF6 signaling pathway. The IkB kinase IKKε is required for the formation of an Act1-TRAF2 and TRAF5 complex [6]. In IKKε- deficient cells, the genes associated with the TRAF2 and TRAF5 pathway are not upregulated in response to IL-17, while the TRAF6-dependent signaling pathway is retained [6]. It has been demonstrated in mice that IKKε phosphorylates Act1 on a specific serine (Ser311) located near the TRAF-binding motif (Figure 1) [6]. Phosphorylation of Act1 induced by IL-17 could be a means of regulating the TRAF2/5 signaling pathway. Computer modeling has been used to analyze how the phosphorylation of serine 311 could affect the interactions between the Act1 and TRAF2/5/6. It was predicted that the phosphorylation of serine 311 could cause the formation of a salt bridge to increase the attraction between the TRAF-binding domains of Act1 and TRAF2/5 [6]. Though not confirmed experimentally, this suggests that phosphorylation plays a role in the balance of TRAF protein interactions with Act1. Mouse models have shown that IKKε is required for IL-17-induced expression of genes encoding inflammatory molecules (Cxcl1, Cxcl2, TNF, IL-6, and Csf3) in epithelial cells. This suggests that IKKε has a function in the inflammatory response to IL-17 [6]. The study also demonstrated that the inhibition of IKKε affected the synergy between IL-17 and TNF. The SNP (rs33980500) decreases the binding between Act1 and TRAF6 and this could also alter TRAF2 and TRAF5 protein interactions within the IL-17R signaling pathway (Figure 5).
Because Act1-binding motifs have been associated with multiple TRAF proteins, the inhibition of the TRAF6 pathway could upregulate the TRAF2/5 pathway, leading to increased neutrophil chemotaxis and an enhanced immune response. Further study is required in order to obtain a clearer understanding of how the signaling pathways function in epithelial cells, in both controls and those with PsA-associated SNPs.
Conclusions
In summary, it is well established that there are multiple IL-17 ligands and receptors, and although each of the proteins is unique, they share some homology and have redundant functions. Act1 is the main adaptor protein that has been associated with IL-17Rs, and there is good evidence to suggest that Act1 plays an important role in immune cell signaling. The structure of the Act1 adaptor protein includes a SEFIR domain, a domain also found on each of the IL-17Rs and through which Act1 is thought to interact. This suggests a functional connection between Act1 and each of the IL-17Rs.
Act1 binds to and activates TRAF6. In the context of TLR4 signaling, the structure, function, and protein interaction partners of TRAF6 have been well characterized. A comparison of the TLR4 and IL-17R pathways (Figure 4) indicates that many proteins are involved in both of the pathways and suggests that they may have similar functions. Notably, TRAF6 has a key role in both signaling pathways, resulting in the release of NF-κB and a pro-inflammatory response. Hence, it is logical to suggest that IL-17R signaling and, in particular, TRAF6 could contribute to the inflammation observed in both skin and synovial tissue in PsA.
However, recent GWASs have identified an SNP (rs33980500) that is in the Act1 gene region and that is associated with PsA but that decreases the binding of TRAF6 to Act1. A decrease in the binding of TRAF6 to Act1 would result in a decreased phosphorylation and ubiquitination of the NF-κB inhibitor protein IkB, reduced NF-κB activation, and a more limited pro-inflammatory response. This is contradictory to the inflammatory role that the IL-17 signaling pathway apparently plays in PsA. An alternative signaling pathway may explain the discrepancy. Here, we suggest that the Act1 SNPs change the balance of the IL-17R signaling through TRAF proteins to increase the role of TRAF2 and TRAF5 (Figure 5) and this could redefine the way in which IL-17 signals in PsA.
It is important to note that, although the Act1 SNPs have been associated with PsA by GWASs, the percentage of patients who express the SNP is low (12%). This raises the important question of whether in PsA there is altered TRAF2/5-mediated signaling only in patients who express the Act1 SNP (rs33980500). It remains an intriguing possibility that in PsA there are mutations/alterations in other components of IL-17R signaling pathways that change the balance of TRAF6 versus TRAF2/5 in response to IL-17. Regardless of this possibility, the GWASs suggest that IL-17R signaling is significant in the development of PsA. We suggest that additional studies focus on the relative roles of the TRAF proteins in IL-17R signaling. Detailed investigation of Act1 and TRAF expression, protein interactions, and post-translational modifications may reveal key information on the mechanisms and pathogenesis of PsA. It is apparent, from clinical and pharmaceutical perspectives, that Act1 and the specific TRAF proteins involved in IL-17R signaling could be targets for therapeutic intervention. Although we have emphasized the key role the IL-17R pathway has in inflammatory responses in PsA, other cytokines, receptors, and signaling pathways also contribute to the inflammatory responses in the disease. Future studies to investigate how PsA-associated SNPs influence Act1's protein interactions and their associated post-trans-lational modifications (including phosphorylation and ubiquitination) should provide a greater insight into pro-inflammatory signaling pathways.
Abbreviations
Act1: nuclear factor-kappa-B activator 1; ASF: alternative splicing factor; GWAS: genome-wide association study; IKK: IkB kinase; IL-17: interleukin-17; IL-17R: interleukin-17 receptor; IRAK4: interleukin receptor-associated kinase 4; Mal: myeloid differentiation primary response gene (MyD88) associate ligand; MEKK: MAP kinase/ERK kinase kinase; MyD88: myeloid differentiation primary response gene; NEMO: nuclear factor-kappa-B essential modulator protein; NF-κB: nuclear factor-kappa-B; PsA: psoriatic arthritis; PsV: psoriatic vulgaris; SEFIR: similar expression to fibroblast growth factor genes/interleukin-17 receptor; SNP: single-nucleotide polymorphism; TAB: transforming growth factor-beta-activated kinase 1 (TAK1)-binding protein; TAK: transforming growth factor-beta-activated kinase; Th: T helper; TIR: Toll/interleukin-1 receptor; TLR: Toll-like receptor; TNF: tumor necrosis factor; TRAF: tumor necrosis factor receptor-associated factor; TRAF3IP2: tumor necrosis factor receptor-associated factor 3-interacting protein 2.
Competing interests
The authors declare that they have no competing interests.
Contributor Information
Matthew S Doyle, Email: matthew.doyle@ucdconnect.ie.
Emily S Collins, Email: emily.collins75@gmail.com.
Oliver M FitzGerald, Email: Oliver.Fitzgerald@ucd.ie.
Stephen R Pennington, Email: Stephen.pennington@ucd.ie.
Acknowledgements
MSD, OMF and SRP acknowledge support from the Health EducationA Ireland under the Molecular and Cellular Underlying Inflammatory Processes Structured PhD programme funded though PRTLI-V.
References
- Hüffmeier U, Uebe S, Ekici AB, Bowes J, Giardina E, Korendowych E, Juneblad K, Apel M, McManus R, Ho P, Bruce IN, Ryan AW, Behrens F, Lascorz J, Böhm B, Traupe H, Lohmann J, Gieger C, Wichmann HE, Herold C, Steff ens M, Klareskog L, Wienker TF, Fitzgerald O, Alenius GM, McHugh NJ, Novelli G, Burkhardt H, Barton A, Reis A. Common variants at TRAF3IP2 are associated with susceptibility to psoriatic arthritis and psoriasis. Nat Genet. 2010;42:996–999. doi: 10.1038/ng.688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryzhakov G, Blazek K, Udalova IA. Evolution of vertebrate immunity: sequence and functional analysis of the SEFIR domain family member Act1. J Mol Evol. 2011;72:521–530. doi: 10.1007/s00239-011-9450-7. [DOI] [PubMed] [Google Scholar]
- Chang SH, Park H, Dong C. Act1 adaptor protein is an immediate and essential signaling component of interleukin-17 receptor. J Biol Chem. 2006;281:35603–35607. doi: 10.1074/jbc.C600256200. [DOI] [PubMed] [Google Scholar]
- Qian Y, Qin J, Cui G, Naram ura M, Snow EC, Ware CF, Fairchild RL, Omori SA, Rickert RC, Scott M, Kotzin BL, Li X. Act1, a negative regulator in CD40- and BAFF-mediated B cell survival. Immunity. 2004;21:575–587. doi: 10.1016/j.immuni.2004.09.001. [DOI] [PubMed] [Google Scholar]
- Ambarus C, Yeremenko N, Tak PP, Baeten D. Pathogenesis of spondyloarthritis: autoimmune or autoinflammatory? Curr Opin Rheumatol. 2012;24:351–358. doi: 10.1097/BOR.0b013e3283534df4. [DOI] [PubMed] [Google Scholar]
- Bulek K, Liu C, Swaidani S, Wang L, Page RC, Gulen MF, Herjan T, Abbadi A, Qian W, Sun D, Lauer M, Hascall V, Misra S, Chance MR, Aronica M, Hamilton T, Li X. The inducible kinase IKKi is required for IL-17-dependent signaling associated with neutrophilia and pulmonary inflammation. Nat Immunol. 2011;12:844–852. doi: 10.1038/ni.2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonder SU, Saret S, Tang W, Sturdevant DE, Porcella SF, Siebenlist U. IL-17-induced NF- B Activation via CIKS/Act1: physiologic significance and signaling mechanisms. J Biol Chem. 2011;286:12881–12890. doi: 10.1074/jbc.M110.199547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Qian W, Qian Y, Gilt iay NV, Lu Y, Swaidani S, Misra S, Deng L, Chen ZJ, Li X. Act1, a U-box E3 ubiquitin ligase for IL-17 signaling. Sci Signal. 2009;2:ra63. doi: 10.1126/scisignal.2000382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian Y. Role of NFkappa B activator Act1 in CD40-mediated signaling in epithelial cells. Proc Natl Acad Sci USA. 2002;99:9386–9391. doi: 10.1073/pnas.142294499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashizaki Y. Discovery of the 'RNA continent' through a contrarian research strategy. Genes Genet Syst. 2011;86:221–229. doi: 10.1266/ggs.86.221. [DOI] [PubMed] [Google Scholar]
- Naidoo N, Pawitan Y, Soong R, Cooper DN, Ku CS. Human genetics and genomics a decade after the release of the draft sequence of the human genome. Hum Genomics. 2011;5:577–622. doi: 10.1186/1479-7364-5-6-577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cookson W, Liang L, Abecasis G, Moffatt M, Lathrop M. Mapping complex disease traits with global gene expression. Nat Rev Genet. 2009;10:184–194. doi: 10.1038/nrg2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke A, McGovern DP, Barrett JC, Wang K, Radford-Smith GL, Ahmad T, Lees CW, Balschun T, Lee J, Roberts R, Anderson CA, Bis JC, Bumpstead S, Ellinghaus D, Festen EM, Georges M, Green T, Haritunians T, Jostins L, Latiano A, Mathew CG, Montgomery GW, Prescott NJ, Raychaudhuri S, Rotter JI, Schumm P, Sharma Y, Simms LA, Taylor KD, Whiteman D. et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn's disease susceptibility loci. Nat Genet. 2010;42:1118–1125. doi: 10.1038/ng.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coenen MJ, Trynka G, Heskamp S, Franke B, van Diemen CC, Smolonska J, van Leeuwen M, Brouwer E, Boezen MH, Postma DS, Platteel M, Zanen P, Lammers JW, Groen HJ, Mali WP, Mulder CJ, Tack GJ, Verbeek WH, Wolters VM, Houwen RH, Mearin ML, van Heel DA, Radstake TR, van Riel PL, Wijmenga C, Barrera P, Zhernakova A. Common and different genetic background for rheumatoid arthritis and coeliac disease. Hum Mol Genet. 2009;18:4195–4203. doi: 10.1093/hmg/ddp365. [DOI] [PubMed] [Google Scholar]
- Hoffjan S, Akkad DA. The genetics of multiple sclerosis: an update 2010. Mol Cell Probes. 2010;24:237–243. doi: 10.1016/j.mcp.2010.04.006. [DOI] [PubMed] [Google Scholar]
- Genetic Analysis of Psoriasis Consortium & the Wellcome Trust Case Control Consortium 2; Strange A, Capon F, Spencer CC, Knight J, Weale ME, Allen MH, Barton A, Band G, Bellenguez C, Bergboer JG, Blackwell JM, Bramon E, Bumpstead SJ, Casas JP, Cork MJ, Corvin A, Deloukas P, Dilthey A, Duncanson A, Edkins S, Estivill X, Fitzgerald O, Freeman C, Giardina E, Gray E, Hofer A, Hüffmeier U, Hunt SE, Irvine AD. et al. A genome-wide association study identifies new psoriasis susceptibility loci and an interaction between HLA-C and ERAP1. Nat Genet. 2010;42:985–990. doi: 10.1038/ng.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellinghaus E, Ellinghaus D, Stuart PE, Nair RP, Debrus S, Raelson JV, Belouchi M, Fournier H, Reinhard C, Ding J, Li Y, Tejasvi T, Gudjonsson J, Stoll SW, Voorhees JJ, Lambert S, Weidinger S, Eberlein B, Kunz M, Rahman P, Gladman DD, Gieger C, Wichmann HE, Karlsen TH, Mayr G, Albrecht M, Kabelitz D, Mrowietz U, Abecasis GR, Elder JT, Schreiber S, Weichenthal M, Franke A. Genome-wide association study identifies a psoriasis susceptibility locus at TRAF3IP2. Nat Genet. 2010;42:991–995. doi: 10.1038/ng.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böhm B, Burkhardt H, Uebe S , Apel M, Behrens F, Reis A, Hüffmeier U. Identification of low-frequency TRAF3IP2 coding variants in psoriatic arthritis patients and functional characterization. Arthritis Res Ther. 2012;14:R84. doi: 10.1186/ar3807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaffen SL. Structure and signalling in the IL-17 receptor family. Nat Rev Immunol. 2009;9:556–567. doi: 10.1038/nri2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaffen S. An overview of IL-17 function and signaling. Cytokine. 2008;43:402–407. doi: 10.1016/j.cyto.2008.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter CA. Act1-ivating IL-17 inflammation. Nature. 2007;8:232–234. doi: 10.1038/ni0307-232. [DOI] [PubMed] [Google Scholar]
- Chang SH, Dong C. IL-17F: regulation, signaling and function in inflammation. Cytokine. 2009;46:7–11. doi: 10.1016/j.cyto.2008.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novatchkova M, Leibbrandt A, Werzowa J, Neubüser A, Eisenhaber F. The STIR-domain superfamily in signal transduction, development and immunity. Trends Biochem Sci. 2003;28:226–229. doi: 10.1016/S0968-0004(03)00067-7. [DOI] [PubMed] [Google Scholar]
- Chang SH, Dong C. Signaling of interleukin-17 family cytokines in immunity and inflammation. Cell Signal. 2011;23:1069–1075. doi: 10.1016/j.cellsig.2010.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- Lin Z, Lu J, Zhou W, Shen Y. Structural insights into TIR domain specificity of the bridging adaptor Mal in TLR4 signaling. PLoS ONE. 2012;7:e34202. doi: 10.1371/journal.pone.0034202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenny EF, O'Neill LA. Signalling adaptors used by Toll-like receptors: an update. Cytokine. 2008;43:342–349. doi: 10.1016/j.cyto.2008.07.010. [DOI] [PubMed] [Google Scholar]
- Gray P, Dunne A, Brikos C, Jefferies CA, Doyle SL, O'Neill LA. MyD88 adapterlike (Mal) is phosphorylated by Bruton's tyrosine kinase during TLR2 and TLR4 signal transduction. J Biol Chem. 2006;281:10489–10495. doi: 10.1074/jbc.M508892200. [DOI] [PubMed] [Google Scholar]
- Moynagh PN. The Pellino family: IRAK E3 ligases with emerging roles in innate immune signalling. Trends Immunol. 2009;30:33–42. doi: 10.1016/j.it.2008.10.001. [DOI] [PubMed] [Google Scholar]
- Smith H, Peggie M, Campbell DG , Vandermoere F, Carrick E, Cohen P. Identification of the phosphorylation sites on the E3 ubiquitin ligase Pellino that are critical for activation by IRAK1 and IRAK4. Proc Natl Acad Sci USA. 2009;106:4584–4590. doi: 10.1073/pnas.0900774106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X. Act1 modulates autoimmunity through its dual functions in CD40L/BAFF and IL-17 signaling. Cytokine. 2008;41:105–113. doi: 10.1016/j.cyto.2007.09.015. [DOI] [PubMed] [Google Scholar]
- Zepp J, Wu L, Li X. IL-17 receptor signaling and T helper 17-mediated autoimmune demyelinating disease. Trends Immunol. 2011;32:232–239. doi: 10.1016/j.it.2011.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Chen ZJ. The novel functions of ubiquitination in signaling. Curr Opin Cell Biol. 2004;16:119–126. doi: 10.1016/j.ceb.2004.02.005. [DOI] [PubMed] [Google Scholar]
- Prickett TD, Ninomiya-Tsuji J, Broglie P, Muratore-Schroeder TL, Shabanowitz J, Hunt DF, Brautigan DL. TAB4 stimulates TAK1-TAB1 phosphorylation and binds polyubiquitin to direct signaling to NF-κB. J Biol Chem. 2008;283:19245–19254. doi: 10.1074/jbc.M800943200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T, Akira S. Signaling to NF-κB by Toll-like receptors. Trends Mol Med. 2007;13:460–469. doi: 10.1016/j.molmed.2007.09.002. [DOI] [PubMed] [Google Scholar]
- Smith H, Liu XY, Dai L, Goh ET, Chan AT, Xi J, Seh CC, Qureshi IA, Lescar J, Ruedl C, Gourlay R, Morton S, Hough J, McIver EG, Cohen P, Cheung PC. The role of TBK1 and IKKε in the expression and activation of Pellino 1. Biochem. 2011. pp. 537–548. [DOI] [PMC free article] [PubMed]
- May MJ. IL-17R signaling new players get in on the Act1. Nat Immunol. 2011;12:813–815. doi: 10.1038/ni.2093. [DOI] [PubMed] [Google Scholar]
- Leonardi A, Chariot A, Claudio E, Cunningham K, Siebenlist U. CIKS, a connection to IκB kinase and stress-activated protein kinase. Proc Natl Acad Sci USA. 2000;97:10494–10499. doi: 10.1073/pnas.190245697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palkowitsch L, Leidner J, Ghosh S, Marienfeld RB. Phosphorylation of serine 68 in the IκB kinase (IKK)-binding domain of NEMO interferes with the structure of the IKK complex and tumor necrosis factor-α-induced NF-κB activity. J Biol Chem. 2008;283:76–86. doi: 10.1074/jbc.M708856200. [DOI] [PubMed] [Google Scholar]
- Matsushima Y, Kikkawa Y, Takada T, Matsuoka K, Seki Y, Yoshida H, Minegishi Y, Karasuyama H, Yonekawa H. An atopic dermatitis-like skin disease with hyper-IgE-emia develops in mice carrying a spontaneous recessive point mutation in the Traf3ip2 (Act1/CIKS) gene. J Immunol. 2010;185:2340–2349. doi: 10.4049/jimmunol.0900694. [DOI] [PubMed] [Google Scholar]
- Noordenbos T, Yeremenko N, Gofita I, van de Sande M, Tak PP, Caňete JD, Baeten D. Interleukin-17-positive mast cells contribute to synovial inflammation in spondylarthritis. Arthritis Rheum. 2012;64:99–109. doi: 10.1002/art.33396. [DOI] [PubMed] [Google Scholar]
- Jandus C, Bioley G, Rivals JP, Dudler J, Speiser D, Romero P. Increased numbers of circulating polyfunctional Th17 memory cells in patients with seronegative spondylarthritides. Arthritis Rheum. 2008;58:2307–2317. doi: 10.1002/art.23655. [DOI] [PubMed] [Google Scholar]
- Raychaudhuri S. Role of IL-17 in psoriasis and psoriatic arthritis. Clin Rev Allergy Immunol. 2012. in press . [DOI] [PubMed]
- Leipe J, Grunke M, Dechant C, Reindl C, Kerzendorf U, Schulze-Koops H, Skapenko A. Role of Th17 cells in human autoimmune arthritis. Arthritis Rheum. 2010;62:2876–2885. doi: 10.1002/art.27622. [DOI] [PubMed] [Google Scholar]
- Chang SH, Reynolds JM, Pappu BP, Chen G, Martinez GJ, Dong C. Interleukin-17C promotes Th17 cell responses and autoimmune disease via interleukin-17 receptor E. Immunity. 2011;35:611–621. doi: 10.1016/j.immuni.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raychaudhuri S, Raychaudhuri S, Genovese M. IL-17 receptor and its functional significance in psoriatic arthritis. Mol Cell Biochem. 2012;359:419–429. doi: 10.1007/s11010-011-1036-6. [DOI] [PubMed] [Google Scholar]
- Benderdour M, Tardif G, Pelletier JP, Di Battista JA, Reboul P, Ranger P, Martel- Pelletier J. Interleukin 17 (IL-17) induces collagenase-3 production in human osteoarthritic chondrocytes via AP-1 dependent activation: differential activation of AP-1 members by IL-17 and IL-1beta. J Rheumatol. 2002;29:1262–1272. [PubMed] [Google Scholar]
- Hartupee J, Liu C, Novotny M, Sun D, Li X, Hamilton TA. IL-17 signaling for mRNA stabilization does not require TNF receptor-associated factor 6. J Immunol. 2009;182:1660–1666. doi: 10.4049/jimmunol.182.3.1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hot A, Lenief V, Miossec P. Combination of IL-17 and TNFα induces a pro-inflammatory, pro-coagulant and pro-thrombotic phenotype in human endothelial cells. Ann Rheum Dis. 2012;71:768–776. doi: 10.1136/annrheumdis-2011-200468. [DOI] [PubMed] [Google Scholar]
- Sun D, Novotny M, Bulek K, Liu C, Li X, Hamilton T. Treatment with IL-17 prolongs the half-life of chemokine CXCL1 mRNA via the adaptor TRAF5 and the splicing-regulatory factor SF2 (ASF) Nat Immunol. 2011;12:853–860. doi: 10.1038/ni.2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha H, Han D, Choi Y. TRAF-mediated TNFR-family signaling. Current Protocols in Immunology. 2009;87:11.9D.1–11.9D.19. doi: 10.1002/0471142735.im1109ds87. [DOI] [PubMed] [Google Scholar]
- Ogilvie AL, Antoni C, Dechant C, Manger B, Kalden JR, Schuler G, Luftl M. Treatment of psoriatic arthritis with antitumour necrosis factor-alpha antibody clears skin lesions of psoriasis resistant to treatment with methotrexate. Br J Dermatol. 2001;144:587–589. doi: 10.1046/j.1365-2133.2001.04089.x. [DOI] [PubMed] [Google Scholar]
- D'Angelo S, Palazzi C, Olivieri I. Psoriatic arthritis: treatment strategies using biologic agents. Reumatismo. 2012;64:113–121. doi: 10.4081/reumatismo.2012.113. [DOI] [PubMed] [Google Scholar]
- Au PY, Yeh WC. In: Advances in Experimental Medicine and Biology. Wu H, editor. Vol. 597. New York: Springer; 2007. Physiological roles and mechanisms of signaling by TRAF2 and TRAF5 TNF receptor associated factors (TRAFs) pp. 32–47. [DOI] [PubMed] [Google Scholar]
- Tada K, Okazaki T, Sakon S, Kobarai T, Kurosawa K, Yamaoka S, Hashimoto H, Mak TW, Yagita H, Okumura K, Yeh WC, Nakano H. Critical roles of TRAF2 and TRAF5 in tumor necrosis factor-induced NF-κB activation and protection from cell death. J Biol Chem. 2001;276:36530–36534. doi: 10.1074/jbc.M104837200. [DOI] [PubMed] [Google Scholar]
- Kruithof E, Baeten D, De Rycke L, Vandooren B, Foell D, Roth J, Canete J, Boots A, Veys E, De Keyser F. Synovial histopathology of psoriatic arthritis, both oligo- and polyarticular, resembles spondyloarthropathy more than it does rheumatoid arthritis. Arthritis Res Ther. 2005;7:R569–R580. doi: 10.1186/ar1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempsey PW, Doyle SE, He JQ, Cheng G. The signaling adaptors and pathways activated by TNF superfamily. Cytokine Growth Factor Rev. 2003;14:193–209. doi: 10.1016/S1359-6101(03)00021-2. [DOI] [PubMed] [Google Scholar]