

Abstract
Immunoglobulin class-switch recombination deficiencies (Ig-CSR-Ds) are rare primary immunodeficiencies characterized by defective switched isotype (IgG/IgA/IgE) production. Depending on the molecular defect in question, the Ig-CSR-D may be combined with an impairment in somatic hypermutation (SHM). Some of the mechanisms underlying Ig-CSR and SHM have been described by studying natural mutants in humans. This approach has revealed that T cell-B cell interaction (resulting in CD40-mediated signaling), intrinsic B-cell mechanisms (activation-induced cytidine deaminase-induced DNA damage), and complex DNA repair machineries (including uracil-N-glycosylase and mismatch repair pathways) are all involved in class-switch recombination and SHM. However, several of the mechanisms required for full antibody maturation have yet to be defined. Elucidation of the molecular defects underlying the diverse set of Ig-CSR-Ds is essential for understanding Ig diversification and has prompted better definition of the clinical spectrum of diseases and the development of increasingly accurate diagnostic and therapeutic approaches.
Introduction
The study of inherited immunoglobulin class-switch recombination deficiencies (Ig-CSR-Ds) has contributed greatly to our understanding of normal antibody maturation processes. These syndromes all involve a defect in Ig-CSR, as demonstrated by normal or elevated serum IgM levels and null or strongly reduced levels of the other Ig isotypes. Somatic hypermutation (SHM) leads to the production of antibodies with a high affinity for antigen. This event usually takes place in the secondary lymphoid organs (the spleen, lymph nodes, and tonsils) in an antigen- and T lymphocyte-dependent manner. When mature but naïve IgM+IgD+ B cells emigrating from the bone marrow (or fetal liver) encounter an antigen that is specifically recognized by their B-cell receptor (BCR), they proliferate vigorously and give rise to a unique lymphoid formation: the germinal center. Here, B cells undergo the two major maturation events required for an efficient humoral response: CSR and SHM (Figure 1a).
Figure 1.
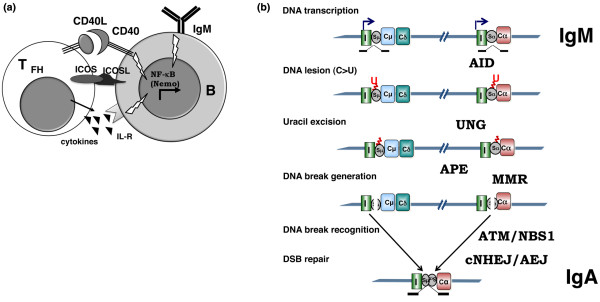
Schematic representation of class-switch recombination in germinal center. (a) T cell-B cell cooperation in immunoglobulin class-switch recombination (Ig-CSR). Impaired function of CD40 and CD40L (CD40 ligand) and, to a lesser extent, inducible co-stimulator (ICOS) and nuclear factor-kappa-B (NF-κB) essential modulator (NEMO) results in class-switch recombination deficiency (CSR-D). B, B cell; IL-R, interleukin receptor; TFH, T follicular helper. (b) Schematic representation of Ig-CSR. Only part of the IgH locus with I (I exon), S (switch region), and C (constant region) is depicted. The intermediate steps of CSR are shown on the left. Impaired function of activation-induced cytidine deaminase (AID), uracil-N-glycosylase (UNG), mismatch repair (MMR), ataxia telangiectasia mutated (ATM), Nijmegen breakage syndrome (NBS1), and conventional non-homologous end joining (cNHEJ) results in variable CSR-D. AEJ, alternative end joining; APE, AP endonuclease; DSB, double-stranded DNA break; U, uracil.
CSR involves DNA recombination between two different switch (S) regions located upstream of the constant regions, whereas the intervening DNA is deleted by formation of an excision circle [1-5]. Replacement of the constant μ region by a downstream constant × region from another class of Ig results in the production of antibodies of different isotypes (IgG, IgA, and IgE) with the same variable (V) region and thus the same antigen specificity and affinity (Figure 1b). The different Ig isotypes vary in terms of their properties (their half-life, affinity for Fc receptors, ability to activate the complement system, and so on) and tissue location (IgA is secreted by mucosal membranes, for example).
SHM introduces missense mutations and (less frequently) deletions or insertions into the V regions of Igs. This process is triggered by activation of the BCR and CD40 [6,7]. The mutations occur at a high frequency within the V regions and their proximal flanking regions (approximately one mutation per 1 × 103 bases). SHM is required as a basis for the selection and proliferation of B cells expressing a BCR with a high affinity for antigen, following interaction with follicular dendritic cells [8,9].
Although CSR and SHM occur simultaneously in B cells in germinal centers after CD40 activation, neither is a prerequisite for the other: in some cases, IgM is mutated when IgG or IgA is not mutated [10,11]. The mechanisms underlying these two antibody maturation processes have been elucidated (at least in part). The first step is transcription of target DNA in the S and V regions. Cytokines induce sterile transcription of the S regions [5,12]. This transcription step enables activation-induced cytidine deaminase (AID) to gain access to the DNA [13-17]. This B-cell molecule controls antibody maturation [18,19] by selectively changing cytosine (C) residues into uracil (U) residues in the transcribed S and V regions [20]. Uracil-N-glycosylase 2 (UNG2) removes the uracil residues created by AID [21] and thus produces an abasic site. In the mouse, abasic sites have been shown to be cleaved eventually by apurinic/apyrimidinic endonucleases (APEs) [22]. This ultimately leads to the formation of single-stranded DNA breaks that, if present on both strands of the DNA, result in the formation of the double-stranded breaks (DSBs) required for CSR. Indeed, it has been shown that AID can deaminate the nontemplate and template strands in transcription bubbles [17]. Recently, the RNA exosome was reported to be involved in AID targeting on both DNA strands [23]. The DNA lesions in the S and V regions are repaired by different mechanisms. In S regions, most of the Ig-CSR-induced DSBs are repaired through the conventional non-homologous end joining (c-NHEJ) pathway [24,25]. However, a recently described alternative end joining (AEJ) pathway can also perform microhomology-mediated repair [26]. The error-prone repair of SHM-induced DNA lesions does not require NHEJ [27] but does involve the MSH2/MSH6 complex - a component of the mismatch repair (MMR) machinery - and error-prone DNA polymerases [28].
Defects in CSR lead to a humoral immunodeficiency that is characterized by normal or elevated production of IgM and the partial or full absence of other isotypes (IgG, IgA, and IgE), resulting in high susceptibility to bacterial infections. Although IgG antibodies against infectious agents or vaccines are not produced, levels of isohemagglutinins and anti-polysaccharide IgM antibodies are normal. Switched IgG+ or IgA+ B cells are absent from the peripheral circulation, and CD27+ B-cell counts are either normal or strongly depressed (Table 1). Depending on the molecular defect, the CSR-D may be combined with an SHM deficiency ([29] and Table 1).
Table 1.
Main characteristics of immunoglobulin class-switch recombination deficiencies
| Defect | Relative frequency, percentage | Transmission | CD27+ B cells | Somatic hypermutation | Complications |
|---|---|---|---|---|---|
| Defect in T cell-B cell cooperation | |||||
| CD40L | 47 | X-L | Decreased | Decreased | Opportunistic infections, liver damage |
| CD40 | 3 | AR | Decreased | Decreased | Opportunistic infections, liver damage |
| NEMOa | 1 | X-L | N or decreased | N or decreased | Opportunistic infections |
| TFH defect? | 10 | ? | N | N | Auto-immunity |
| Intrinsic B-cell defect | |||||
| AID | 10 | AR | N | Absent | Lymphadenopathies, auto-immunity |
| AID co-factor? | 12 | ? | N | N | Lymphadenopathies, auto-immunity |
| DNA repair defect | |||||
| Uracil-N-glycosylase | 1 | AR | N | N (skewed pattern) | Auto-immunity, B lymphomas |
| Part of a known repair defect: PMS2a | 2 | AR | Decreased | N | Cancers |
| Ataxia telangiectasiaa | 2 | AR | N | N | Lymphomas, neurological deterioration |
| DNA repair? | 11 | AR | Decreased | Decreased | Auto-immunity, B lymphomas, leukemias |
aFrequency could be underestimated because it was sequenced only in patients with obvious symptoms. AID, activation-induced cytidine deaminase; N, normal; NEMO, nuclear factor-kappa-B essential modulator; PMS2, post-meiotic segregation 2; Tfh, T follicular helper.
I. CSR-Ds caused by a defect in T cell-B cell cooperation
A. X-linked CSR-D due to CD40L deficiency
X-linked CSR-D due to CD40L deficiency is the most frequent CSR-D and is caused by mutations in the gene encoding CD40L [30,31] (Online Mendelian Inheritance in Man (OMIM) #308230). Patients display little or no CD40L at the surface of in vitro activated CD4+ T cells, making diagnosis of this syndrome straightforward. However, owing to immaturity of the immune system, neonatal diagnosis cannot be made through CD40L expression analysis. A CD40 trans-activation defect means that the B cells of the patient cannot form germinal centers in secondary lymphoid organs in vivo and results in impaired CSR and SHM. However, B cells can undergo CSR in vitro following activation with CD40L and appropriate cytokines [32]. Most (but not all) affected patients present low memory CD27+ B-cell counts and a low frequency of SHM [33]. However, the detection of serum IgA and SHM in some patients suggests that alternative diversification pathways can occur: (a) IgA production following activation of B cells in the gut lamina propria by CpG or the proliferation-inducing ligand (APRIL) [34] and (b) SHM, possibly as an innate defense mechanism in the spleen marginal zone [35].
Impaired CD40L expression leads to defective T-cell interactions with monocytes and dendritic cells. This results in an abnormal cellular immune response and thus severe susceptibility to opportunistic infections with Pneumocystis jiroveci or Cryptosporidium. The latter cannot be controlled by Ig replacement therapy and is associated with a worse prognosis. Liver disease is very common. Sclerosing cholangitis (often associated with Cryptosporidium infection) is particularly severe and may lead to terminal liver damage.
Intermittent or chronic neutropenia is also a common feature of X-linked CD40L deficiency and may result from defective 'stress'-induced CD40-dependent granulopoiesis since myeloid progenitors express CD40 molecules [36]. Complications such as auto-immune manifestations or cancer have been reported but are not frequent.
Although mutations affect the entire CD40L gene, they are not distributed evenly; the majority are found within exon 5, which comprises most of the tumor necrosis factor homology domain [37]. A strict relationship between genotype and phenotype has not been established. Since the CD40L gene is located on the × chromosome, female patients are not affected, except in the case of a skewed pattern of × inactivation [38] or chromosomal translocation [39].
B. Autosomal recessive CSR-D due to CD40 deficiency
In a very few patients, the CD40 defect has been reported as part of an inherited autosomal recessive disease (OMIM #606843) and was diagnosed on the basis of a lack of CD40 expression at the surface of B lymphocytes and monocytes [40]. However, we recently observed three patients (from two different families) with normal CD40 expression but a defective response to CD40 activation and homozygous mutations in the CD40 gene: an amino acid substitution (E144K) and a stop codon disruption that resulted in an extra 30 amino acids (A Durandy, unpublished results). The clinical and immuno- logical profile of CD40-deficient patients is identical to that reported in CD40L-deficient patients, except that B cells are unable to undergo in vitro CSR following activation with CD40L and appropriate cytokines.
Despite efficient Ig replacement therapy and antibiotic prophylaxis, the long-term prognosis in both CD40L and CD40 deficiencies is poor; death can occur early in life (due to infections) or later on (due to severe liver damage). Thus, hematopoietic stem cell transplantation (HSCT) should be advocated if an HLA-identical sibling or matched unrelated donor is available [41]. Nevertheless, for patients already infected with Cryptosporidium, the risk of complications (sepsis and death) is extremely high.
C. X-linked CSR-D due to defective NF-κB activation
Cross-linking of CD40 activates the NF-κB signaling pathway. The latter is critical in CSR, as shown by the description of patients with ectodermal dysplasia associated with immunodeficiency (EDA-ID) [42,43] (OMIM #300291). Although this syndrome is heterogeneous, it is often characterized by normal to elevated IgM levels, low serum IgG and IgA levels, and impaired antibody responses (particularly to polysaccharide antigens). Susceptibility to Mycobacteria infections is elevated. The EDA-ID is inherited as an X-linked trait. The deficiency is caused by hypomorphic mutations in the zinc-finger domain of the NF-κB essential modulator (NEMO, also known as IκB kinase gamma, or IKKγ), a scaffold protein that binds to IKKα and IKKβ kinases (both of which are required for NF-κB nuclear activation and translocation). In vitro CSR and SHM may be normal or defective - probably as a result of genetic heterogeneity [44,45] (A Durandy, unpublished results). However, the deficiency is not restricted to CD40-B cell activation, since NF-κB nuclear translocation is required for many signaling pathways (including the T- and B-cell receptor pathways). The hallmark EDA-ID dysplasia also results from the NEMO deficiency since the ectodysplasin receptor expressed on ectoderm-derived tissues activates NF-κB via the IKKα/β NEMO complex [43]. Depending on the clinical phenotype, HLA-matched HSCT can be advocated.
D. Autosomal recessive CSR-D due to ICOS molecule deficiency
Although firstly described as common variable immunodeficiency, inducible co-stimulator (ICOS) molecule deficiency generally leads to a CSR-D, as shown by the increased or normal IgM levels observed in patients [46] (OMIM #607594). The ICOS molecule is involved in the generation and function (cytokine production) of follicular T helper cells in germinal centers [47]. Residual levels of IgG and IgA are not uncommon. The SHM process is impaired in CD27+ B cells (the number of which is very low). A T-cell defect has been reported in some (but not all) patients [48].
E. Uncharacterized Ig-CSR-D with normal in vitro CSR
The involvement of CD40-L and ICOS molecule can be ruled out by the observation of normal protein expression or gene sequences (or both) in several CSR-deficient patients whose B cells display in vitro CSR following activation with CD40L and appropriate cytokines. Hence, there is no B-cell defect in this pathway. In fact, the phenotype of these patients is quite different from that of CD40L-deficient patients, since there is no susceptibility to opportunistic infections. In contrast, there is susceptibility to bacterial infections but the latter are well controlled by Ig replacement therapy. Moreover, lymphadenopathies with enlarged germinal centers are observed and SHM is normal in terms of the frequency and pattern. Several possible causes have been ruled out, such as congenital rubella (in which defective T-cell activation leads to low CD40L expression on CD4+ T cells [49]) and major histocompatibility complex class II deficiency (in which diminished expression of CD40L by activated CD4+ T cells can also be responsible for an in vivo CSR-D [50]). A defect in the generation of T follicular helper cells or in their activation or interaction with follicular B cells can be suspected in this CSR-D.
II. CSR-Ds caused by an intrinsic B-cell defect
Other CSR-Ds are caused by an intrinsic B-cell defect, resulting in increased susceptibility to bacterial infections (but not opportunistic infections) that can be easily controlled by regular Ig replacement therapy. SHM may be normal or defective, depending on the molecular defect in question.
A. Autosomal recessive activation-induced cytidine deaminase deficiency
Activation-induced cytidine deaminase deficiency is the most frequent autosomal recessive CSR-D (OMIM #605258) and is caused by mutations in the AICDA gene. In fact, AID plays a crucial role in B-cell terminal differentiation by inducing DNA lesions in both the S and V regions. A lack of AID leads to the complete absence of CSR and SHM, even though the proportion of CD27+ B cells remains normal in affected individuals [19].
In addition to bacterial infections of the respiratory and digestive tracts, lymphoid hyperplasia is a prominent feature of this disease and is caused by massive enlargement of germinal centers - probably as a consequence of microbial infection [51]. Auto-immunity (hemolytic anemia, thrombocytopenia, hepatitis, and systemic lupus erythematosus) affects about 20% of the patients, and IgM auto-antibodies are present [52].
Mutations in AICDA are scattered throughout the gene (with no peculiar hotspots) and lead to a defect in both CSR and SHM [53]. However, mutations located in the C-terminal part of the AICDA gene result in a complete lack of CSR but do not affect SHM [54]. The C-terminal mutated enzyme has been found to retain normal cytidine deaminase activity in vitro or in Escherichia coli. This suggests that, in CSR, AID has an activity in addition to its cytidine deaminase activity - probably as a docking protein for CSR-specific co-factor(s). Although AID is normally detected only in the cytoplasm, these mutant proteins that lack a functional nuclear export signal (NES) are localized in the nucleus [54,55]. These observations suggest that wild-type AID could be retained in the cytoplasm by a CSR-specific co-factor [56,57]. Some data also suggest that this co-factor could be important for AID stability since AID is rapidly degraded by the proteasome in the nucleus [58].
Another unexpected finding [59] is that heterozygous nonsense mutations in the C-terminal domain - resulting in the loss of the last 11 (V186X) or nine (R190X) amino acids of NES (AIDΔNES) - lead to a variable, autosomal dominant CSR-D. Serum IgG levels are low and IgA is absent. No other heterozygous mutation located outside the C-terminal domain led to any haploinsufficiency (although weak effects on Ig levels have been reported in mice [60]).
B. Uncharacterized Ig-CSR-D caused by an intrinsic B-cell deficiency
Half of Ig-CSR-Ds due to an intrinsic B-cell deficiency may be related to a lack of cytoplasmic co-factor of AID rather than to AID deficiency itself. Although most of the observed cases are sporadic, the mode of inheritance observed in a few multiplex or consanguineous families is compatible with an autosomal recessive pattern. The clinical phenotype is similar to that of AID deficiency and includes increased susceptibility to bacterial infections of the respiratory and gastrointestinal tracts. Lymphoid hyperplasia is milder and less frequent (50%) and consists of moderate follicular hyperplasia but without the giant germinal centers that are typical of AID deficiency. Autoimmune manifestations have been reported [61]. The CSR-D appears to be milder than in AID deficiency since low serum levels of IgG can be detected in some patients. The sequence and expression of AID protein are normal. The deficiency is restricted to CSR since SHM in the CD27+ B-cell subset is normal in terms of both frequency and pattern and CD27+ B-cell counts are normal. Hence, the phenotype is similar to that of patients carrying mutations in the C-terminal part of AID and strongly suggests the involvement of a co-factor in the pathogenesis of this CSR-D.
III. Ig-CSR-Ds caused by a DNA repair deficiency
A. Autosomal recessive uracil-N-glycosylase deficiency
Introduction of lesions into DNA by AID activates the repair process. The first step consists of the recognition and removal of the uracil residues present in the DNA by the nuclear UNG2. An UNG2 deficiency results in an autosomal recessive CSR-D (OMIM #608106) that appears to be very rare: to the best of our knowledge, only three cases have been reported to date [62]. UNG is capable of deglycosylating uracil residues that are misintegrated into DNA by AID. Following removal of uracil residues by UNG, abasic sites that eventually can be attacked by APEs are created, leading to single-stranded DNA breaks. The processing and repair of the DNA lesions complete both CSR and SHM [21]. The absence of UNG results in abnormal CSR-D and SHM (confirmed in all three patients) [62]. SHM is biased toward transitions on G:C residues that probably arise from the replication of unprocessed U:G lesions in the absence of U removal. MMR enzymes may also recognize and repair these mismatches, introducing mutations on neighboring nucleotides that result in both transitions and transversions on A:T residues [20,21,62].
Patients are well controlled by Ig replacement therapy. However, UNG is part of the DNA base excision repair involved in the repair of spontaneously occurring base lesions and therefore constitutes a tool in the cells' antimutagenic defenses. UNG-deficient mice develop B-cell lymphomas as they age [63] and so UNG deficiency may predispose patients to tumorigenesis in adulthood.
B. Ig-CSR-D as part of a known DNA repair deficiency
Some CSR-D can occur in patients with a defined DNA repair deficiency. Given that CSR and SHM use different repair pathways, these CSR-Ds are associated with normal SHM. Although the occurrence of cancers is the main symptom of the disease, the CSR-D may be the main phenotypic feature for several years in some patients. Hence, DNA repair deficiencies should be investigated when a CSR-D lacks a molecular definition.
1. Autosomal recessive post-meiotic segregation 2 deficiency
Mono-allelic mutations in genes encoding MMR enzymes lead to a hereditary predisposition to non-poly-posis colon carcinoma (also known as Lynch syndrome) (OMIM #120435 [64]). Bi-allelic mutations in one of these enzymes lead to the early occurrence of a variety of cancers during childhood [65]. Recently, we showed that a lack of the MMR enzyme post-meiotic segregation 2 (PMS2) can also result in a CSR-D. Indeed, the CSR-D may be the main symptom for several years [66]. The conditions of four of the nine PMS2-deficient patients we observed were diagnosed as primary humoral immunodeficiency because of the recurrence of bacterial infections, and Ig replacement therapy was prescribed in three individuals. Serum IgM levels are normal or elevated. Low levels of serum IgG2 and IgG4 were observed in all nine patients, and reduced total IgG or IgA levels or both were found in the four symptomatic patients. IgG and IgA levels rose with age - probably because of the accumulation of long-lived plasma cells. In vitro, B cells are unable to undergo CSR following activation with CD40L and appropriate cytokines. SHM has a normal nucleotide substitution pattern in all patients, but the peripheral blood CD27+ B-cell count is low.
The PMS2 protein is part of the MMR pathway that recognizes and repairs mismatched nucleotides on DNA (provided that a nick on the same DNA strand is already present) [67]. Thus, one can hypothesize that the MMR acts downstream from UNG-APE activity and repairs the remaining U:G mismatches (that is, those not processed by UNG). In the absence of a nick close to the mismatch site, PMS2 is able to create one through its endonuclease activity [68]. Hence, the MMR enzymes in general and PMS2 in particular may play a specific role in the CSR-induced generation of DNA breaks in S regions [69].
The main symptom of PMS2 deficiency is the occurrence of cancers during childhood. Nevertheless, the CSR-D (which appears to be present in all of the patients studied to date - at least in vitro) might lead to even greater susceptibility to infections during cancer chemotherapy. The CSR-D may well be the most prominent feature for several years and this diagnosis should be considered in patients with a CSR-D that lacks a molecular definition. A non-specific but suggestive symptom is the presence of café-au-lait skin spots.
2. Autosomal recessive ataxia telangiectasia
Ataxia telangiectasia (AT), caused by bi-allelic mutations in the ATM gene, is a severe disease that combines progressive neurodegeneration (ataxia), cutaneous abnormalities (telangiectasia), a predisposition to malignancy (lymphoma), and an immunodeficiency (OMIM #120435 [70]). Patients with AT present frequently with a progressive T-cell defect predisposing them to viral and opportunistic infections. Some patients with AT could also present with a typical CSR-D, elevated IgM (sometimes at very high levels), and a contrastingly strong decrease in IgG and IgA levels, requiring Ig replacement therapy [71-73]. This CSR-D may be the main symptom during early childhood (diagnosis in the oldest patient was made at 7 years; A Durandy, personal communication). B cells are generally (but not always) unable to undergo CSR in vitro. Analysis of the recombined switch junctions in Ig gene loci indicates the failure of DNA repair during CSR and suggests a role for the product of the ATM gene in CSR-induced DSB repair in S regions [74]. DNA repair during CSR is a complex process that leads to the recombination of two S regions (Sμ and Sx) that may be very far away from each other. CSR activation has been shown to change the conformation of the Ig locus and bring the Sμ-Sx regions together within a synapsis [75]. The maintenance of this synapsis requires a multimolecular complex, including molecules phosphory-lated- by ATM (such as the histone H2AX, 53BP1, and MDC1) [76]. This synapsis is essential for S region repair via the NHEJ pathway. In the absence of ATM activity, the synapsis is not maintained and the repair fails, leading to CSR-D. In contrast, normal SHM generation and pattern (in a normal CD27+ B-cell subpopulation) confirm that ATM is not essential for DNA repair in V regions.
The Ig-CSR-D observed in AT is very similar to that caused by the defect in the putative AID co-factor, and AT should be screened for (with a fetoprotein assay) in molecularly undefined CSR-Ds with a normal CD27+ cell count and normal SHM. As with ATM, the MRE11/RAD50/NBS1 complex is also involved in the intra-switch region synapsis, a CSR-D is not unexpected in MRE11 and NBS1 deficiencies (AT-like disease and Nijmegen breakage syndrome, respectively).
C. Ig-CSR-Ds associated with an unknown DNA repair deficiency
We have described another subset of patients who very probably have an autosomal recessive CSR-D, as suggested by the gender ratio and the pedigree trees. Apart from having a susceptibility to bacterial infections, lymphadenopathies, and auto-immune manifestations, these patients present a greater incidence of tumors; in our series of 45 patients, five developed a non-Epstein-Barr virus (EBV)-induced B-cell lymphoma, one developed acute lymphoid leukemia, and another developed acute myeloid leukemia, strongly suggesting that they have a DNA repair deficiency. The CSR-D in vivo is often partial with residual IgG or IgA levels, contrasting with a strong reduction of in vitro CSR activity [77]. Moreover, the observation of a strong decrease in the CD27+ B-cell count, abnormal switch junction repair (with preferential use of microhomology), and particularly elevated radio-sensitivity in fibroblasts and EBV B-cell lines argues strongly in favor of a DNA repair deficiency [78]. The SHM level and pattern (on decreased CD27+ B cells) are normal, again emphasizing the fact that S and V regions use different repair pathways.
For CSR-induced DSBs in S regions, DNA repair is achieved in the Sμ-Sx synapsis by the NHEJ pathway. The process is initiated by DNA-PKcs and the Ku70/Ku80 [24,25,79] complex and then completed by molecules such as Artemis, ligase IV, and Cernunnos [80,81]. None of these molecules was found to be involved in our patients. Recently, an AEJ pathway was found to perform DNA repair in S regions [26]. However, its involvement in the CSR-D observed in the patients is doubtful since AEJ leads to repair through microhomology usage. Hence, one or more presently unidentified components of CSR-induced DNA repair of S regions are likely to be deficient in these patients. Although this condition has not yet been defined in molecular terms, it should be screened for, so that affected patients can receive appropriate follow-up (in view of the risk of tumorigenesis).
V. Concluding remarks
The precise description of the various Ig-CSR-Ds is essential from a medical point of view since the patients' prognosis and follow-up vary from one deficiency to another. Some Ig-CSR-Ds are associated with impaired cellular immune responses (for example, CD40L, CD40, and NEMO deficiencies). The only curative treatment for these severe conditions is HLA-identical HSCT, when possible. CSR-Ds caused by an intrinsic B-cell defect lead to a specific humoral deficiency that is easily controlled by Ig replacement therapy. However, the auto-immune complications (some of which are clearly related to IgM auto-antibodies) can be life-threatening. Auto-immunity is probably not simply related to high IgM serum levels, because auto-immune manifestations are significantly less frequent in CD40L deficiency in which IgM levels are equally elevated. Likewise, auto-immunity does not appear to be related to SHM since the latter occurs with the same frequency in both AID deficiency (with a lack of SHM) and other Ig-CSR-Ds with normal SHM generation. Occurrence of cancer is another special concern in Ig-CSR-Ds that are related to a DNA repair deficiency. Indeed, phenotypic data in mice suggest that B-cell lymphomas might occur in UNG-deficient patients. We have also observed a CSR-D that is very probably associated with a DNA repair factor deficiency, and that leads to the occurrence of lymphomas and leukemia. Although the molecular basis of this condition has not yet been defined, it is essential to diagnose the problem and then provide patients with appropriate follow-up. Lastly, a CSR-D may be the first long-lasting symptom of well-known DNA repair deficiencies, such as MMR deficiency and AT. These two conditions must be screened for whenever the CSR-D lacks a defined molecular basis. Thus, the accurate diagnosis and characterization of the CSR-D aid the establishment of a prognosis and the initiation of appropriate treatment.
In addition to the clinical interest of understanding CSR-Ds, the ongoing description of inherited CSR-Ds is shedding new light on the complex molecular mechanisms involved in antibody maturation in humans. The genetic definition of the X-linked CD40L and NEMO deficiencies (which were determined before the generation of the corresponding engineered mutant mice) provided clear evidence of the essential role of the CD40 signaling pathway in antibody maturation. The phenotypes of AID-deficient patients and mice (described at the same time) have demonstrated the master role of this newly described B-cell molecule in both CSR and SHM antibody maturation events. The description of an Ig-CSR-D caused by UNG deficiency provides strong in vivo evidence for a DNA-editing activity for AID first suggested by in vitro data. Additional data have shown that AID could act in antibody maturation not only through its cytidine deaminase activity but also as a potential docking protein for co-factors. The study of CSR-Ds associated with a DNA repair deficiency has revealed the role of the MMR pathway and the ATM protein in human CSR. Lastly, the study of nonelucidated Ig-CSR-Ds could also help to resolve some of the issues that remain obscure in CSR: the nature of the CSR-specific AID co-factor(s) and the complex mechanism involved in inter-switch region repair. Thus, studies of primary immune deficiencies appear to be critical for improving our understanding of the immune response.
Note
This article is part of the series on Adult immunodeficiency, edited by Hans-Hartmut Peter. Other articles in this series can be found at http://arthritis-research.com/series/immunodeficiency
Abbreviations
AEJ: alternative end joining; AID: activation-induced cytidine deaminase; APE: apurinic/apyrimidinic endonuclease; AT: ataxia telangiectasia; BCR: B-cell receptor; CSR: class-switch recombination; CSR-D: class-switch recombination deficiency; DSB: double-stranded DNA break; EBV: Epstein-Barr virus; EDA-ID: ectodermal dysplasia associated with immunodeficiency; HSCT: hematopoietic stem cell transplantation; ICOS: inducible co-stimulator; Ig: immunoglobulin; IKK: IκB kinase; MMR: mismatch repair; NEMO: nuclear factor-kappa-B essential modulator; NES: nuclear export signal; NF-κB: nuclear factor-kappa-B; NHEJ: non-homologous end joining; OMIM: Online Mendelian Inheritance in Man; PMS2: post-meiotic segregation 2; S: switch; SHM: somatic hypermutation; UNG: uracil-N-glycosylase; V: variable.
Competing interests
The authors declare that they have no competing interests.
Contributor Information
Anne Durandy, Email: anne.durandy@inserm.fr.
Sven Kracker, Email: sven.kracker@inserm.fr.
Acknowledgements
We thank Monique Forveille and Aminata Diabate for excellent technical assistance. This work was funded by grants from Institut National de la Santé et de la Recherche Médicale, the 7th RTD Framework Programme of the European Union (EUROPAD contract 201549 and PID-IMMUNE, ERC, contract 232809), Association Contre Le Cancer, and Agence Nationale de la Recherche (ANR) Blanc 2010-CSRD. SK is funded by the ANR.
References
- Iwasato T, Shimizu A, Honjo T, Yamagishi H. Circular DNA is excised by immunoglobulin class switch recombination. Cell. 1990;62:143–149. doi: 10.1016/0092-8674(90)90248-D. [DOI] [PubMed] [Google Scholar]
- Matsuoka M, Yoshida K, Maeda T, Usuda S, Sakano H. Switch circular DNA formed in cytokine-treated mouse splenocytes: evidence for intramolecular DNA deletion in immunoglobulin class switching. Cell. 1990;62:135–142. doi: 10.1016/0092-8674(90)90247-C. [DOI] [PubMed] [Google Scholar]
- von Schwedler U, Jack HM, Wabl M. Circular DNA is a product of the immunoglobulin class switch rearrangement. Nature. 1990;345:452–456. doi: 10.1038/345452a0. [DOI] [PubMed] [Google Scholar]
- Kinoshita K, Honjo T. Unique and unprecedented recombination mechanisms in class switching. Curr Opin Immunol. 2000;12:195–198. doi: 10.1016/S0952-7915(99)00072-2. [DOI] [PubMed] [Google Scholar]
- Manis JP, Tian M, Alt FW. Mechanism and control of class-switch recombination. Trends Immunol. 2002;23:31–39. doi: 10.1016/S1471-4906(01)02111-1. [DOI] [PubMed] [Google Scholar]
- Storb U, Peters A, Klotz E, Kim N, Shen HM, Kage K, Rogerson B, Martin TE. Somatic hypermutation of immunoglobulin genes is linked to transcription. Curr Top Microbiol Immunol. 1998;229:11–19. doi: 10.1007/978-3-642-71984-4_2. [DOI] [PubMed] [Google Scholar]
- Jacobs H, Bross L. Towards an understanding of somatic hypermutation. Curr Opin Immunol. 2001;13:208–218. doi: 10.1016/S0952-7915(00)00206-5. [DOI] [PubMed] [Google Scholar]
- Rajewsky K. Clonal selection and learning in the antibody system. Nature. 1996;381:751–758. doi: 10.1038/381751a0. [DOI] [PubMed] [Google Scholar]
- Frazer JK, LeGros J, de Bouteiller O, Liu YJ, Banchereau J, Pascual V, Capra JD. Identification and cloning of genes expressed by human tonsillar B lymphocyte subsets. Ann N Y Acad Sci. 1997;815:316–318. doi: 10.1111/j.1749-6632.1997.tb52073.x. [DOI] [PubMed] [Google Scholar]
- Kaartinen M, Griffiths GM, Markham AF, Milstein C. mRNA sequences define an unusually restricted IgG response to 2-phenyloxazolone and its early diversification. Nature. 1983;304:320–324. doi: 10.1038/304320a0. [DOI] [PubMed] [Google Scholar]
- Jacob J, Kelsoe G. In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl)acetyl. II. A common clonal origin for periarteriolar lymphoid sheath-associated foci and germinal centers. J Exp Med. 1992;176:679–687. doi: 10.1084/jem.176.3.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storb U, Peters A, Klotz E, Kim N, Shen HM, Hackett J, Rogerson B, Martin TE. Cis-acting sequences that affect somatic hypermutation of Ig genes. Immunol Rev. 1998;162:153–160. doi: 10.1111/j.1600-065X.1998.tb01438.x. [DOI] [PubMed] [Google Scholar]
- Chaudhuri J, Tian M, Khuong C, Chua K, Pinaud E, Alt FW. Transcription-targeted DNA deamination by the AID antibody diversification enzyme. Nature. 2003;422:726–730. doi: 10.1038/nature01574. [DOI] [PubMed] [Google Scholar]
- Dickerson SK, Market E, Besmer E, Papavasiliou FN. AID mediates hypermutation by deaminating single stranded DNA. J Exp Med. 2003;197:1291–1296. doi: 10.1084/jem.20030481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramiro AR, Stavropoulos P, Jankovic M, Nussenzweig MC. Transcription enhances AID-mediated cytidine deamination by exposing single-stranded DNA on the nontemplate strand. Nat Immunol. 2003;4:452–456. doi: 10.1038/ni920. [DOI] [PubMed] [Google Scholar]
- Yu K, Chedin F, Hsieh CL, Wilson TE, Lieber MR. R-loops at immunoglobulin class switch regions in the chromosomes of stimulated B cells. Nat Immunol. 2003;4:442–451. doi: 10.1038/ni919. [DOI] [PubMed] [Google Scholar]
- Bransteitter R, Pham P, Scharff MD, Goodman MF. Activation-induced cytidine deaminase deaminates deoxycytidine on single-stranded DNA but requires the action of RNase. Proc Natl Acad Sci USA. 2003;100:4102–4107. doi: 10.1073/pnas.0730835100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102:553–563. doi: 10.1016/S0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- Revy P, Muto T, Levy Y, Geissmann F, Plebani A, Sanal O, Catalan N, Forveille M, Dufourcq-Labelouse R, Gennery A, Tezcan I, Ersoy F, Kayserili H, Ugazio AG, Brousse N, Muramatsu M, Notarangelo LD, Kinoshita K, Honjo T, Fischer A, Durandy A. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the Hyper-IgM syndrome (HIGM2) Cell. 2000;102:565–575. doi: 10.1016/S0092-8674(00)00079-9. [DOI] [PubMed] [Google Scholar]
- Petersen-Mahrt SK, Harris RS, Neuberger MS. AID mutates E. coli suggesting a DNA deamination mechanism for antibody diversification. Nature. 2002;418:99–104. doi: 10.1038/nature00862. [DOI] [PubMed] [Google Scholar]
- Rada C, Williams GT, Nilsen H, Barnes DE, Lindahl T, Neuberger MS. Immunoglobulin isotype switching is inhibited and somatic hypermutation perturbed in UNG-deficient mice. Curr Biol. 2002;12:1748–1755. doi: 10.1016/S0960-9822(02)01215-0. [DOI] [PubMed] [Google Scholar]
- Guikema JE, Linehan EK, Tsuchimoto D, Nakabeppu Y, Strauss PR, Stavnezer J, Schrader CE. APE1- and APE2-dependent DNA breaks in immunoglobulin class switch recombination. J Exp Med. 2007;204:3017–3026. doi: 10.1084/jem.20071289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu U, Meng FL, Keim C, Grinstein V, Pefanis E, Eccleston J, Zhang T, Myers D, Wasserman CR, Wesemann DR, Januszyk K, Gregory RI, Deng H, Lima CD, Alt FW. The RNA exosome targets the AID cytidine deaminase to both strands of transcribed duplex DNA substrates. Cell. 2011;144:353–363. doi: 10.1016/j.cell.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manis JP, Gu Y, Lansford R, Sonoda E, Ferrini R, Davidson L, Rajewsky K, Alt FW. Ku70 is required for late B cell development and immunoglobulin heavy chain class switching. J Exp Med. 1998;187:2081–2089. doi: 10.1084/jem.187.12.2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casellas R, Nussenzweig A, Wuerffel R, Pelanda R, Reichlin A, Suh H, Qin XF, Besmer E, Kenter A, Rajewsky K, Nussenzweig MC. Ku80 is required for immunoglobulin isotype switching. Embo J. 1998;17:2404–2411. doi: 10.1093/emboj/17.8.2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan CT, Boboila C, Souza EK, Franco S, Hickernell TR, Murphy M, Gumaste S, Geyer M, Zarrin AA, Manis JP, Rajewsky K, Alt FW. IgH class switching and translocations use a robust non-classical end-joining pathway. Nature. 2007;449:478–482. doi: 10.1038/nature06020. [DOI] [PubMed] [Google Scholar]
- Bemark M, Sale JE, Kim HJ, Berek C, Cosgrove RA, Neuberger MS. Somatic hypermutation in the absence of DNA-dependent protein kinase catalytic subunit (DNA-PK(cs)) or recombination-activating gene (RAG)1 activity. J Exp Med. 2000;192:1509–1514. doi: 10.1084/jem.192.10.1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cascalho M, Wong J, Steinberg C, Wabl M. Mismatch repair co-opted by hypermutation. Science. 1998;279:1207–1210. doi: 10.1126/science.279.5354.1207. [DOI] [PubMed] [Google Scholar]
- Kracker S, Gardes P, Mazerolles F, Durandy A. Immunoglobulin class switch recombination deficiencies. Clin Immunol. 2010;135:193–203. doi: 10.1016/j.clim.2010.01.012. [DOI] [PubMed] [Google Scholar]
- Korthäuer U, Graf D, Mages HW, Brière F, Padayachee M, Malcolm S, Ugazio AG, Notarangelo LD, Levinsky RJ, Kroczek RA. Defective expression of T-cell CD40 ligand causes X-linked immunodeficiency with hyper-IgM. Nature. 1993;361:539–541. doi: 10.1038/361539a0. [DOI] [PubMed] [Google Scholar]
- Aruffo A, Farrington M, Hollenbaugh D, Li X, Milatovich A, Nonoyama S, Bajorath J, Grosmaire LS, Stenkamp R, Neubauer M, Roberts RL, Noelle RJ, Ledbetter JA, Francke U, Ochs HD. The CD40 ligand, gp39, is defective in activated T cells from patients with X-linked hyper-IgM syndrome. Cell. 1993;72:291–300. doi: 10.1016/0092-8674(93)90668-G. [DOI] [PubMed] [Google Scholar]
- Durandy A, Schiff C, Bonnefoy JY, Forveille M, Rousset F, Mazzei G, Milili M, Fischer A. Induction by anti-CD40 antibody or soluble CD40 ligand and cytokines of IgG, IgA and IgE production by B cells from patients with X-linked hyper IgM syndrome. Eur J Immunol. 1993;23:2294–2299. doi: 10.1002/eji.1830230936. [DOI] [PubMed] [Google Scholar]
- Agematsu K, Nagumo H, Shinozaki K, Hokibara S, Yasui K, Terada K, Kawamura N, Toba T, Nonoyama S, Ochs HD, Komiyama A. Absence of IgD-CD27(+) memory B cell population in X-linked hyper-IgM syndrome. J Clin Invest. 1998;102:853–860. doi: 10.1172/JCI3409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He B, Xu W, Santini PA, Polydorides AD, Chiu A, Estrella J, Shan M, Chadburn A, Villanacci V, Plebani A, Knowles DM, Rescigno M, Cerutti A. Intestinal bacteria trigger T cell-independent immunoglobulin A(2) class switching by inducing epithelial-cell secretion of the cytokine APRIL. Immunity. 2007;26:812–826. doi: 10.1016/j.immuni.2007.04.014. [DOI] [PubMed] [Google Scholar]
- Scheeren FA, Nagasawa M, Weijer K, Cupedo T, Kirberg J, Legrand N, Spits H. T cell-independent development and induction of somatic hypermutation in human IgM+ IgD+ CD27+ B cells. J Exp Med. 2008;205:2033–2042. doi: 10.1084/jem.20070447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroczek RA, Graf D, Brugnoni D, Giliani S, Korthüer U, Ugazio A, Senger G, Mages HW, Villa A, Notarangelo LD. Defective expression of CD40 ligand on T cells causes 'X-linked immunodeficiency with hyper-IgM (HIGM1)'. Immunol Rev. 1994;138:39–59. doi: 10.1111/j.1600-065X.1994.tb00846.x. [DOI] [PubMed] [Google Scholar]
- Hollenbaugh D, Grosmaire LS, Kullas CD, Chalupny NJ, Braesch-Andersen S, Noelle RJ, Stamenkovic I, Ledbetter JA, Aruffo A. The human T cell antigen gp39, a member of the TNF gene family, is a ligand for the CD40 receptor: expression of a soluble form of gp39 with B cell co-stimulatory activity. Embo J. 1992;11:4313–4321. doi: 10.1002/j.1460-2075.1992.tb05530.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Saint Basile G, Tabone MD, Durandy A, Phan F, Fischer A, Le Deist F. CD40 ligand expression deficiency in a female carrier of the X-linked hyper-IgM syndrome as a result of × chromosome lyonization. Eur J Immunol. 1999;29:367–373. doi: 10.1002/(SICI)1521-4141(199901)29:01<367::AID-IMMU367>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Imai K, Shimadzu M, Kubota T, Morio T, Matsunaga T, Park YD, Yoshioka A, Nonoyama S. Female hyper IgM syndrome type 1 with a chromosomal translocation disrupting CD40LG. Biochim Biophys Acta. 2006;1762:335–340. doi: 10.1016/j.bbadis.2005.10.003. [DOI] [PubMed] [Google Scholar]
- Ferrari S, Giliani S, Insalaco A, Al-Ghonaium A, Soresina AR, Loubser M, Avanzini MA, Marconi M, Badolato R, Ugazio AG, Levy Y, Catalan N, Durandy A, Tbakhi A, Notarangelo LD, Plebani A. Mutations of CD40 gene cause an autosomal recessive form of immunodeficiency with hyper IgM. Proc Natl Acad Sci USA. 2001;98:12614–12619. doi: 10.1073/pnas.221456898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gennery AR, Cant AJ. Advances in hematopoietic stem cell transplantation for primary immunodeficiency. Immunol Allergy Clin North Am. 2008;28:439–456. doi: 10.1016/j.iac.2008.01.006. x-xi. [DOI] [PubMed] [Google Scholar]
- Zonana J, Elder ME, Schneider LC, Orlow SJ, Moss C, Golabi M, Shapira SK, Farndon PA, Wara DW, Emmal SA, Ferguson BM. A novel X-linked disorder of immune deficiency and hypohidrotic ectodermal dysplasia is allelic to incontinentia pigmenti and due to mutations in IKK-gamma (NEMO) Am J Hum Genet. 2000;67:6. doi: 10.1086/316914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Döffinger R, Smahi A, Bessia C, Geissmann F, Feinberg J, Durandy A, Bodemer C, Kenwrick S, Dupuis-Girod S, Blanche S, Wood P, Rabia SH, Headon DJ, Overbeek PA, Le Deist F, Holland SM, Belani K, Kumararatne DS, Fischer A, Shapiro R, Conley ME, Reimund E, Kalhoff H, Abinun M, Munnich A, Israël A, Courtois G, Casanova JL. X-linked anhidrotic ectodermal dysplasia with immunodeficiency is caused by impaired NF-kappaB signaling. Nat Genet. 2001;27:277–285. doi: 10.1038/85837. [DOI] [PubMed] [Google Scholar]
- Jain A, Ma CA, Lopez-Granados E, Means G, Brady W, Orange JS, Liu S, Holland S, Derry JM. Specific NEMO mutations impair CD40-mediated c-Rel activation and B cell terminal differentiation. J Clin Invest. 2004;114:1593–1602. doi: 10.1172/JCI21345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson EP, Monaco-Shawver L, Solt LA, Madge LA, Banerjee PP, May MJ, Orange JS. Hypomorphic nuclear factor-kappaB essential modulator mutation database and reconstitution system identifies phenotypic and immunologic diversity. J Allergy Clin Immunol. 2008;122:1169–1177 e1116. doi: 10.1016/j.jaci.2008.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnatz K, Bossaller L, Salzer U, Skrabl-Baumgartner A, Schwinger W, van der Burg M, van Dongen JJ, Orlowska-Volk M, Knoth R, Durandy A, Draeger R, Schlesier M, Peter HH, Grimbacher B. Human ICOS deficiency abrogates the germinal center reaction and provides a monogenic model for common variable immunodeficiency. Blood. 2006;107:3045–3052. doi: 10.1182/blood-2005-07-2955. [DOI] [PubMed] [Google Scholar]
- Bossaller L, Burger J, Draeger R, Grimbacher B, Knoth R, Plebani A, Durandy A, Baumann U, Schlesier M, Welcher AA, Peter HH, Warnatz K. ICOS deficiency is associated with a severe reduction of CXCR5+CD4 germinal center Th cells. J Immunol. 2006;177:4927–4932. doi: 10.4049/jimmunol.177.7.4927. [DOI] [PubMed] [Google Scholar]
- Takahashi N, Matsumoto K, Saito H, Nanki T, Miyasaka N, Kobata T, Azuma M, Lee SK, Mizutani S, Morio T. Impaired CD4 and CD8 effector function and decreased memory T cell populations in ICOS-deficient patients. J Immunol. 2009;182:5515–5527. doi: 10.4049/jimmunol.0803256. [DOI] [PubMed] [Google Scholar]
- Kawamura N, Okamura A, Furuta H, Katow S, Yamada M, Kobayashi I, Okano M, Kobayashi K, Sakiyama Y. Improved dysgammaglobulinaemia in congenital rubella syndrome after immunoglobulin therapy: correlation with CD154 expression. Eur J Pediatr. 2000;159:764–766. doi: 10.1007/PL00008342. [DOI] [PubMed] [Google Scholar]
- Nonoyama S, Tsukada S, Yamadori T, Miyawaki T, Jin YZ, Watanabe C, Morio T, Yata J, Ochs HD. Functional analysis of peripheral blood B cells in patients with X-linked agammaglobulinemia. J Immunol. 1998;161:3925–3929. [PubMed] [Google Scholar]
- Fagarasan S, Honjo T. T-Independent immune response: new aspects of B cell biology. Science. 2000;290:89–92. doi: 10.1126/science.290.5489.89. [DOI] [PubMed] [Google Scholar]
- Quartier P, Bustamante J, Sanal O, Plebani A, Debré M, Deville A, Litzman J, Levy J, Fermand JP, Lane P, Horneff G, Aksu G, Yalçin I, Davies G, Tezcan I, Ersoy F, Catalan N, Imai K, Fischer A, Durandy A. Clinical, immunologic and genetic analysis of 29 patients with autosomal recessive hyper-IgM syndrome due to activation-induced cytidine deaminase deficiency. Clin Immunol. 2004;110:22–29. doi: 10.1016/j.clim.2003.10.007. [DOI] [PubMed] [Google Scholar]
- Durandy A, Peron S, Taubenheim N, Fischer A. Activation-induced cytidine deaminase: structure-function relationship as based on the study of mutants. Hum Mutat. 2006;27:1185–1191. doi: 10.1002/humu.20414. [DOI] [PubMed] [Google Scholar]
- Ta VT, Nagaoka H, Catalan N, Durandy A, Fischer A, Imai K, Nonoyama S, Tashiro J, Ikegawa M, Ito S, Kinoshita K, Muramatsu M, Honjo T. AID mutant analyses indicate requirement for class-switch-specific cofactors. Nat Immunol. 2003;4:843–848. doi: 10.1038/ni964. [DOI] [PubMed] [Google Scholar]
- Geisberger R, Rada C, Neuberger MS. The stability of AID and its function in class-switching are critically sensitive to the identity of its nuclear-export sequence. Proc Natl Acad Sci USA. 2009;106:6736–6741. doi: 10.1073/pnas.0810808106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doi T, Kato L, Ito S, Shinkura R, Wei M, Nagaoka H, Wang J, Honjo T. The C terminal region of activation-induced cytidine deaminase is responsible for a recombination function other than DNA cleavage in class switch recombination. Proc Natl Acad Sci USA. 2009;106:2758–2763. doi: 10.1073/pnas.0813253106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patenaude AM, Orthwein A, Hu Y, Campo VA, Kavli B, Buschiazzo A, Di Noia JM. Active nuclear import and cytoplasmic retention of activation-induced deaminase. Nat Struct Mol Biol. 2009;16:517–527. doi: 10.1038/nsmb.1598. [DOI] [PubMed] [Google Scholar]
- Aoufouchi S, Faili A, Zober C, D'Orlando O, Weller S, Weill JC, Reynaud CA. Proteasomal degradation restricts the nuclear lifespan of AID. J Exp Med. 2008;205:1357–1368. doi: 10.1084/jem.20070950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai K, Zhu Y, Revy P, Morio T, Mizutani S, Fischer A, Nonoyama S, Durandy A. Analysis of class switch recombination and somatic hypermutation in patients affected with autosomal dominant hyper-IgM syndrome type 2. Clin Immunol. 2005;115:277–285. doi: 10.1016/j.clim.2005.02.003. [DOI] [PubMed] [Google Scholar]
- Takizawa M, Tolarová H, Li Z, Dubois W, Lim S, Callen E, Franco S, Mosaico M, Feigenbaum L, Alt FW, Nussenzweig A, Potter M, Casellas R. AID expression levels determine the extent of cMyc oncogenic translocations and the incidence of B cell tumor development. J Exp Med. 2008;205:1949–1957. doi: 10.1084/jem.20081007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durandy A, Taubenheim N, Peron S, Fischer A. Pathophysiology of B-cell intrinsic immunoglobulin class switch recombination deficiencies. Adv Immunol. 2007;94:275–306. doi: 10.1016/S0065-2776(06)94009-7. [DOI] [PubMed] [Google Scholar]
- Imai K, Slupphaug G, Lee WI, Revy P, Nonoyama S, Catalan N, Yel L, Forveille M, Kavli B, Krokan HE, Ochs HD, Fischer A, Durandy A. Human uracil-DNA glycosylase deficiency associated with profoundly impaired immunoglobulin class-switch recombination. Nat Immunol. 2003;4:1023–1028. doi: 10.1038/ni974. [DOI] [PubMed] [Google Scholar]
- Nilsen H, Stamp G, Andersen S, Hrivnak G, Krokan HE, Lindahl T, Barnes DE. Gene-targeted mice lacking the Ung uracil-DNA glycosylase develop B-cell lymphomas. Oncogene. 2003;22:5381–5386. doi: 10.1038/sj.onc.1206860. [DOI] [PubMed] [Google Scholar]
- Niessen RC, Berends MJ, Wu Y, Sijmons RH, Hollema H, Ligtenberg MJ, de Walle HE, de Vries EG, Karrenbeld A, Buys CH, van der Zee AG, Hofstra RM, Kleibeuker JH. Identification of mismatch repair gene mutations in young patients with colorectal cancer and in patients with multiple tumours associated with hereditary non-polyposis colorectal cancer. Gut. 2006;55:1781–1788. doi: 10.1136/gut.2005.090159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa H, Lockman JC, Frankel WL, Hampel H, Steenblock K, Burgart LJ, Thibodeau SN, de la Chapelle A. Mismatch repair gene PMS2: disease-causing germline mutations are frequent in patients whose tumors stain negative for PMS2 protein, but paralogous genes obscure mutation detection and interpretation. Cancer Res. 2004;64:4721–4727. doi: 10.1158/0008-5472.CAN-03-2879. [DOI] [PubMed] [Google Scholar]
- Péron S, Metin A, Gardès P, Alyanakian MA, Sheridan E, Kratz CP, Fischer A, Durandy A. Human PMS2 deficiency is associated with impaired immunoglobulin class switch recombination. J Exp Med. 2008;205:2465–2472. doi: 10.1084/jem.20080789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadyrov FA, Dzantiev L, Constantin N, Modrich P. Endonucleolytic function of MutLalpha in human mismatch repair. Cell. 2006;126:297–308. doi: 10.1016/j.cell.2006.05.039. [DOI] [PubMed] [Google Scholar]
- van Oers JM, Roa S, Werling U, Liu Y, Genschel J, Hou H Jr, Sellers RS, Modrich P, Scharff MD, Edelmann W. PMS2 endonuclease activity has distinct biological functions and is essential for genome maintenance. Proc Natl Acad Sci USA. 2010;107:13384–13389. doi: 10.1073/pnas.1008589107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavnezer J, Schrader CE. Mismatch repair converts AID-instigated nicks to double-strand breaks for antibody class-switch recombination. Trends Genet. 2006;22:23–28. doi: 10.1016/j.tig.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Savitsky K, Bar-Shira A, Gilad S, Rotman G, Ziv Y, Vanagaite L, Tagle DA, Smith S, Uziel T, Sfez S, Ashkenazi M, Pecker I, Frydman M, Harnik R, Patanjali SR, Simmons A, Clines GA, Sartiel A, Gatti RA, Chessa L, Sanal O, Lavin MF, Jaspers NG, Taylor AM, Arlett CF, Miki T, Weissman SM, Lovett M, Collins FS, Shiloh Y. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science. 1995;268:1749–1753. doi: 10.1126/science.7792600. [DOI] [PubMed] [Google Scholar]
- Etzioni A, Ben-Barak A, Peron S, Durandy A. Ataxia-telangiectasia in twins presenting as autosomal recessive hyper-immunoglobulin M syndrome. Isr Med Assoc J. 2007;9:406–407. [PubMed] [Google Scholar]
- Meyts I, Weemaes C, De Wolf-Peeters C, Proesmans M, Renard M, Uyttebroeck A, De Boeck K. Unusual and severe disease course in a child with ataxia-telangiectasia. Pediatr Allergy Immunol. 2003;14:330–333. doi: 10.1034/j.1399-3038.2003.00037.x. [DOI] [PubMed] [Google Scholar]
- Soresina A, Meini A, Lougaris V, Cattaneo G, Pellegrino S, Piane M, Darra F, Plebani A. Different clinical and immunological presentation of ataxia-telangiectasia within the same family. Neuropediatrics. 2008;39:43–45. doi: 10.1055/s-2008-1076736. [DOI] [PubMed] [Google Scholar]
- Pan Q, Petit-Frere C, Lahdesmaki A, Gregorek H, Chrzanowska KH, Hammarstrom L. Alternative end joining during switch recombination in patients with ataxia-telangiectasia. Eur J Immunol. 2002;32:1300–1308. doi: 10.1002/1521-4141(200205)32:5<1300::AID-IMMU1300>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Wuerffel R, Wang L, Grigera F, Manis J, Selsing E, Perlot T, Alt FW, Cogne M, Pinaud E, Kenter AL. S-S synapsis during class switch recombination is promoted by distantly located transcriptional elements and activation-induced deaminase. Immunity. 2007;27:711–722. doi: 10.1016/j.immuni.2007.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavnezer J. Complex regulation and function of activation-induced cytidine deaminase. Trends Immunol. 2011;32:194–201. doi: 10.1016/j.it.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai K, Catalan N, Plebani A, Maródi L, Sanal O, Kumaki S, Nagendran V, Wood P, Glastre C, Sarrot-Reynauld F, Hermine O, Forveille M, Revy P, Fischer A, Durandy A. Hyper-IgM syndrome type 4 with a B lymphocyte-intrinsic selective deficiency in Ig class-switch recombination. J Clin Invest. 2003;112:136–142. doi: 10.1172/JCI18161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Péron S, Pan-Hammarström Q, Imai K, Du L, Taubenheim N, Sanal O, Marodi L, Bergelin-Besançon A, Benkerrou M, de Villartay JP, Fischer A, Revy P, Durandy A. A primary immunodeficiency characterized by defective immunoglobulin class switch recombination and impaired DNA repair. J Exp Med. 2007;204:1207–1216. doi: 10.1084/jem.20070087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolink A, Melchers F, Andersson J. The SCID but not the RAG-2 gene product is required for S mu-S epsilon heavy chain class switching. Immunity. 1996;5:319–330. doi: 10.1016/S1074-7613(00)80258-7. [DOI] [PubMed] [Google Scholar]
- Buck D, Malivert L, de Chasseval R, Barraud A, Fondanèche MC, Sanal O, Plebani A, Stéphan JL, Hufnagel M, le Deist F, Fischer A, Durandy A, de Villartay JP, Revy P. Cernunnos, a novel nonhomologous end-joining factor, is mutated in human immunodeficiency with microcephaly. Cell. 2006;124:287–299. doi: 10.1016/j.cell.2005.12.030. [DOI] [PubMed] [Google Scholar]
- Pan-Hammarström Q, Jones AM, Lähdesmäki A, Zhou W, Gatti RA, Hammarström L, Gennery AR, Ehrenstein MR. Impact of DNA ligase IV on nonhomologous end joining pathways during class switch recombination in human cells. J Exp Med. 2005;201:189–194. doi: 10.1084/jem.20040772. [DOI] [PMC free article] [PubMed] [Google Scholar]