

Abstract
Adenosylcobalamin (coenzyme B12) serves as the cofactor for a group of enzymes that catalyze unusual rearrangement or elimination reactions. The role of the cofactor as the initiator of reactive free radicals needed for these reactions is well established. Less clear is how these enzymes activate the coenzyme towards homolysis and control the radicals once generated. The availability of high resolution X-ray structures combined with detailed kinetic and spectroscopic analyses have allowed several adenosylcobalamin enzymes to be computationally modeled in some detail. Computer simulations have generally obtained good agreement with experimental data and provided valuable insight into the mechanisms of these unusual reactions. Importantly, atomistic modeling of the enzymes has allowed the role of specific interactions between protein, substrate and coenzyme to be explored, leading to mechanistic predictions that can now be tested experimentally. This article is part of a Special Issue entitled: Radical SAM enzymes and Radical Enzymology.
Keywords: Coenzyme B12, Free radical, Enzyme mechanism
1. Introduction
Adenosylcobalamin, also known as coenzyme-B12, (Fig. 1) is a biologically active form of vitamin B12 [1,2]. It serves as the cofactor for a relatively small group of enzymes, only 12 have so far been identified, that catalyze unusual rearrangement or elimination reactions involving the interchange of a hydrogen atom on one carbon with an electron-withdrawing group, X, on an adjacent carbon (Scheme 1).
Fig. 1.

The structure of AdoCbl. The adenosyl moiety is highlighted in red; the corrin ring and nucleotide tail are in blue. Free radicals are generated by homolytic cleavage of the bond between the adenosyl group and the central cobalt atom.
Scheme 1.

These reactions proceed through free radical mechanisms. The role of the cofactor is to serve as a source of organic radicals that are unmasked by homolysis of the cobalt–carbon bond to form cob(II) alamin and a highly reactive 5′-deoxyadenosyl radical (Ado•). This allows enzymes to exploit the reactivity of free radicals to catalyze reactions on otherwise un-reactive molecules.
This review aims to provide the reader first with an overview of the reactions catalyzed by AdoCbl enzymes, and then focus on some of the more recent advances in understanding their mechanisms. In particular, computational studies have recently become an important tool to investigate questions regarding the energetics and mechanisms of free radical reactions, and so are given some emphasis here. It would be impossible to provide a comprehensive review of all the work in this area within the limited space of this article and we have not attempted to do so! We apologize in advance to colleagues whose work we may not have cited, and refer the reader to several recent reviews that discuss various aspects of AdoCbl biochemistry in more detail than space permits here [1–9].
AdoCbl-dependent enzymes may be divided into three subgroups. i. Carbon-skeleton mutases catalyze the interchange of a hydrogen atom on one carbon with a carbon-containing group on an adjacent carbon to effect a skeletal rearrangement (Fig. 2). Members of this subgroup include glutamate mutase, 2-methyleneglutarate mutase, and methylmalonyl-CoA mutase [7,8,10,11], the only AdoCbl-dependent enzyme found in animals. Closely related to methylmalonyl-CoA mutase are isobutyrl-CoA mutase [12] and the recently identified hydroxybutyrl-CoA mutase [13] and ethylmalonyl-CoA mutase enzymes [14]. ii. The eliminase subgroup catalyze 1,2-rearrangements of either 1,2-diols to 1,1-diols or 1,2-amino-alcohols to 1,1-amino-alcohols, which then undergo enzyme-catalyzed elimination of water or ammonia to give aldehydes as the final product (Fig. 3). iii. Aminomutases catalyze 1,2-migrations of amino groups and additionally require pyridoxal phosphate as a cofactor; the two known enzymes in this subgroup are ornithine-4,5-aminomutase [15,16] and lysine-5,6-aminomutase [17,18] (Fig. 4). The enzymes in this subgroup are ethanolamine ammonialyase, diol dehydrase and glycerol dehydrase [3]. Lastly, class II (AdoCbl dependent) ribonucleotide reductase [19,20] appears not to fit into any of these subgroups, as it catalyzes a reduction rather than a rearrangement (Fig. 3). However, AdoCbl serves the same function in this enzyme and the mechanism by which the 2′-OH group is removed parallels the rearrangements catalyzed by the eliminases.
Fig. 2.

The reactions catalyzed by carbon skeleton mutases. The migrating hydrogen is shown in red and the migrating carbon fragment in blue.
Fig. 3.

The reactions catalyzed by AdoCbl-dependent eliminases and class II ribonucleotide reductase. The migrating hydrogen is shown in red and the migrating –OH or NH3 + groups in blue. After rearrangement, the intermediate amino alcohols or 1,1-diols undergo enzyme-catalyzed elimination. For ribonucleotide reductase, the C–H bond (red) on the carbon adjacent to the 2′-OH (blue) is transiently cleaved and the 2′-OH eliminated, followed by reduction of the 2′-carbon.
Fig. 4.

The reactions catalyzed by aminomutases. The migrating hydrogen is shown in red and the migrating amino group in blue.
2. Mechanistic overview
Enzymes have evolved various mechanisms to generate reactive radical species that can be used for C–H bond activation. It is useful to note the close mechanistic parallels between AdoCbl-dependent enzymes and the more recently recognized but much larger class of radical S-adenosylmethionine (SAM) enzymes [21,22]. Whereas the Co–C bond in AdoCbl has a relatively low bond dissociation energy (~30 kcal/mol), the higher bond dissociation energy of the C–S bond in SAM (~60 kcal/mol) means that direct homolytic cleavage is not feasible. Therefore these enzymes generate Ado• through a single-electron reduction of SAM complexed to a reduced iron-sulfur cluster (Fig. 5).
Fig. 5.

Generation of 5′-deoxyadenosyl radical can be accomplished either by homolysis of AdoCbl (left) or reductive cleavage of S-adenosylmethionine (right).
Whereas AdoCbl always serves as a cofactor, Ado• generated from SAM may be used catalytically as a true cofactor, but in many enzymes is consumed during turn-over as a co-substrate. The similarity of AdoCbl and radical SAM enzymes is illustrated by the aminomutases. Lysine-2,3-aminomutase is a well characterized radical SAM enzyme [6] that shares a requirement for pyridoxal phosphate and catalyzes a rearrangement chemically identical to those catalyzed by AdoCbl-dependent lysine and ornithine aminomutases. Thus both cofactors function to generate Ado•; the chemical reaction catalyzed is then independent of the cofactor used to generate free radicals.
A significant difference between AdoCbl and radical-SAM enzymes is their oxygen sensitivity. Whereas AdoCbl enzymes are not especially oxygen sensitive, all radical-SAM enzymes studied to date must be handled under rigorously anaerobic conditions to maintain their activity. Reactive oxygen species rapidly oxidize and destroy the iron–sulfur clusters in these enzymes. Oxygen sensitivity may have provided the pressure for the evolution of AdoCbl-dependent enzymes; however the discovery of genes for radical-SAM enzymes in aerobic organisms hints that these enzymes may be functional in the microenvironment of a cell even under aerobic conditions [21].
A general mechanism that accounts for the rearrangements catalyzed by AdoCbl-dependent enzymes is shown in Fig. 6. Following homolysis, Ado• abstracts the migrating hydrogen atom from the substrate to generate a substrate-based radical and 5′-deoxyadenosine (Ado-H). The migrating group (-X in Fig. 6) then moves from one carbon to the other to effect the rearrangement step and form a product-like radical. The mechanistic details of this step vary dependent upon the nature of -X. Lastly, a hydrogen from the methyl group of Ado-H is re-abstracted by the product-radical to give the product and regenerate Ado•, which immediately recombines with cob(II)alamin to reform AdoCbl.
Fig. 6.

A minimal mechanistic scheme describing the 1,2-rearrangement reactions catalyzed by AdoCbl-dependent isomerases. X may be –OH, NH3 + or a carbon-containing fragment.
The reactions catalyzed by the carbon-skeleton mutases and aminomutases are fully reversible. However, in the eliminases the gem-diol or 1,1-amino-alcohol initially formed by radical rearrangement subsequently undergoes irreversible, enzyme-catalyzed elimination of water or ammonia to form an aldehyde (Fig. 3). Although the reduction of ribonucleotide triphosphates catalyzed by AdoCbl-dependent ribonucleotide reductase appears to be quite different, the chemistry is very similar to that for the 1,2-migration of –OH catalyzed by diol dehydrase. In this case, AdoCbl is used to reversibly generate an active site cysteinyl radical [19,20]. The cysteinyl radical, in turn, abstracts the 3′-H adjacent to the site of reduction and this activates the 2′-OH to become a good leaving group (Fig. 7). In fact, this essential radical chemistry, including the cysteinyl radical, is conserved in both the aerobic tyrosyl radical-dependent and anaerobic glycyl radical-dependent ribonucleotide reductases [19,20].
Fig. 7.

In the reaction catalyzed by AdoCbl-dependent ribonucleotide reductase the 3′-hydrogen is abstracted through an intermediate cysteinyl radical. This activates the substrate towards elimination of the 3′-OH group; subsequent reduction of the 2′-carbon and replacement of the 3′-hydrogen generates the reduced nucleotide.
Whereas activation of the substrate by abstraction of a non-reactive hydrogen atom is the common mechanistic step in all AdoCbl enzymes, the mechanisms by which the substrate radicals rearrange are dependent on the nature of the migrating chemical group. Here we briefly summarize the rearrangement mechanisms, which are discussed in more detail later for selected enzymes.
For the carbon skeleton mutases two distinct mechanisms appear to operate (Fig. 8). For reactions catalyzed by 2-methyleneglutarate mutase, methylmalonyl-CoA mutase and related acyl-CoA mutases, the inter-conversion of substrate and product radicals most likely occurs through a cyclopropylcarbenyl radical intermediate, a mechanism supported by model chemical reactions [23,24]. In contrast, it has been shown that the rearrangement catalyzed by glutamate mutase involves fragmentation of the glutamyl radical to form a glycyl radical and acrylate followed by recombination to form the methylaspartyl radical [25].
Fig. 8.

The mechanisms for the carbon skeleton rearrangements catalyzed by methyleneglutarate mutase and the acyl-CoA mutases most likely proceed through an associative addition–elimination mechanism involving a cyclic intermediate, whereas the rearrangement catalyzed by glutamate mutase proceeds through a dissociative fragmentation–recombination mechanism involving a glycyl radical and acrylate as intermediates.
The aminomutases require pyridoxal phosphate as a cofactor. Lysine-2,3-aminomutase (LAM), which is actually a radical SAM enzyme, serves as a paradigm for the role of pyridoxal phosphate in this reaction. EPR experiments using isotopically-labeled substrates and lysine analogs that preferentially stabilize the different radicals formed during the rearrangement have allowed each of the substrate radical species proposed in the mechanism to be identified [26–28]. A similar approach has recently been used to study the mechanism of lysine-5,6-aminomutase [18]. During the reaction (Fig. 9), the α-amino group of lysine forms an external aldimine with pyridoxal phosphate [29], rendering the nitrogen sp2 hybridized. This allows the 1,2-nitrogen migration to occur through a cyclic azacyclopropylcarbinyl radical intermediate transition state in which nitrogen is bonded to both C-2 and C-3 of lysine and the unpaired electron is situated on the 4′-carbon of pyridoxal and stabilized by the adjacent π system.
Fig. 9.

The role of pyridoxal phosphate in facilitating the rearrangements of amino groups in the aminomutase-catalyzed reactions.
The AdoCbl-dependent diol dehydrase, glycerol dehydrase and ethanolamine deaminase catalyze elimination reactions that first involve a 1,2-migration of –OH or –NH3 + followed by dehydration or deamination of the resulting 1,1-diols or amino-alcohol to give aldehydes [3]. Here it appears that the charge state of the migrating oxygen or nitrogen atom is important (Fig. 10). Theoretical studies, discussed in more detail below, suggest that a migration pathway involving a positively charged cyclic transition state structure is favored [30,31]. Although it was long thought that an active site potassium ion was likely to provide the positive charge, recent evidence now points to calcium as the catalytic metal [32].
Fig. 10.

The mechanism for the migration of –OH groups catalyzed by diol and glycerol dehydrases involves polarization of the C–O bond by an active site metal ion.
3. Structures of AdoCbl dependent enzymes
A detailed discussion of enzyme structure is beyond the scope of this review, however we briefly describe the common structural features of these enzymes, as knowledge of the structure has allowed the mechanistic roles of active site residues to be tested and is essential for accurate computational modeling. To date, X-ray structures are known for methylmalonyl-CoA mutase [33], glutamate mutase [34], diol dehydrase [35], glycerol dehydrase [36], ethanol-amine ammonialyase [37], lysine-5,6-aminomutase [17], ornithine-4,5-aminomutase [38] and ribonucleotide reductase [39]. Despite their diversity in both subunit composition and quaternary structures, the enzymes all comprise two major domains—a (β/α)8 barrel (TIM barrel) domain which encompasses the substrate binding site and binds the “upper” face of the corrin ring and the adenosyl moiety (in RNR this is a (β/α)10 barrel). A second cobalaminbinding domain binds the “lower” face of the corrin ring and the dimethylbenzimidazole ‘tail’ of the coenzyme that provides the lower ligand to the central cobalt atom.
There are two binding modes for binding cobalamins that differ in the identity of the axial nitrogen ligand to cobalt. In the carbon skeleton mutases and aminomutases the cobalamin-binding domain adopts an α/β (Rossmann-like) fold, with the corrin ring of cobalamin sitting at the C-terminal end. A histidine residue, part of a conserved motif [40] from the protein, replaces the dimethylbenzimidazole tail as the lower ligand to cobalt, which is instead extended down into the core of the protein. In contrast, in the eliminases and ribonucleotide reductase AdoCbl is bound with the dimethylbenzimidazole base coordinated to cobalt, as it is in free solution. In this case the coenzyme rests on the side of a β-sheet which is part of an α/β fold. For comparison, the structures of representative enzymes from each binding mode, diol dehydrase and glutamate mutase, are presented in Fig. 11. Why dimethylbenzimidazole is replaced with histidine in some proteins, and not others, remains unclear.
Fig. 11.
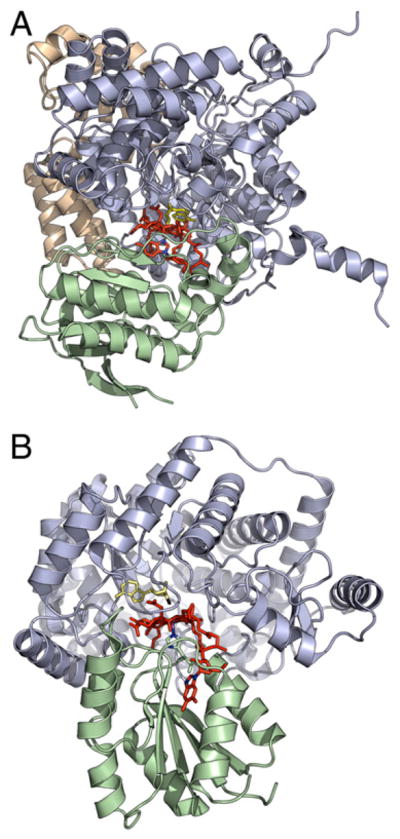
A: Structure of diol dehydrase (PDB 1EEX) with adenylpentanyl-cobalamin bound in ‘base-on’ mode. The catalytic subunit is shown in blue and the B12-binding subunit in green (the purely structural γ-subunit is in gold); the adenylpentanyl ligand is in yellow and the cobalamin portion in red. B: Structure of glutamate mutase (PDB 1I9C) with AdoCbl bound in ‘base-off’ mode. The catalytic E subunit is in blue and the B12-binding S subunit, with Co-coordinating histidine side-chain shown, is in green; the adenosyl ligand is in yellow (the bond between the ligand and cobalt is broken in the structure) and the cobalamin portion in red.
For several enzymes, structures are available of both substrate-bound and substrate-free enzyme forms, e.g. methylmalonyl-CoA mutase [41] and diol dehydrase [42]. These show that significant changes to the conformation of the protein occur on substrate binding. Obviously, such changes may be important to activate the coenzyme towards homolysis. However, a difficulty encountered in all the enzyme structures is that it has not been possible to obtain a good structure of the Michaelis complex of the enzyme poised for catalysis. Either the coenzyme does not survive intact in the structure, i.e. the Co–C bond is cleaved and the cobalt is oxidized to the 3+ form, or the enzyme has to be crystallized with an inactive analog of AdoCbl such as adeninylpentyl-Cbl. In other cases, such as lysine and ornithine aminomutases, the enzyme crystallizes in a conformation that places the coenzyme and substrate too far apart for reaction to occur and so clearly cannot be the catalytically active conformation.
4. Mechanism of adenosyl radical formation
The first step in the mechanisms of all AdoCbl enzymes involves homolytic fission of the coenzyme and subsequent substrate radical formation. An important aspect of the mechanism for generating radicals is that the energetics of forming Ado• are extremely unfavorable, as the bond dissociation energy of the Co–C bond, in free solution, is 32 kcal/mol [43]. Ado• is such a reactive species that it has never been directly observed in an enzyme, although it is generally accepted as the key intermediate. Yet in response to substrate binding, AdoCbl readily undergoes homolytic cleavage and substrate-based radicals accumulate on the enzyme during turn-over, implying that the equilibrium constant for homolysis is now close to unity [15,44–47]. How enzymes stabilize highly reactive radical species and harness them towards catalysis is an important and fundamental question, the answer to which has implications for all enzymes that catalyze radical reactions. Only recently, as insights from structure, mechanistic experiments and computational modeling have converged, have the answers to this question begun to emerge.
It has been generally assumed that a protein-induced distortion of the coenzyme, possibly triggered by substrate binding, weakens the Co–C bond sufficiently to promote homolysis; i.e. the enzyme uses binding energy to offset the unfavorable bond dissociation energy. Attempts to verify this hypothesis experimentally have largely failed to find evidence to support this type of activation mechanism. The X-ray structures of the enzymes do not show any major structural distortion of the corrin ring. However, as noted above, there is not a good structure for the pre-homolysis Michaelis complex. And it is unlikely that a crystal structure would trap reactive, strained species if indeed they were formed. Complementary studies on several enzymes using resonance Raman, Uv–Visible and magnetic circular dichroism spectroscopies, which are sensitive to changes in the electronic or vibrational properties of the Co–C bond, have similarly failed to detect signicicant distortion of the coenzyme when AdoCbl binds to the enzyme [48–52]. Computational studies, discussed below, have been particularly useful in understanding this aspect of AdoCbl reactions, highlighting the role that electrostatic interactions may play in facilitating homolysis of the coenzyme.
A key feature of AdoCbl homolysis is that it is closely coupled to abstraction of a non-acidic hydrogen atom from the substrate by Ado•. No homolysis is observed until the substrate binds to the enzyme, so that reactive free radicals are only generated when substrate is present to react. EPR experiments with isotopically-labeled substrates have established that it is the substrate-based radical, not Ado•, that accumulates on the enzyme [4,53–56]. The coupling of homolysis and hydrogen abstraction has been investigated by pre-steady state stopped-flow experiments [15,44–47]. The cleavage of the Co–C bond can be followed directly by monitoring changes to the Uv–Visible spectrum that are associated with the conversion of AdoCbl to cob(II)alamin and Ado•. The observed rate of homolysis was found to be significantly slower when deuterated substrates were used to initiate the reaction, i.e. there was a significant KIE operating, even though this is not formally an isotopically sensitive step.
This result can be interpreted as a formally concerted mechanism, in which the Co–C bond cleavage occurs as the hydrogen atom is transferred from the substrate to 5′-deoxyadenosine. Alternatively, a kinetically coupled mechanism may be operating, in which a rapidly established but unfavorable equilibrium between AdoCbl and Ado• is first formed. Then Ado• is siphoned off by the subsequent slow and isotopically sensitive reaction with the substrate to form the more stable substrate-based radical. The net result is that AdoCbl is depleted at a rate that depends on the rate of hydrogen atom transfer. Both mechanisms, illustrated in Fig. 12, predict a kinetic isotope effect, and experimentally they are very hard to distinguish between.
Fig. 12.
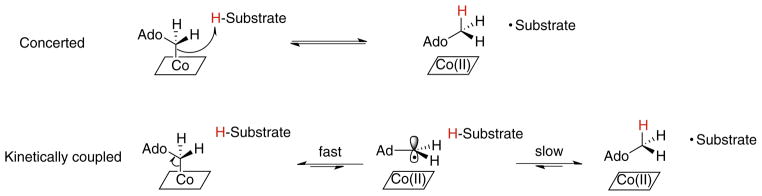
Distinction between a concerted mechanism for AdoCbl homolysis and hydrogen transfer from the substrate (top) and a kinetically coupled mechanism for AdoCbl homolysis and hydrogen transfer from the substrate (bottom). In the latter mechanism, an isotope effect on the second, slower step results in a slower apparent rate constant for Co–C bond cleavage in the first step.
On balance, a kinetically coupled mechanism appears more likely. Analysis of the X-ray structures for enzymes indicated that the substrate-binding sites and the 5′-carbon of AdoCbl appear to be too far apart to permit a formally concerted reaction to occur without a major reorganization of the protein’s conformation. Further support comes from an experiment using a ribonucleotide reductase in which the catalytic cysteine residue was mutated. It was found that the crippled enzyme could catalyze epimerization of AdoCbl stereospecifically deuterium-labeled at the 5′-carbon in the presence of an allosteric activator (Fig. 13), thereby providing evidence for the transient formation of Ado• [57].
Fig. 13.

Epimerization of stereospecifically deuterated AdoCbl by a catalytically crippled ribonucleotide reductase mutant provides evidence for the transient cleavage of the Co–C bond in the absence of hydrogen transfer.
Interestingly, AdoCbl homolysis is also, potentially, susceptible to external magnetic fields. The magnetic field affects the rate at which the initially-formed singlet radical pair, which can recombine to reform the Co–C bond, converts to the triplet state, which cannot recombine. Photolysis of AdoCbl bound to ethanolamine ammonia lyase was observed to be sensitive to magnetic fields, whereas substrate-initiated (thermal) cleavage of the coenzyme was not [58,59]. This result was attributed to the rapid removal of the alkyl radical immediately after homolysis, such that there was inadequate time for radical pair recombination to occur. This is consistent with the idea that the coupling of homolysis and hydrogen abstraction steps, and subsequent radical pair stabilization, contribute to the acceleration of Co–C bond homolysis observed in the enzymes.
The observation of kinetic isotope effects associated with transfer of hydrogen between substrate and Ado• has allowed the transition state for this unusual reaction to be investigated in more detail. For methylmalonyl-CoA mutase, the magnitude and temperature dependence of the pre-steady state kinetic isotope effects (measured spectroscopically by following the apparent KIE on Co–C bond cleavage) both indicate that hydrogen transfer between the substrate and Ado• occurs through quantum tunneling rather classical motion. The deuterium kinetic isotope effect decreases from 50 at 5 °C to 36 at 20 °C. These values are clearly much larger than the semi-classical limit of ~7. Furthermore, the steep temperature dependence of the KIE indicates a reaction in which tunneling dominates the transfer of protium between substrate and coenzyme whereas deuterium transfer occurs semi-classically [60,61].
Interestingly, very similar KIEs were measured for the non-enzymatic reaction of AdoCbl with ethylene glycol [62]. In these experiments, the rates of reaction were compared for the formation of Ado-H by thermolysis of AdoCbl in either ethylene glycol or perdeutrated ethylene glycol. During the reaction, the transiently formed Ado• abstracts hydrogen from ethylene glycol and thus may be considered a model for the initial step of the diol dehydrase reaction. The KIEs measured in this experiment were also unusually large (~12 at 80 °C) and showed strong temperature dependence. Although the reaction was too slow to measure KIEs at lower temperatures, which would have allowed direct comparisons with measurements on the enzymes, subsequent experiments using the more reactive neopentylcobalamin allowed the measurements to be extended to physiological temperatures, confirming the high temperature results [63]. From these data it was argued that the degree of tunneling was the same in both enzymatic and non-enzymatic reactions and thus tunneling should be discounted as a source of the ~1012-fold rate acceleration for the homolysis of AdoCbl by the enzyme. However, more recently developed theoretical treatments of tunneling in enzyme reactions, so called “full tunneling models” [60,61], which emphasize the importance of protein motions in catalysis, suggest that temperature-dependent KIEs cannot be interpreted as simply “more” or “less” tunneling. This is because both hydrogen tunneling and low frequency protein motions that are believed to promote catalysis (so-called gating motions) contribute to the magnitude and temperature dependence of the KIE.
Further insights into the nature of the hydrogen transfer step have come from experiments on glutamate mutase in which the intrinsic primary and secondary KIEs on Ado-H formation were measured. In particular, secondary isotope effects are sensitive to changes in bond stiffness during reactions and therefore can provide valuable insights into transition states [64]. Using rapid quench pre-steady state techniques, both secondary tritium kinetic isotope effects and secondary tritium equilibrium isotope effects associated with AdoCbl homolysis and hydrogen abstraction in glutamate mutase were measured [65]. The secondary equilibrium tritium isotope effect reports on both breaking of the AdoCbl cobalt–carbon bond and transfer of hydrogen from substrate to Ado-H, whereas the secondary kinetic isotope effect is dominated by the latter step, which primary isotope effect measurements indicate is relatively slow. Surprisingly, a large inverse equilibrium isotope effect of 0.72±0.04 was found for the overall reaction. This indicates that the 5′-C–H bonds become significantly stiffer in going from AdoCbl to Ado-H, even though the 5′-carbon remains formally sp3 hybridized [66].
The secondary kinetic isotope effect for the formation of Ado-H was 0.76±0.02. This large, inverse secondary KIE would usually be interpreted as a “late” transition state for the reaction [64]. However, when the isotope effect measurements were repeated using deuterated substrate, so that Ado-D was formed, the secondary kinetic isotope effect deflated to a value close to unity, 1.05±0.08, whereas the equilibrium secondary isotope effect remained unchanged [67]. This observation represents a breakdown in the “Rule of the Geometric Mean” which in essence states that there are no isotope effects on isotope effects [68]. The change in secondary KIE due to the primary KIE is consistent with concerted motion in the transition state of the 5′-hydrogen atoms adjacent to the hydrogen that is transferred between substrate and coenzyme (Fig. 14), in a reaction that involves a large degree of quantum tunneling.
Fig. 14.
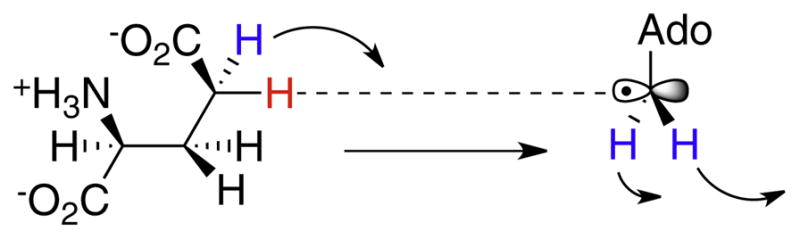
Illustration of the coupled motions of primary (red) and secondary (blue) hydrogen atoms involved in the transition state for hydrogen transfer between 5′-dA radical and glutamate, as deduced from secondary isotope effect measurements.
Further evidence for hydrogen tunneling in glutamate mutase comes from temperature dependence studies on the primary deuterium KIE for hydrogen transfer from methylaspartate to Ado-H [69,70]. In this experiment the KIE was measured by setting up an intra-molecular competition between hydrogen and deuterium on the same methyl group of methylaspartate and analyzing the isotopic composition of the Ado-H formed. The experiment has the important advantage that the isotope effect can be measured, even when the isotopically sensitive step is not rate-determining. This is because the protium and deuterium atoms on the methyl group remain chemically equivalent, even in the enzyme active site, due to the rapid rotation of the methyl group.
The intrinsic deuterium KIE for glutamate mutase is much smaller than those measured for either MMCM or the AdoCbl model studies [47,62,63]. At 10 °C the KIE is 4.1, which is well within the semi-classical limit. However, the isotope effect still exhibits strong temperature dependence indicative of hydrogen tunneling, consistent with secondary KIE measurements. This argues strongly that glutamate mutase plays a role in modulating the transition state for hydrogen transfer, and thereby changing the isotope effect. Indeed, one expected consequence of extensive coupling between the motions of the primary and secondary hydrogen atoms, noted above, is that the primary isotope effect will remain quite small even if extensive hydrogen tunneling is occurring.
5. Computational studies of AdoCbl enzymes
Computational approaches are playing an increasingly important role in understanding the mechanisms of enzymes. Facilitated by increases in computational speed and the development of QM/MM methods that can reliably model both the chemical reaction and the enzyme structure, computational modeling has advanced to the point where, rather than simply rationalizing experimental results, it can provide insights into the role of the protein in catalysis that can be tested by experiment. Computational studies of AdoCbl reactions are particularly interesting because the unusual nature of these rearrangements raises many mechanistic questions that cannot easily be addressed by experiments. For example, experimental approaches have failed to adequately explain the origin of the ~1012-fold rate acceleration for AdoCbl homolysis or the remarkable ability of enzymes to stabilize radical intermediates; however, recent computational studies have highlighted the potential importance of electrostatic interactions in facilitating homolysis [71].
Computational chemistry is a rapidly evolving field, with different research groups pursuing different approaches to modeling enzyme reactions, e.g. density functional theory or valence bond methods, with each methodology possessing its own strengths and weaknesses. It is not the purpose of this review to provide a critical analysis of the methodology used, (see Ref. [9] for an excellent review on computational studies) but rather to concentrate on what insights the computational studies have provided into the mechanisms of AdoCbl enzymes. We briefly note, though, that accurately calculating the BDE of the AdoCbl cobalt–carbon bond has proved particularly challenging; because of this, many studies have focused on the steps that come after homolysis. One factor that has recently been recognized as important for obtaining accurate cobalt–carbon BDE is the inclusion of dispersion forces between the corrin ring and adenosyl moiety of the coenzyme, which are missing from most standard DFT calculations [72].
The use of computational methods to study AdoCbl reactions has been pioneered by Radom and co-workers, who have investigated the mechanisms of all three classes of enzymes [73]. Their work has focused on evaluating the energetic feasibility of various mechanisms proposed for the 1,2 migrations of radical species using high level computational approaches on small molecule models (i.e. the protein is not considered). These studies were the first to highlight the importance of electrostatic interactions in facilitating substrate radical rearrangements, which was previously not appreciated. The protonation state of the migrating groups was found to significantly influence the energetic feasibility of the reaction, implying that enzymes can facilitate the rearrangement of substrate radicals by either general acid–general base catalysis or hydrogen bonding interactions. This early insight has been borne out by subsequent QM/MM studies using the full enzyme structures, as discussed below, in which electrostatic interactions between coenzyme, substrate and protein make energetic contributions to catalysis. Here we discuss how computational studies on three enzymes, glutamate mutase, MMCM and diol dehydrase, have contributed to our understanding of their mechanisms.
Small molecule computational studies were initially used to investigate the feasibility of fragmentation–recombination mechanism for the rearrangement of the glutamyl radical to methylaspartyl radical in glutamate mutase [74,75]. The mechanism is supported experimentally by the observation that acrylate is formed as a kinetically competent intermediate during turn-over [25]. Fragmentation of the glutamyl radical to form a glycyl radical and acrylate was calculated to be unfavorable by 44 kcal/mol when the amino group of glutamate was protonated. However, when deprotonated, the energy is dramatically reduced to 8.8 kcal/mol above that of the glutamyl radical. The lower energy of the intermediate fragments was attributed to the capto-dative stabilization of the glycyl radical, involving the strong π-donor (amino) and strong π-acceptor (carboxylic acid) substituents adjacent to the unpaired electron. Mutagenesis studies have demonstrated that Glu171 in glutamate mutase, which makes a hydrogen bonding interaction with the amino group of the substrate, is important for catalysis [76,77]. This supports the idea that the enzyme may fully or partially (by hydrogen bonding) deprotonate the substrate to facilitate the rearrangement.
Important insights into the mechanism by which the protein activates the coenzyme bond towards homolysis and achieves the remarkable stabilization of radical intermediates have come from computational studies on the full glutamate mutase system using QM/MM methods. A particularly detailed study by Jensen and Ryde on glutamate mutase was one of the first to explicitly include the protein in the calculations and obtain results that closely matched experiments [78]. The study compared AdoCbl homolysis in ‘vacuum’ and in the enzyme. In the protein environment the Co–C bond dissociation energy was only ~4 kcal/mol, compared with 34 kcal/mol calculated in vacuum. When the energetics of the subsequent hydrogen transfer step were considered, the overall energy for the reaction of AdoCbl and glutamate to give glutamyl radical, Ado-H and Cbl(II) was favorable by ~2 kcal/mol—in good agreement with experimental data showing the equilibrium constant for this reaction to be close to unity.
The study also provided some detailed analysis into how Co–C bond is cleaved enzymatically. Some of the catalytic effect arises from the enzyme maintaining the Ado• in close proximity to the corrin ring, essentially a caging effect. The largest effect is attributed to distortion of the coenzyme by the protein, primarily through interactions with the ribosyl moiety which distorts the Co–C5′– C4′ angle. This distortion could not be discerned from the X-ray structure of GM as the Co–C bond is cleaved in the structure [10]. Further stabilization is afforded by differential electrostatic interactions with the ribosyl group that are present in the Cbl(II) state, but not in the ground state. Lastly, a minor component appears to be stabilization of the protein structure itself in the Cbl(II) state from favorable van der Waals and electrostatic interactions. Thus a subtle combination of factors appears to explain the overall rather large, (~30 kcal/mol) stabilization of radical species by the enzyme.
A subsequent study of the glutamate mutase reaction employed an all QM approach to a truncated model system, without the protein [79]. It was found that a transition state involving concerted Co–C bond homolysis and hydrogen transfer was more energetically favorable than a step-wise mechanism, which appears more likely to occur in the enzyme. In this case the overall energetics of the reaction are highly unfavorable; formation of substrate radical, Ado-H, and Cbl(II) from AdoCbl and substrate requires 22 kcal/mol. This illustrates the importance of the protein, both in influencing the reaction mechanism and energetics.
Most recently, the full glutamate mutase reaction pathway (but excluding AdoCbl homolysis) has been modeled using the complete protein and compared to the reaction occurring in the absence of protein [80]. The calculations identify the hydrogen atom transfer steps as rate-determining in the overall rearrangement, and qualitatively reproduce the free energy profile for the reaction determined experimentally [81]. The study also investigated the role of a number of protein side-chains in the reaction, both at the active site and further away, that appear to significantly alter the free energy profile of the reaction, mainly though electrostatic effects. Some of these residues had previously been investigated by mutagenesis, allowing the calculations to be compared with experimental data. In particular, Glu-171, (discussed above) was found to assist in catalysis by deprotonating the amino group of the substrate and thereby facilitate rearrangement of the substrate. This nicely agrees with earlier mechanistic proposals based on the pH vs rate profile for the enzyme combined with kinetic analysis of Glu-171 mutants [76,77].
Small molecule computational studies have been effectively used to investigate the mechanism of methylmalonyl-CoA mutase [73,82]. One question is whether the rearrangement of the substrate radical involves a fragmentation–recombination mechanism, in which acrylate and a thioacyl-CoA radical would be intermediates, or an addition–elimination mechanism, which involves the transient formation of a cyclopropyl intermediate (Fig. 8). Neither species has been directly observed in the enzyme, although studies on model compounds support the latter mechanism [23]. Using the rearrangement of the 3-propanal radical as a minimal model for the methylmalonyl-CoA mutase reaction, the addition-elimination mechanism was found to be the more favorable mechanism by ~12 kcal/mol. The thioester carbonyl oxygen of methylmalonyl-CoA can potentially form a hydrogen bond to the enzyme through His242 that may stabilize the cyclopropyl-radical intermediate. In the model reaction, protonation of the migrating carbonyl group was found to significantly lower the reaction barrier: with H3O+ as the protonating species (a strong acid in the gas phase) the energy barrier for rearrangement was only 2.5 kcal/mol.
Various QM/MM computational studies have examined aspects of the methylmalonyl-CoA mutase reaction using either the full enzyme structure or a truncated structure. Similar to glutamate mutase, methylmalonyl-CoA mutase has been found to strongly stabilize the dissociated form of AdoCbl in the conformation that binds substrate, with changes in electrostatic interactions between substrate and protein appearing to play a significant role [83,84]. Again a step-wise, rather than concerted pathway for coenzyme homolysis and/or hydrogen abstraction appears most energetically feasible.
The influence of hydrogen tunneling in the methylmalonyl-CoA mutase reaction has also been addressed computationally [85]. The very large KIEs observed for hydrogen abstraction in methylmalonyl-CoA mutase could be successfully modeled using a multidimensional tunneling model that emphasizes the coupling of hydrogen tunneling to active site motions of the protein. In this enzyme, tunneling appears to increase the rate of hydrogen transfer by two orders of magnitude.
The reaction catalyzed by diol dehydrase involves the energetically unfavorable 1,2-migration of a hydroxyl group to for a gem-diol intermediate (Fig. 3). Computational studies on the rearrangement of the ethylene glycol radical suggested that protonation (or partial protonation by hydrogen bonding) of the migrating hydroxyl would lead to significant decrease in the barrier height for this step [86]. It was subsequently found that the energy barrier could be lowered further by (partial) deprotonation of the spectator hydroxyl to only ~2 kcal/mol [87]. The synergistic effect of protonation with deprotonation has been referred to as a “push–pull” mechanism with protonation of the migrating –OH providing the “push” and deprotonation of the spectator –OH providing the “pull”.
The diol dehydrase reaction mechanism has also been computationally modeled using the complete structure of the enzyme [88,89]. The studies provide a nice illustration of the usefulness of computational studies to inform the design of experiments; in this case leading to a re-evaluation of the role of essential metal ions in the reaction. The enzyme requires potassium ions for activity and the crystal structure revealed two metal ions bound at the active site [42]. One K+ ion is bound close to the adenine moiety of the coenzyme, and may be important for activating the coenzyme towards homolysis. The other metal ion coordinates the two hydroxyl groups of the substrate. Other important substrate-protein interactions are made by the carboxylate of Glu170, which forms a hydrogen bond to the C-1 (spectator) –OH, and His143 and Asp335 that hydrogen to the C-2 (migrating) –OH.
The relative energies and structures of the radical intermediates and transition states for the reaction were calculated for diol dehydrase using QM/MM methods [88,89]. The calculations appear to qualitatively reproduce experimentally determine features of the reaction mechanism; importantly, the highest energy transition state is associated with abstraction of hydrogen from the substrate by adenosyl radical, which is consistent with a large deuterium kinetic isotope effect for the overall reaction. The calculations also evaluated the role of the active site residues in catalysis. Calculations on enzyme indicated that Glu170 appears to facilitate –OH migration by deprotonating the spectator –OH, consistent with it providing “pull” and also with mutagenesis studies indicating its important role in catalysis. On the other hand, calculations predicted that protonation of the migrating –OH by His143 (to provide some “push”) would lead to its loss as water from the substrate and direct formation of a propanalyl radical. This result contradicted labeling studies that require 1,1-propanediol to be formed as an intermediate [90]. Interestingly, the propanalyl radical is calculated to be too stable to re-abstract hydrogen from Ado-H, so the enzyme could not complete the catalytic cycle. Presumably the enzyme has learned not to push too hard!
Although it was generally accepted that K+ was the only metal required by diol dehydrase, the metal-oxygen bond lengths determined from the crystal structure were significantly shorter than those calculated from the QM/MM model of diol dehydrase. The crystallographic bond lengths averaged 2.4 A as opposed to 2.7 A obtained by calculation, assuming K+. To investigate this discrepancy computational modeling was used to replace K+ in-silico with other metals and the structures re-calculated [91]. Using Na+, K+, Mg2+ and Ca2+, only calcium resulted in metal–oxygen bond lengths that were in agreement with the experimental data. Subsequent biochemical investigations determined that Ca2+ is indeed required for activity [32]. The metal is tightly bound by the protein and its removal with chelating agents results in unfolding of the protein. Because of this, the essential role of Ca2+ in catalysis had been over-looked for more than 40 years. K+ is still required for activity and is presumably bound at the other metal site.
6. Conclusions
Although restricted in their biological functions, AdoCbl enzymes serve as a paradigm for understanding how enzymes generate and control highly reactive free radicals. The remarkable stabilization of radical species by these enzymes is central to their catalytic efficiency and has proved challenging to explain. Despite the fact that free radicals are electrically neutral species, various lines of experimental and theoretic evidence now point to the importance of electrostatic interactions between the protein, substrate and coenzymes in stabilizing radical intermediates. As in other enzymes, hydrogen tunneling has emerged as dominant mechanism by which the hydrogen atom moves between substrate and coenzyme. Computer modeling of AdoCbl-enzymes, using either the complete structure of the enzyme or a structure truncated around the active site, has provided new insight into their mechanisms and allowed detailed predictions to be made as to the role of specific protein–substrate and protein–coenzyme interactions in catalyzing the reaction; in the case of diol dehydrase, modeling led to a reevaluation of the metal requirements for the enzyme. It should now be possible to test experimentally, e.g. by kinetic analysis of mutant enzymes and/or modified substrates and coenzymes, the importance of protein–coenzyme and protein–substrate interactions identified by QM/MM simulations, many of which are non-obvious from inspection of the structure.
AdoCbl provides an elegant method of generating reactive radicals at the enzyme active site in response to substrate binding. Moreover, there are many reactions, both enzymic and non-enzymic, that proceed through radical mechanisms that could in principle be catalyzed by AdoCbl. Advances in our understanding of the principles by which these enzymes generate and control free radicals holds forth the prospect of re-designing them to catalyze a broader range of free radical chemistry.
Acknowledgments
Research in the authors’ laboratory on AdoCbl enzymes is supported by NIH grant GM 093088 to E.N.G.M.
Footnotes
This article is part of a Special Issue entitled: Radical SAM enzymes and Radical Enzymology.
References
- 1.Brown KL. Chemistry and enzymology of vitamin B12. Chem Rev. 2005;105:2075–2149. doi: 10.1021/cr030720z. [DOI] [PubMed] [Google Scholar]
- 2.Matthews RG. Cobalamin- and corrinoid-dependent enzymes. Met Ions Life Sci. 2009:53–114. doi: 10.1039/BK9781847559159-00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Toraya T. Radical catalysis in coenzyme B12-dependent isomerization (eliminating) reactions. Chem Rev. 2003;103:2095–2127. doi: 10.1021/cr020428b. [DOI] [PubMed] [Google Scholar]
- 4.Reed GR. Radical mechanisms in adenosylcobalamin-dependent enzymes. Curr Opin Chem Biol. 2004;8:477–483. doi: 10.1016/j.cbpa.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 5.Marsh ENG, Drennan CL. Adenosylcobalamin-dependent isomerases: new insights into structure and mechanism. Curr Opin Chem Biol. 2001;5:499–505. doi: 10.1016/s1367-5931(00)00238-6. [DOI] [PubMed] [Google Scholar]
- 6.Frey PA, Hegerman AD, Reed GH. Free radical mechanisms in enzymology. Chem Rev. 2006;106:3302–3316. doi: 10.1021/cr050292s. [DOI] [PubMed] [Google Scholar]
- 7.Buckel W, Golding BT. Radical enzymes in anaerobes. Annu Rev Microbiol. 2006;60:27–49. doi: 10.1146/annurev.micro.60.080805.142216. [DOI] [PubMed] [Google Scholar]
- 8.Banerjee R. Radical carbon skeleton rearrangements: catalysis by coenzyme B12-dependent mutases. Chem Rev. 2003;103:2083–2094. doi: 10.1021/cr0204395. [DOI] [PubMed] [Google Scholar]
- 9.Jensen KP, Ryde U. Cobalamins uncovered by modern electronic structure calculations. Coord Chem Rev. 2009;253:769–778. [Google Scholar]
- 10.Gruber K, Kratky C. Coenzyme B12 dependent glutamate mutase. Curr Opin Chem Biol. 2002;6:598–603. doi: 10.1016/s1367-5931(02)00368-x. [DOI] [PubMed] [Google Scholar]
- 11.Marsh ENG. Coenzyme B12-dependent glutamate mutase. Bioorg Chem. 2000;28:176–189. doi: 10.1006/bioo.2000.1168. [DOI] [PubMed] [Google Scholar]
- 12.Zerbe-Burkhardt K, Ratnatilleke A, Philippon N, Birch A, Leiser A, Vrijbloed JW, Hess D, Hunziker P, Robinson JA. Cloning, sequencing, expression, and insertional inactivation of the gene for the large subunit of the coenzyme B12-dependent isobutyryl-CoA mutase from Streptomyces cinnamonensis. J Biol Chem. 1998;273:6508–6517. doi: 10.1074/jbc.273.11.6508. [DOI] [PubMed] [Google Scholar]
- 13.Rohwerder T, Breuer U, Benndorf D, Lechner U, Muller RH. The alkyl tert-butyl ether intermediate 2-hydroxyisobutyrate is degraded via a novel cobalamin-dependent mutase pathway. Appl Environ Microbiol. 2006;72:4128–4135. doi: 10.1128/AEM.00080-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Erb TJ, Retey J, Fuchs G, Alber BE. Ethylmalonyl-CoA mutase from Rhodobacter sphaeroides defines a new subclade of coenzyme B12-dependent acyl-CoA mutases. J Biol Chem. 2008;283:32283–32293. doi: 10.1074/jbc.M805527200. [DOI] [PubMed] [Google Scholar]
- 15.Wolthers KR, Rigby SEJ, Scrutton NS. Mechanism of radical-based catalysis in the reaction catalyzed by adenosylcobalamin-dependent ornithine 4,5-amino-mutase. J Biol Chem. 2008;283:34615–34625. doi: 10.1074/jbc.M807911200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen HP, Hsui FC, Lin LY, Ren CT, Wu SH. Coexpression, purification and characterization of the E and S subunits of coenzyme B12 and B-6 dependent Clostridium sticklandii D-ornithine aminomutase in Escherichia coli. Eur J Biochem. 2004;271:4293–4297. doi: 10.1111/j.1432-1033.2004.04369.x. [DOI] [PubMed] [Google Scholar]
- 17.Berkovitch F, Behshad E, Tang KH, Enns EA, Frey PA, Drennan CL. Locking mechanism preventing radical damage in the absence of substrate as revealed by the X-ray structure of lysine 5,6-aminomutase. Proc Natl Acad Sci U S A. 2004;101:15870–15875. doi: 10.1073/pnas.0407074101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tang KH, Mansoorabadi SO, Reed GH, Frey PA. Radical triplets and suicide inhibition in reactions of 4-thia-D- and 4-thia-L-lysine with lysine 5,6-aminomutase. Biochemistry. 2009;48:8151–8160. doi: 10.1021/bi900828f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stubbe J. Radicals with a controlled lifestyle. Chem Commun. 2003:2511–2513. doi: 10.1039/b307617m. [DOI] [PubMed] [Google Scholar]
- 20.Reichard P. Ribonucleotide reductases: the evolution of allosteric regulation. Arch Biochem Biophys. 2002;397:149–155. doi: 10.1006/abbi.2001.2637. [DOI] [PubMed] [Google Scholar]
- 21.Marsh ENG, Patterson DP, Li L. Adenosyl radical: reagent and catalyst in enzyme reactions. Chembiochem. 2010;11:604–621. doi: 10.1002/cbic.200900777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Frey PA, Hegeman AD, Ruzicka FJ. The radical SAM superfamily. Crit Rev Biochem Mol Biol. 2008;43:63–88. doi: 10.1080/10409230701829169. [DOI] [PubMed] [Google Scholar]
- 23.He M, Dowd P. Mechanism of action of vitamin B12. Ultrafast radical clocks provide no evidence for radical intermediates in cyclopropane models for the methylmalonyl-CoA to succinyl-CoA carbon skeleton rearrangement. J Am Chem Soc. 1998;120:1133–1137. [Google Scholar]
- 24.Ashwell S, Davies AG, Golding BT, Haymotherwell R, Mwesigyekibende S. Model experiments pertaining to the mechanism of action of vitamin-B12- dependent alpha-methyleneglutarate mutase. J Chem Soc Chem Commun. 1989:1483–1485. [Google Scholar]
- 25.Chih H-W, Marsh ENG. Mechanism of glutamate mutase: identification and kinetic competence of acrylate and glycyl radical as intermediates in the rearrangement of glutamate to methylaspartate. J Am Chem Soc. 2000;122:10732–10733. [Google Scholar]
- 26.Wu WM, Booker S, Lieder KW, Bandarian V, Reed GH, Frey PA. Lysine 2,3-aminomutase and trans-4,5-dehydrolysine: characterization of an allylic analogue of a substrate-based radical in the catalytic mechanism. Biochemistry. 2000;39:9561–9570. doi: 10.1021/bi000658p. [DOI] [PubMed] [Google Scholar]
- 27.Wu WM, Lieder KW, Reed GH, Frey PA. Observation of a second substrate radical intermediate in the reaction of lysine 2,3-aminomutase—a radical centered on the beta-carbon of the alternative substrate, 4-thia-L-lysine. Biochemistry. 1995;34:10532–10537. doi: 10.1021/bi00033a027. [DOI] [PubMed] [Google Scholar]
- 28.Ballinger MD, Reed GH, Frey PA. An organic radical in the lysine 2,3-amino-mutase reaction. Biochemistry. 1992;31:949–953. doi: 10.1021/bi00119a001. [DOI] [PubMed] [Google Scholar]
- 29.Tang KH, Harms A, Frey PA. Identification of a novel pyridoxal 5′-phosphate binding site in adenosylcobalamin-dependent lysine 5,6-aminomutase from Porphyromonas gingivalis. Biochemistry. 2002;41:8767–8776. doi: 10.1021/bi020255k. [DOI] [PubMed] [Google Scholar]
- 30.Sandala GM, Smith DM, Coote ML, Golding BT, Radom L. Insights into the hydrogen-abstraction reactions of diol dehydratase: relevance to the catalytic mechanism and suicide inactivation. J Am Chem Soc. 2006;128:3433–3444. doi: 10.1021/ja057902q. [DOI] [PubMed] [Google Scholar]
- 31.Wetmore SD, Smith DM, Bennett JT, Radom L. Understanding the mechanism of action of B12-dependent ethanolamine ammonialyase: synergistic interactions at play. J Am Chem Soc. 2002;124:14054–14065. doi: 10.1021/ja027579g. [DOI] [PubMed] [Google Scholar]
- 32.Toraya T, Honda S, Mori K. Coenzyme B12-dependent diol dehydratase is a potassium ion-requiring calcium metalloenzyme: evidence that the substrate-coordinated metal ion is calcium. Biochemistry. 2010;49:7210–7217. doi: 10.1021/bi100561m. [DOI] [PubMed] [Google Scholar]
- 33.Mancia F, Keep NH, Nakagawa A, Leadlay PF, McSweeney S, Rasmussen B, Bosecke P, Diat O, Evans PR. How coenzyme B12 radicals are generated: the crystal structure of methylmalonyl-coenzyme A mutase at 2 A resolution. Structure. 1996;4:339–350. doi: 10.1016/s0969-2126(96)00037-8. [DOI] [PubMed] [Google Scholar]
- 34.Reitzer R, Gruber K, Jogl G, Wagner UG, Bothe H, Buckel W, Kratky C. Structure of coenzyme B12 dependent enzyme glutamate mutase from Clostridium cochlearium. Structure. 1999;7:891–902. doi: 10.1016/s0969-2126(99)80116-6. [DOI] [PubMed] [Google Scholar]
- 35.Masuda J, Shibata N, Morimoto Y, Toraya T, Yasuoka N. How a protein generates a catalytic radical from coenzyme B12: X-ray structure of a dioldehydratase-adeninylpentylcobalamin complex. Struct Fold Des. 2000;8:775–788. doi: 10.1016/s0969-2126(00)00164-7. [DOI] [PubMed] [Google Scholar]
- 36.Yamanishi M, Yunoki M, Tobimatsu T, Sato H, Matsui J, Dokiya A, Iuchi Y, Oe K, Suto K, Shibata N, Morimoto Y, Yasuoka N, Toraya T. The crystal structure of coenzyme B12-dependent glycerol dehydratase in complex with cobalamin and propane-1,2-diol. Eur J Biochem. 2002;269:4484–4494. doi: 10.1046/j.1432-1033.2002.03151.x. [DOI] [PubMed] [Google Scholar]
- 37.Shibata N, Tamagaki H, Hieda N, Akita K, Komori H, Shomura Y, Terawaki S, Mori K, Yasuoka N, Higuchi Y, Toraya T. Crystal structures of ethanolamine ammonialyase complexed with coenzyme B12 analogs and substrates. J Biol Chem. 2010;285:26484–26493. doi: 10.1074/jbc.M110.125112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wolthers KR, Levy C, Scrutton NS, Leys D. Large-scale domain dynamics and adenosylcobalamin reorientation orchestrate radical catalysis in ornithine 4,5-aminomutase. J Biol Chem. 2010;285:13942–13950. doi: 10.1074/jbc.M109.068908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sintchak MD, Arjara G, Kellogg BA, Stubbe J, Drennan CL. The crystal structure of class II ribonucleotide reductase reveals how an allosterically regulated monomer mimics a dimer. Nat Struct Biol. 2002;9:293–300. doi: 10.1038/nsb774. [DOI] [PubMed] [Google Scholar]
- 40.Marsh ENG, Holloway DE. Cloning and sequencing of glutamate mutase component S from Clostridium tetanomorphum. Homologies with other cobalamin-dependent enzymes. FEBS Lett. 1992;310:167–170. doi: 10.1016/0014-5793(92)81321-c. [DOI] [PubMed] [Google Scholar]
- 41.Mancia F, Evans PR. Conformational changes on substrate binding to methylmalonyl CoA mutase and new insights into the free radical mechanism. Structure. 1998;6:711–720. doi: 10.1016/s0969-2126(98)00073-2. [DOI] [PubMed] [Google Scholar]
- 42.Shibata N, Masuda J, Morimoto Y, Yasuoka N, Toraya T. Substrate-induced conformational change of a coenzyme B12-dependent enzyme: crystal structure of the substrate-free form of diol dehydratase. Biochemistry. 2002;41:12607–12617. doi: 10.1021/bi026104z. [DOI] [PubMed] [Google Scholar]
- 43.Hay BP, Finke RG. Thermolysis of the Co–C bond in adenosylcorrins. 3. Quantification of the axial base effect in adenosylcobalamin by the synthesis and thermolysis of axial base-free adenosylcobinamide. Insights Into the energetics of enzyme-assisted cobalt-carbon bond homolysis. J Am Chem Soc. 1987;109:8012–8018. [Google Scholar]
- 44.Marsh ENG, Ballou DP. Coupling of cobalt–carbon bond homolysis and hydrogen atom abstraction in adenosylcobalamin-dependent glutamate mutase. Biochemistry. 1998;37:11864–11872. doi: 10.1021/bi980512e. [DOI] [PubMed] [Google Scholar]
- 45.Bandarian V, Reed GH. Isotope effects in the transient phases of the reaction catalyzed by ethanolamine ammonialyase: determination of the number of exchangeable hydrogens in the enzyme-cofactor complex. Biochemistry. 2000;39:12069–12075. doi: 10.1021/bi001014k. [DOI] [PubMed] [Google Scholar]
- 46.Licht SS, Lawrence CC, Stubbe J. Thermodynamic and kinetic studies on cobalt–carbon bond homolysis by ribonucleotide triphosphate reductase: the importance of entropy in catalysis. Biochemistry. 1999;38:1234–1242. doi: 10.1021/bi981886a. [DOI] [PubMed] [Google Scholar]
- 47.Chowdhury S, Banerjee R. Evidence for quantum mechanical tunneling in the coupled cobalt–carbon bond homolysis-substrate radical generation reaction catalyzed by methylmalonyl-CoA mutase. J Am Chem Soc. 2000;122:5417–5418. [Google Scholar]
- 48.Dong SL, Padmakumar R, Banerjee R, Spiro TG. Co–C bond activation in B12-dependent enzymes: cryogenic resonance Raman studies of methylmalonyl-coenzyme A mutase. J Am Chem Soc. 1999;121:7063–7070. [Google Scholar]
- 49.Huhta MS, Chen H-P, Hemann C, Hille CR, Marsh ENG. Protein–coenzyme interactions in adenosylcobalamin-dependent glutamate mutase. Biochem J. 2001;355:131–137. doi: 10.1042/0264-6021:3550131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brooks AJ, Vlasie M, Banerjee R, Brunold TC. Spectroscopic and computational studies on the adenosylcobalamin-dependent methylmalonyl-CoA mutase: evaluation of enzymatic contributions to Co–C bond activation in the Co3+ ground state. J Am Chem Soc. 2004;126:8167–8180. doi: 10.1021/ja039114b. [DOI] [PubMed] [Google Scholar]
- 51.Brooks AJ, Fox CC, Marsh ENG, Vlasie M, Banerjee R, Brunold TC. Electronic structure studies of the adenosylcobalamin cofactor in glutamate mutase. Biochemistry. 2005;44:15167–15181. doi: 10.1021/bi051094y. [DOI] [PubMed] [Google Scholar]
- 52.Robertson WD, Wang M, Warncke K. Characterization of protein contributions to cobalt–carbon bond cleavage catalysis in adenosylcobalamin-dependent ethanolamine ammonialyase by using photolysis in the ternary complex. J Am Chem Soc. 2011;133:6968–6977. doi: 10.1021/ja107052p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Warncke K. Characterization of the product radical structure in the CoII-product radical pair state of coenzyme B12-dependent ethanolamine deaminase by using three-pulse 2H ESEEM spectroscopy. Biochemistry. 2005;44:3184–3193. doi: 10.1021/bi048196t. [DOI] [PubMed] [Google Scholar]
- 54.Bender G, Poyner RR, Reed GH. Identification of the substrate radical intermediate derived from ethanolamine during catalysis by ethanolamine ammonialyase. Biochemistry. 2008;47:11360–11366. doi: 10.1021/bi801316v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mansoorabadi SO, Padmakumar R, Fazliddinova N, Vlasie M, Banerjee R, Reed GH. Characterization of a succinyl-CoA radical-cob(II)alamin spin triplet intermediate in the reaction catalyzed by adenosylcobalamin-dependent methylmalonyl-CoA mutase. Biochemistry. 2005;44:3153–3158. doi: 10.1021/bi0482102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yoon M, Patwardhan A, Qiao CH, Mansoorabadi SO, Menefee AL, Reed GH, Marsh ENG. Reaction of adenosylcobalamin-dependent glutamate mutase with 2-thiolglutarate. Biochemistry. 2006;45:11650–11657. doi: 10.1021/bi061067n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen DW, Abend A, Stubbe J, Frey PA. Epimerization at carbon-5′ of (5′R)-[5′, H-2]adenosylcobalamin by ribonucleoside triphosphate reductase: cysteine 408-independent cleavage of the Co\C5′ bond. Biochemistry. 2003;42:4578–4584. doi: 10.1021/bi030018x. [DOI] [PubMed] [Google Scholar]
- 58.Jones AR, Hay S, Woodward JR, Scrutton NS. Magnetic field effect studies indicate reduced geminate recombination of the radical pair in substrate-bound adenosylcobalamin-dependent ethanolamine ammonia lyase. J Am Chem Soc. 2007;129:15718–15727. doi: 10.1021/ja077124x. [DOI] [PubMed] [Google Scholar]
- 59.Jones AR, Woodward JR, Scrutton NS. Continuous wave photolysis magnetic field effect investigations with free and protein-bound alkylcobalamins. J Am Chem Soc. 2009;131:17246–17253. doi: 10.1021/ja9059238. [DOI] [PubMed] [Google Scholar]
- 60.Kohen A. Kinetic isotope effects as probes for hydrogen tunneling, coupled motion and dynamics contributions to enzyme catalysis. Prog React Kinet Mech. 2003;28:119–156. [Google Scholar]
- 61.Nagel ZD, Klinman JP. Tunneling and dynamics in enzymatic hydride transfer. Chem Rev. 2006;106:3095–3118. doi: 10.1021/cr050301x. [DOI] [PubMed] [Google Scholar]
- 62.Doll KM, Bender BR, Finke RG. The first experimental test of the hypothesis that enzymes have evolved to enhance hydrogen tunneling. J Am Chem Soc. 2003;125:10877–10884. doi: 10.1021/ja030120h. [DOI] [PubMed] [Google Scholar]
- 63.Doll KM, Finke RG. A compelling experimental test of the hypothesis that enzymes have evolved to enhance quantum mechanical tunneling in hydrogen transfer reactions: the neopentylcobalamin system combined with prior adocobalamin data. Inorg Chem. 2003;42:4849–4856. doi: 10.1021/ic0300722. [DOI] [PubMed] [Google Scholar]
- 64.Schramm VL. Transition state variation in enzymatic reactions. Curr Opin Chem Biol. 2001;5:556–563. doi: 10.1016/s1367-5931(00)00246-5. [DOI] [PubMed] [Google Scholar]
- 65.Cheng M-C, Marsh ENG. Isotope effects for deuterium transfer between substrate and coenzyme in adenosylcobalamin-dependent glutatmate mutase. Biochemistry. 2005;44:2686–2691. doi: 10.1021/bi047662b. [DOI] [PubMed] [Google Scholar]
- 66.Cheng M-C, Marsh ENG. Pre-steady state measurement of intrinsic secondary tritium isotope effects associated with the homolysis of adenosylcobalamin and the formation of 5′-deoxyadensosine in glutamate mutase. Biochemistry. 2004;43:2155–2158. doi: 10.1021/bi036122w. [DOI] [PubMed] [Google Scholar]
- 67.Cheng MC, Marsh ENG. Evidence for coupled motion and hydrogen tunneling of the reaction catalyzed by glutamate mutase. Biochemistry. 2007;46:883–889. doi: 10.1021/bi0616908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Huskey WP, Schowen RL. Reaction coordinate tunneling in hydride transfer reactions. J Am Chem Soc. 1983;105:5704–5704. [Google Scholar]
- 69.Yoon M, Kalli A, Lee H-Y, Hakensson K, Marsh ENG. Intrinsic deuterium kinetic isotope effects in glutamate mutase measured by an intramolecular competition experiment. Angew Chem. 2007;46:8455–8459. doi: 10.1002/anie.200702448. [DOI] [PubMed] [Google Scholar]
- 70.Yoon M, Song H, Hakansson K, Marsh ENG. Hydrogen tunneling in adenosylcobalamin-dependent glutamate mutase: evidence from intrinsic kinetic isotope effects measured by intramolecular competition. Biochemistry. 2010;49:3168–3173. doi: 10.1021/bi1001695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sharma PK, Chu ZT, Olsson MHM, Warshel A. A new paradigm for electrostatic catalysis of radical reactions in vitamin B12 enzymes. Proc Natl Acad Sci U S A. 2007;104:9661–9666. doi: 10.1073/pnas.0702238104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Siegbahn PEM, Blomberg MRA, Chen SL. Significant van der Waals effects in transition metal complexes. J Chem Theory Comput. 2010;6:2040–2044. doi: 10.1021/ct100213e. [DOI] [PubMed] [Google Scholar]
- 73.Sandala GM, Smith DM, Radow L. Modeling the reactions catalyzed by coenzyme B12-dependent enzymes. Acc Chem Res. 2010;43:642–651. doi: 10.1021/ar900260c. [DOI] [PubMed] [Google Scholar]
- 74.Sandala GM, Smith DM, Marsh ENG, Radom L. Toward an improved understanding of the glutamate mutase system. J Am Chem Soc. 2007;129:1623–1633. doi: 10.1021/ja066432c. [DOI] [PubMed] [Google Scholar]
- 75.Wetmore SD, Smith DM, Golding BT, Radom L. Interconversion of (S)-glutamate and (2S,3S)-3-methylaspartate: a distinctive B12-dependent carbon-skeleton re-arrangement. J Am Chem Soc. 2001;123:7963–7972. doi: 10.1021/ja004246f. [DOI] [PubMed] [Google Scholar]
- 76.Madhavapeddi P, Ballou DP, Marsh ENG. Pre-steady state kinetic studies on the Gly171Gln active site mutant of adenosylcobalamin-dependent glutamate mutase. Biochemistry. 2002;41:15803–15809. doi: 10.1021/bi020596y. [DOI] [PubMed] [Google Scholar]
- 77.Madhavapeddi P, Marsh ENG. The role of the active site glutamate in the rearrangement of glutamate to 3-methylaspartate catalyzed by adenosylcobalamin-dependent glutamate mutase. Chem Biol. 2001;8:1143–1149. doi: 10.1016/s1074-5521(01)00081-3. [DOI] [PubMed] [Google Scholar]
- 78.Ryde U, Jensen KP. How the Co – C bond is cleaved in coenzyme B12 enzymes: a theoretical study. J Am Chem Soc. 2005;127:9117–9128. doi: 10.1021/ja050744i. [DOI] [PubMed] [Google Scholar]
- 79.Kozlowski PM, Kamachi T, Toraya T, Yoshizawa K. Does Cob(II)alamin act as a conductor in coenzyme B12 dependent mutases? Angew Chem Int Ed. 2007;46:980–983. doi: 10.1002/anie.200602977. [DOI] [PubMed] [Google Scholar]
- 80.Rommel JB, Kaestner J. The fragmentation-recombination mechanism of the enzyme glutamate mutase studied by QM/MM simulations. J Am Chem Soc. 2011;133:10195–10203. doi: 10.1021/ja202312d. [DOI] [PubMed] [Google Scholar]
- 81.Chih H-W, Marsh ENG. Tritium partitioning and isotope effects in adenosylcobalamin-dependent glutamate mutase. Biochemistry. 2001;40:13060–13067. doi: 10.1021/bi011298o. [DOI] [PubMed] [Google Scholar]
- 82.Wetmore SD, Smith DM, Radom L. Catalysis by mutants of methylmalonyl-CoA mutase: a theoretical rationalization for a change in the rate-determining step. Chembiochem. 2001;2:919–922. doi: 10.1002/1439-7633(20011203)2:12<919::AID-CBIC919>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 83.Kwiecien RA, Khavrutskii IV, Musaev DG, Morokuma K, Banerjee R, Paneth P. Computational insights into the mechanism of radical generation in B12-dependent methylmalonyl-CoA mutase. J Am Chem Soc. 2006;128:1287–1292. doi: 10.1021/ja056333j. [DOI] [PubMed] [Google Scholar]
- 84.Li X, Chung LW, Paneth P, Morokuma K. DFT and ONIOM(DFT:MM) Studies on Co – C bond cleavage and hydrogen transfer in B12-dependent methylmalonyl- CoA mutase. Stepwise or concerted mechanism? J Am Chem Soc. 2009;131:5115–5125. doi: 10.1021/ja807677z. [DOI] [PubMed] [Google Scholar]
- 85.Dybala-Defratyka A, Paneth P, Banerjee R, Truhlar DG. Coupling of hydrogenic tunneling to active-site motion in the hydrogen radical transfer catalyzed by a coenzyme B12-dependent mutase. Proc Natl Acad Sci U S A. 2007;104:10774–10779. doi: 10.1073/pnas.0702188104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Smith DM, Golding BT, Radom L. Toward a consistent mechanism for diol dehydratase catalyzed reactions: an application of the partial-proton-transfer concept. J Am Chem Soc. 1999;121:5700–5704. [Google Scholar]
- 87.Smith DM, Golding BT, Radom L. Understanding the mechanism of B12- dependent diol dehydratase: a synergistic retro-push–pull proposal. J Am Chem Soc. 2001;123:1664–1675. doi: 10.1021/ja001454z. [DOI] [PubMed] [Google Scholar]
- 88.Kamachi T, Toraya T, Yoshizawa K. Catalytic roles of active-site amino acid residues of coenzyme B12-dependent diol dehydratase: protonation state of histidine and pull effect of glutamate. J Am Chem Soc. 2004;126:16207–16216. doi: 10.1021/ja045572o. [DOI] [PubMed] [Google Scholar]
- 89.Kamachi T, Toraya T, Yoshizawa K. Computational mutation analysis of hydrogen abstraction and radical rearrangement steps in the catalysis of coenzyme B12-dependent diol dehydratase. Chem Eur J. 2007;13:7864–7873. doi: 10.1002/chem.200601466. [DOI] [PubMed] [Google Scholar]
- 90.Retey J, Umaniron A, Seibl J, Arigoni D. Zum Mechanismus Der Propandioldehydrasf-Reaktion. Experientia. 1966;22:502–503. doi: 10.1007/BF01898652. [DOI] [PubMed] [Google Scholar]
- 91.Kamachi T, Takahata M, Toraya T, Yoshizawa K. What is the identity of the metal ions in the active sites of coenzyme B12-dependent diol dehydratase? A computational mutation analysis. J Phys Chem B. 2009;113:8435–8438. doi: 10.1021/jp9001737. [DOI] [PubMed] [Google Scholar]