

Abstract
Glutamate is the primary excitatory amino acid neurotransmitter in the CNS. The concentration of glutamate in the synaptic cleft is tightly controlled by interplay between glutamate release and glutamate clearance. Abnormal glutamate release and/or dysfunction of glutamate clearance can cause overstimulation of glutamate receptors and result in neuronal injury known as excitotoxicity. The glial glutamate transporter EAAT2 plays a major role in glutamate clearance. Dysfunction or reduced expression of EAAT2 has been documented in many neurodegenerative diseases. In addition, many studies in animal models of disease indicate that increased EAAT2 expression provides neuronal protection. Here, we summarize these studies and suggest that EAAT2 is a potential target for the prevention of excitotoxicity. EAAT2 can be upregulated by transcriptional or translational activation. We discuss current progress in the search for EAAT2 activators, which is a promising direction for the treatment of neurodegenerative diseases.
Excitotoxicity: a common problem in neurodegenerative diseases
Glutamate is the main excitatory neurotransmitter in the CNS, responsible for fast excitatory neurotransmission (Figure 1A). In nerve terminals, glutamate is stored in synaptic vesicles from which glutamate is released in a calcium-dependent manner upon depolarization of the nerve terminal. Glutamate release causes a significant increase of glutamate concentration (~1000-fold) in the synaptic cleft. Released glutamate binds to ionotropic glutamate receptors (NMDA-R and AMPA-R in Figure 1A) on postsynaptic neurons, which stimulates an influx of Na+ and Ca2+ ions into neurons. This leads to depolarization and generation of action potentials. Glutamate is then quickly removed from the synaptic cleft by glutamate transporters (EAAT2 in Figure 1A) to prevent glutamate receptor overstimulation. Glutamate taken up by perisynaptic astrocytes is then converted to glutamine by glutamine synthetase. Following this, glutamine is transferred to neurons where it is converted into glutamate by glutaminase and then taken by vesicular glutamate transporters into synaptic vesicles, where it is available for use in excitatory neurotransmission [1].
Figure 1. Glutamatergic neurotransmission and excitotoxicity.

(A) Under normal conditions, glutamate released from the presynaptic neuron activates ionotropic glutamate receptors (NMDA and AMPA receptors) present on the postsynaptic neuron. This results in the influx of Na+ and Ca2+ ions into the cell, leading to depolarization and generation of an action potential. (B) Acute elevations of glutamate (when the release of glutamate from presynaptic terminals and/or glial cells is increased) in conditions such as stroke, neurotrauma and epilepsy cause severe neuronal injury or death. (C) Chronic and mild elevations of glutamate (when the glutamate reuptake function is impaired) such as Alzheimer’s disease, Parkinson’s disease and amylotrophic lateral sclerosis cause neuronal damage. (D) Increased EAAT2 is a potential therapeutic strategy for the prevention of excitotoxicity.
AMPA: α-amino-3-hydroxy-5-methyl-4 isoxazolepropionate; NMDA: N-methyl-D-aspartate.
Elevated extracellular glutamate concentrations can occur under disease conditions when the release of glutamate from presynaptic terminals and/or glial cells is increased (Figure 1B) or when the glutamate reuptake function is impaired (Figure 1C). Activated microglia and reactive astrocytes release large amounts of glutamate under pathological conditions [2–3]. Excessive glutamate can cause overstimulation of glutamate receptors and lead to an increased intracellular concentration of Na+ and Ca2+ ions. This can result in a form of neuronal injury or death known as ‘excitotoxicity’ [4]. Excitotoxicity is thought to contribute to a wide range of acute and chronic neurodegenerative diseases. Acute elevations of glutamate in conditions such as stroke, neurotrauma and epilepsy cause severe neuronal damage [5–7]. Chronic and mild elevations of glutamate underlie excitotoxicity in chronic neurodegenerative diseases such as Alzheimer’s disease (AD), Parkinson’s disease (PD) and amyotrophic lateral sclerosis (ALS) [8–10]. Therefore, the glutamate-induced excitotoxicity pathway has been a therapeutic strategy for neurodegenerative diseases.
N-methyl-D-aspartate (NMDA) and Ca2+-permeable α-amino-3-hydroxy-5-methyl-4 isoxazolepropionate (AMPA) receptors are considered to be mainly responsible for excitotoxicity [4]. Blocking these receptors, especially the NMDA receptor, has been a therapeutic strategy. However, the majority of NMDA antagonists are competitive antagonists that cause side effects including hallucinations and schizophrenia-type symptoms in patients [11]. The side effects likely result from the competitive antagonists blocking physiological functions of the receptor. Memantine is a noncompetitive NMDA receptor antagonist that can decrease pathological activation of NMDA-receptors without affecting physiological NMDA receptor activity. This drug is available for treating the advanced stages of AD. Memantine is a relatively safe drug with few side effects, but only has mild clinically relevant effects on cognition, global functioning and activities of daily living [12]. Riluzole is another marketed drug that has anti-excitotoxic properties. This drug inhibits the release of glutamate and also blocks some of the postsynaptic effects of glutamate by inhibition of NMDA and AMPA receptors [13]. Riluzole is the only approved pharmacologic treatment for ALS. Riluzole is a relatively safe drug for ALS patients that prolongs survival by approximately 2–3 months [14]. It appears that the beneficial effects of these available anti-excitotoxic drugs are very limited. Therefore, there is a need for better therapeutics.
EAAT2: a potential target for prevention of excitotoxicity
Another potential approach to preventing excitotoxicity is enhanced glutamate reuptake. The concentration of glutamate in the synaptic cleft and the resultant activity of the postsynaptic glutamate receptors are tightly regulated by the interplay between glutamate release and glutamate clearance. Glutamate clearance is facilitated by a family of Na+- and K+-coupled excitatory amino acid transporters (EAATs). Five mammalian EAATs have been cloned and characterized: EAAT1 (known as GLAST in rodents) [15], EAAT2 (known as GLT-1 in rodents) [16], EAAT3 (known as EAAC1 in rodents) [17], EAAT4 [18] and EAAT5 [19]. In the CNS, while EAAT1 and EAAT2 are found primarily in astrocytes, EAAT3 and EAAT4 are found principally in neurons [18,20]. EAAT5 is expressed primarily in the retina [19]. However, a recent study reported that astrocytes express all EAATs, and microglia also express all EAATs except EAAT4 [21].
The glial glutamate transporters, EAAT1 and EAAT2, are found primarily on perisynaptic processes of astrocytes closely associated with excitatory synaptic contacts and are responsible for maintaining low extracellular glutamate concentrations [22]. This is particularly the case for EAAT2, which may be responsible for up to 80–90% of all extracellular glutamate uptake activity [23]. Mice deficient in EAAT2 develop lethal spontaneous seizures and show increased susceptibility to acute cortical injury [24]. Dysfunction or reduced expression of EAAT2 has been documented in both chronic and acute neurodegenerative disorders. In addition, several lines of evidence in animal models of disease suggest that upregulation of EAAT2 is a potential therapeutic strategy for the prevention of excitotoxicity (Figure 1D). These studies are described below.
Alzheimer’s disease
AD is a progressive neurodegenerative disease in which patients have declarative memory impairments and progressing dementia. Early in the disease process, cortical and hippocampal synapse density is significantly reduced. This loss of synapses strongly correlates with memory impairments [25]. Several reports have demonstrated that glutamate transport function is significantly reduced in AD brains. This decrease correlates with loss of synapse and neuronal death [26–30]. Reduced glutamate transport function was found to be associated with decreased EAAT2 protein expression in AD brains [30,31]. The mechanism underlying the loss of EAAT2 in AD is unclear. It appears that this loss is due to disturbances at the post-transcriptional level because EAAT2 mRNA is not decreased [30]. It has been shown that EAAT2 is oxidatively modified by the lipid peroxidation product 4-hydroxy-2-nonenal in AD brains [32]. Oxidative damage of EAAT2 proteins could result in their degradation and loss of function. In addition, a recent study showed that a disturbance of cholesterol metabolism, known to occur in AD, can cause a reduction of membrane cholesterol levels. This reduction results in dissociation of EAAT2 from lipid raft microdomains of the plasma membrane and subsequently leads to loss of EAAT2 and glutamate transport function [33,34].
Several studies of glutamate uptake inhibition in vivo indicate that impaired glutamate transport function can cause neuronal death. Injection of a glutamate uptake blocker, DL-threo-β-hydroxyaspartate, into rat striatum caused neuronal degeneration [35]. Chronic inhibition of glutamate transport with DL-threo-β-hydroxyaspartate leads to slow neuronal death that can be prevented by glutamate-receptor antagonists [36]. Intrahippocampal injections of the glutamate transport inhibitor dihydokainate induce neuronal damage. This effect can be prevented by the glutamate receptor antagonists [37].
Furthermore, our laboratory recently investigated the role of EAAT2 dysfunction in the pathogenesis of AD in APPSw/Ind mice, a transgenic mouse of AD. Loss of EAAT2 protein and associated functions was also found in these transgenic mice. We crossed APPSw/Ind mice with the transgenic mice overexpressing human EAAT2 (EAAT2 transgenic mice), which have a 1.5- to two-fold increase in EAAT2 protein levels compared with their nontransgenic counterparts, to investigate whether supplementation of the loss of EAAT2 would have beneficial effects on disease progression. EAAT2 protein levels and the associated glutamate-uptake functions were restored in APP/EAAT2 double transgenic mice. Several important phenomena were observed in APP/EAAT2 mice compared with their APP littermates. First, premature death was significantly reduced. Second, learning and memory capacity was significantly improved. Third, several Alzheimer’s-like pathological changes were significantly improved, including reduced amyloid plaques, increased synaptic density and reduced glial activation [Kong Q et al., Unpublished Data]. These results indicate that loss of EAAT2 contributes to the pathogenesis in an AD mouse model and suggest that enhanced glutamate uptake by increasing EAAT2 expression may be a potential therapeutic strategy for AD.
Amyotrophic lateral sclerosis
ALS is a late-onset and fatal neurodegenerative disorder that is characterized by progressive degeneration of motor neurons in the spinal cord, motor cortex and brainstem. This typically results in muscle weakness, atrophy, spasticity and progressive paralysis until death, which usually occurs within 2–5 years of symptom onset. There is currently no effective treatment for ALS. A number of mechanisms have been implicated in the pathogenesis of ALS, including aberrant RNA metabolism, glutamate excitotoxicity, oxidative damage, mitochondrial dysfunction, impaired axonal transport, growth factor deficiency and accumulation of intracellular aggregates [38]. In addition, ALS is not only a multifactorial disease. but also a multisystemic disease that affects several cell types [39].
Approximately 60–70% of ALS patients have a 30–95% loss of the EAAT2 protein in the motor cortex and spinal cord [40]. The loss of EAAT2 protein has also been observed in transgenic mice or rats expressing familial ALS-linked mutant SOD1 [41–43]. Studies indicate that the reduction in EAAT2 protein is correlated with neuronal loss. This raises an important question as to whether loss of EAAT2 protein is a contributing factor or is simply a consequence of motor neuron degeneration. Guo and colleagues addressed this question by crossing transgenic mice overexpressing human mutant SOD1(G93A) with the EAAT2 transgenic mice [44]. The EAAT2/SOD1(G93A) double transgenic mice showed a significant delay in motor function decline and motor neuron degeneration, but not in the life span, when compared with SOD1(G93A) littermates. In addition, Rothstein and colleagues discovered that β-lactam antibiotics were able to induce EAAT2 protein expression. Treating SOD1(G93A) mice with ceftriaxone (Figure 2), a β-lactam antibiotic, significantly ameliorated symptoms and prolonged survival [45]. These studies demonstrate that loss of EAAT2 does contribute to disease progression in ALS mice and suggest that a therapeutic approach by inducing EAAT2 protein expression may have beneficial effects to ALS patients.
Figure 2.
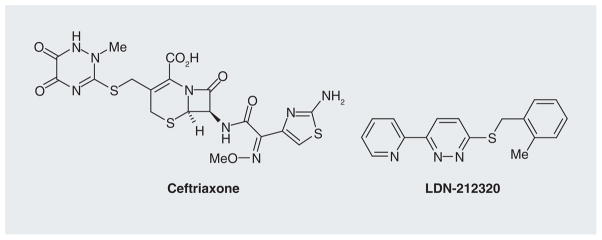
Ceftriaxone and LDN-212320.
Parkinson disease
PD is a common age-related neurodegenerative disease that is caused by a progressive degeneration of dopaminergic neurons in the pars compacta of substantia nigra (SNc). Degeneration of SNc neurons deprives striatal neurons of their dopaminergic innervations, which triggers complex functional modifications within the basal ganglia circuitry. This ultimately leads to development of the motor symptoms, including bradykinesia, tremor and rigidity [46]. Glutamate is also the predominant excitatory transmitter in the basal ganglia. In addition to sending glutamatergic projections to the striatum, the cortex also sends projections to the subthalamic nucleus (STN) and SNc [47]. The SNc receives further glutamatergic innervation from the STN. The dopaminergic projections from the SNc to various nuclei in the basal ganglia circuit perform important regulatory functions. Under disease conditions, the reduction in the dopaminergic message to the striatum, as seen in PD, results in an increase in firing of STN neurons. This increase functions as a compensatory mechanism to stimulate the release of dopamine from the surviving dopaminergic neurons in the SNc in order to maintain dopamine homeostasis [48]. However, a sustained increase in glutamate release into already compromised dopaminergic neurons could evoke excitotoxicity and potentiate neurodegeneration.
There is growing evidence that supports the important role of glutamatergic pathways in the pathogenesis of PD. Evidence from mouse and primate models of PD demonstrated that certain NMDA- and AMPA-receptor antagonists have antiparkinsonian action [49–51]. It has been shown that mGlu2/3 receptor agonists can attenuate 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced dopaminergic neuron degeneration in mice [52,53]. Treatment with mGlu5 receptor antagonists alleviated motor symptoms in parkinsonian animals [54–56]. Furthermore, ablation of the STN by lesion or inactivation of the STN by deep-brain stimulation leads to a reduction in firing of the STN and blunted release of glutamate onto the SNc. This has been shown to ameliorate the motor symptoms associated with PD and increase survival of dopaminergic neurons in SNc [57–59]. These neuroprotections are probably due to a reduction in glutamate excitotoxicity, as a result of the loss or reduction of the STN input to the SNc. Therefore, antiglutamate approaches are promising.
Changes of EAAT2 expression have been reported in animal models of PD. Chung et al. reported downregulation of EAAT2 after 6-hydroxydopamine injection into the nigrostriatal pathway [60]. Holmer et al. reported a decrease in striatal EAAT2 immunolabeling in the acute 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model [61]. However, it has not been reported whether increased EAAT2 expression could ameliorate PD symptoms.
Epilepsy
Epilepsy is an array of diverse chronic neurological disorders characterized by seizures. Abnormal hyper-synchronous neuronal activity in the brain causes seizures. There are two broad categories of epilepsy syndromes: partial and generalized [62]. Partial seizures are the most common seizure disorder in adults, often originating from lesions such as head trauma, strokes and tumors [7]. The most prevalent of these syndromes features complex partial seizures arising from the mesial temporal lobe, termed temporal lobe epilepsy (TLE). There is a latent period of several years between the initial lesion and the emergence of chronic TLE. The initial lesion causes complex molecular, biochemical and structural changes that over time result in the development of spontaneous recurrent seizures. This process is referred to as epileptogenesis [63]. The most common lesion in patients with TLE is hippocampal sclerosis. There is selective loss of neurons in the dentate hilus and the hippocampal pyramidal-cell layer; thus, patients with TLE often show impairments in learning, memory and other cognitive functions [64].
Glutamate plays a crucial role in the initiation of seizures and their propagation. In vivo microdialysis studies of patients with epilepsy show a significant increase in extracellular glutamate levels, which reach neurotoxic concentrations in the hippocampus before and during seizure onset [65,66]. It is believed that abnormal glutamate released by astrocytes plays a causal role in the synchronous firing of large populations of neurons during seizures [3,67–69]. In addition, glutamate transport dysfunction may contribute to high extracellular glutamate levels in the epileptogenic hippocampus [70,71]. Brain injuries that result from seizures occur in a dynamic process. Intense seizure activity causes excessive glutamate release and overstimulation of glutamate receptors, which leads to massive influxes of calcium and subsequently triggers acute neuronal cell death. Massive neuronal death consequently causes complex modifications including synaptic plasticity, aberrant reorganization of neuronal circuitry, alterations in interneuron number and function, and change in dentate neurogenesis. These epileptogenic changes over time result in the development of spontaneous recurrent seizures. Therefore, reducing glutamate-mediated excitotoxicity may potentially prevent seizure-induced epileptogenesis. Kong et al. recently investigated pilocarpine-induced status epilepticus in EAAT2 transgenic mice [72]. They observed that increased EAAT2 expression can significantly prevent seizure-induced neuronal death, epileptogenesis and subsequent recurrent seizures. This study suggests that increased EAAT2 protein expression is a potential therapeutic approach for epilepsy.
Multiple sclerosis
Multiple sclerosis (MS) is primarily an inflammatory disease in which focal lymphocytic infiltration results in damage of myelin sheaths around the axons of the brain and spinal cord, leading to a broad spectrum of symptoms [73]. The early course of the disease is characterized by episodes of neurological dysfunction that usually recover; however, over time the pathological changes become dominated by wide-spread microglial activation, astrogliosis, axonal degeneration and chronic neurodegeneration. MS often progresses to physical and cognitive disabilities.
There is growing evidence that glutamate plays a role in the pathology of MS and experimental autoimmune encephalomyelitis (EAE), the animal model of MS. In vivo magnetic resonance spectroscopy has shown that glutamate is elevated in MS normal-appearing white matter and acute white matter lesions [74]. Cerebrospinal fluid glutamate levels are significantly increased during the relapse period and correlate with disease severity and course [75]. Oligodendrocytes and neurons are highly vulnerable to glutamate-mediated excitotoxic damage [76]. Blockade of AMPA/kainate receptors with antagonists, NBQX, MPQX, GYKI52466 or GYKI5377 resulted in marked reduction of neurological deficits in the EAE model [77]. In addition, NMDA-receptor antagonists, memantine, amantadine and MK-801 all reduce neurological deficits in the EAE model [78,79]. A study focused on EAAT2 expression in different regions of rat brain during the course of EAE revealed that EAAT2 protein levels in the cerebellum were significantly reduced, although EAAT2 mRNAs were upregulated, both immediately before the acute phase and later during the recovery phase [80]. These results suggest post-translational disturbances associated with EAE conditions may lead to insufficient protection against glutamate excitotoxicity. There is currently no study that reports whether increased EAAT2 expression could ameliorate MS symptoms.
Stroke
Stroke, a leading cause of adult disability, is caused by ischemia (lack of blood flow) or a hemorrhage (leakage of blood). The injury process begins following the interruption of blood flow to brain tissue. If blood flow is not recovered within a short period of time, cell death within the ischemic area will occur. Consequently, the affected area of the brain loses function, resulting in disability or even death.
Substantial evidence indicates that glutamatemediated excitotoxicity is an important contributor in the neuropathology of stroke. Rapidly following interruption of blood flow, mitochondrial inhibition of ATP synthesis leads to the depletion of ATP stores in the affected area of a stroke, which quickly leads to neuronal plasma membrane depolarization, release of potassium into the extracellular space and entry of sodium into cells [81]. Excessive membrane depolarization and accumulation of sodium inside cells ultimately causes glutamate release in high concentrations at the core of the infarct [82]. This causes hyperactivation of glutamate receptors and subsequently raises intracellular calcium to high levels, leading to excitotoxic cell death.
Glutamate release via reversal of glutamate transport plays a critical role in ischemic pathology [83,84]. Glutamate is released from neurons rather than glia in ischemia [83], possibly because their higher internal glutamate concentrations drive reversed uptake more readily. Dihydrokainate (DHK), a nontransported blocker of EAAT2, has no effect on the glutamate release in a hippocampal slice model of ischemia [83]. Therefore, increased EAAT2 protein and function may not cause more glutamate release, but rather augment glutamate clearance. Several studies suggest that uptake of glutamate by EAAT2 plays a neuroprotective role after ischemia. Rao et al. reported that knockdown of EAAT2 exacerbates the transient middle cerebral artery occlusion-induced neuronal damage [85]. Weller et al. showed that overexpression of EAAT2 enhances neuroprotection following a moderate oxygen glucose deprivation in rat hippocampal slice cultures [86]. Chu et al. reported that ceftriaxone treatment in rats upregulated EAAT2 expression and reduced infarct volume in the middle cerebral artery occlusion model of ischemia [87]. A recent study shows that focal overexpression of EAAT2 significantly reduces ischemia-induced glutamate overflow, decreases cell death and improves behavioral recovery in a rat model of stroke [88]. These studies suggest that EAAT2 can be a therapeutic target for stroke.
Traumatic brain injury
Traumatic brain injury (TBI) is one of the leading causes of death and disability among children and young adults. Patients with brain injury suffer from surface contusion, focal or diffuse intracranial hemorrhages, and diffuse axonal injuries. These primary injuries can lead to more serious secondary events, which include alterations in cerebral blood flow and the pressure within the skull. These secondary processes contribute substantially to the damage from the initial injury. Glutamate excitotoxicity plays an important role in the development of secondary injuries and contributes significantly to expansion of the total volume of injury [5].
Following TBI, levels of extracellular glutamate increase acutely [89–91]. Glutamate may move into the brain following disruption of the blood–brain barrier [92]. Excessive synaptic release of glutamate occurs following the injury and leads to glutamate overflow into extra-synaptic regions [93]. Decreased glutamate transporter activity, due either to functional impairment or decreased EAATs protein expression can also contribute to the accumulation of extracellular glutamate [94]. Furthermore, glutamate transporter reversal could also result in a rise in extracellular glutamate levels. Currently, there are no reports that have specifically investigated the role of increased EAAT2 expression in TBI animal models.
Searching for EAAT2 activators: a promising approach for the treatment of neurodegenerative diseases
The mechanisms underlying regulation of EAAT2 expression have been extensively studied [95–99]. Expression of EAAT2 is regulated at both the transcriptional and translational level. Many growth factors, including EGF, TGF-α, PDGF and pituitary adenylate cyclase-activating polypeptide, can increase EAAT2 expression through increased transcription of the EAAT2 gene [95,96]. Expression of EAAT2 can also be regulated through increased translation of EAAT2 transcripts [97]. Several factors including corticosterone and retinol are able to increase translation of EAAT2 transcripts. Moreover, several disease-associated insults affect the efficiency of EAAT2 translation, suggesting that translational dysregulation may be involved in the loss of EAAT2 in neurodegenerative diseases. Therefore, two approaches can be used to search for EAAT2 activators: transcriptional or translational activation.
Searching for EAAT2 transcriptional activators
One approach to identifying small-molecule activators of EAAT2 expression would be to utilize a reporter gene assay using the EAAT2 promoter to drive expression of a reporter, such as luciferase. The human EAAT2 promoter has been cloned and characterized [100]. Rothstein et al. have generated primary human fetal astrocytes that stably express firefly luciferase driven by the human EAAT2 promoter. They demonstrated that the human EAAT2 promoter was significantly activated by ceftriaxone, amoxicillin and dibutyryl cyclic AMP [45]. Increased expression of EAAT2 was observed 48 h after treatment. The results of these experiments suggest that this system may be used to screen for compounds that activate EAAT2 promoter activity. However, at this time, there is no report if this approach successfully identifies EAAT2 transcriptional activators.
Searching for EAAT2 translational activators
A second approach would be to screen for compounds that increase EAAT2 translation. Our laboratories have taken this approach because of the following two reasons: loss of EAAT2 protein in patients is probably due to disturbances at the post-transcriptional level because EAAT2 mRNA is not decreased; and, in the acute conditions such as stroke, TBI and epilepsy, immediately upregulated EAAT2 protein and function is critical. The progress of this drug-discovery project is briefly described below.
We developed a cell-based enzyme-linked immunosorbent assay using a primary astrocyte line stably transfected with a vector designed to identify modulators of EAAT2 translation [101]. This assay was optimized for high-throughput screening, and a library of approximately 140,000 compounds was tested. In the initial screening, 293 compounds were identified as hits (increased activation by >60%), for an initial 0.2% hit rate. After rescreening the 293 initial hits, a total of 61 compounds were selected that showed a dose-dependent increase of activation. Of the 61 confirmed compounds, 16 unique structural classes were identified. The effects of these compounds on increasing EAAT2 protein level, glutamate transport function, EAAT2 protein localization, and with no change in EAAT2 mRNA level were confirmed. These compounds provide an adequate degree of structural diversity necessary for the foundation of a drug-discovery project.
We then analyzed the structure tractability of these compounds, and six classes were prioritized based on structure. The compounds of these six classes were further evaluated in mouse primary cortical neurons and astrocytes mixed cultures and then in wild-type mice by intrathecal administration of compounds. After intensive studies with these compounds, two lead series were selected for further investigation. Many analogs with better potency were identified. Two analogs with higher potency (one for each lead series) were selected for in vivo pharmacokinetics, in vivo pharmacodynamics, in vivo acute and chronic maximum tolerated dose studies, and in vitro profiling studies. One of the lead series, a pyridazine derivative, was further selected for SAR studies [102]. We then performed efficacy studies in two animal models of disease including the ALS (SOD1[G93A] transgenic mouse model, chronic excitotoxicity) and epilepsy (pilocarpine-induced limbic epilepsy model, acute excitotoxicity) using the analog LDN-212320 (Figure 2). Our results indicate that this compound had significant protective effects in these animal models of disease [Kong Q et al., Unpublished Data], providing support for this approach. Further investigation into these lead compound series is underway.
Future perspective
Glutamate-mediated excitotoxicity is involved in a wide range of acute and chronic neurodegenerative diseases. However, there is currently no safe and effective drug for the prevention of excitotoxicity. Glutamate-receptor antagonist treatment has not been a very successful strategy in humans because it affects normal brain function and produces negative side effects. There is a need for better therapeutics. Loss of EAAT2 protein and function is commonly found in chronic neurodegenerative diseases such as ALS and AD and may be the main cause of excitotoxicity in these diseases. Restored EAAT2 protein levels and function may provide therapeutic benefit. In disease conditions such as epilepsy, stroke and TBI, a dramatic increase in extracellular glutamate levels cause severe neuronal damage. Immediate upregulation of EAAT2 protein may reduce extracellular glutamate levels and thereby prevent damage to neurons. Therefore, increased EAAT2 expression is a potential approach to preventing excitotoxicity in both acute and chronic neurodegenerative diseases. EAAT2 can be upregulated by transcriptional or translational activation. β-lactam antibiotics, such as ceftriaxone, were identified as transcriptional activators of EAAT2. Ceftriaxone has been tested in many disease models and is capable of providing neuronal protection. This drug is currently in human clinical trials for ALS. It may be possible to develop new derivatives of ceftriaxone with enhanced pharmacological and bioactivity properties. In addition, new EAAT2 transcriptional activators may be identified in the near future. However, the transcriptional activation approach may not work well for acute conditions, such as those stated above, because the process from transcription to protein production takes time. For instance, the release of glutamate occurs within minutes of ischemic onset; therefore therapeutic drugs targeted at blocking excitotoxicity must not only be administered rapidly, but also need to take effect quickly.
Our laboratories have identified several series of compounds that can increase EAAT2 expression through translational activation. Our current results indicate that these EAAT2 translational activators have significant protective effects in ALS and epilepsy animal models, suggesting that this translational activation approach works well in mice and that these drug-like compounds are worth further pursuit. In addition, a thorough understanding of the mechanisms underlying translational regulation of EAAT2, such as identifying the molecular targets of the compounds, signaling pathways involved in the regulation, and translational activation processes, are very important for this novel drug-development effort. Furthermore, a more detailed understanding of the therapeutic window for EAAT2 upregulation that is safe for humans is also needed.
We are still in the early stages of developing EAAT2 activator treatment as a therapeutic strategy for preventing excitotoxicity. In our opinion, we expect that in the coming years, more EAAT2 activators will be identified and evaluated in animal models of disease. The already identified compounds, such as ceftriaxone and LDN-212320, will be further developed to improve their pharmacological and bioactivity properties. The outcomes of ceftriaxone trials in ALS patients will be revealed soon. The continuous development of LDN-212320 may lead to a more potent and safe analog for human clinical trials. In addition, we expect that LDN-212320 or its analog will be tested in more animal models of disease, which will provide valuable information about the potential of these compounds for multiple diseases. We also expect that the molecular mechanisms underlying LDN-212320-activated EAAT2 translation will be uncovered in the coming years. Transition from preclinical mouse studies to human clinical trials is difficult for most diseases; successful trails in mice often fail in subsequent human trials. It is our great hope that the EAAT2 treatment strategy will be successful and greatly benefit patients suffering from neurodegenerative diseases.
Executive summary.
Excitotoxicity: a common problem in neurodegenerative diseases
Under disease conditions, elevated extracellular glutamate concentrations can occur, which result in neuronal injury or death.
The beneficial effects of currently available anti-excitotoxic drugs are very limited. There is a need for better therapeutics.
EAAT2: a potential target for prevention of excitotoxicity
Several lines of evidence indicate that upregulation of EAAT2 is a potential therapeutic strategy for the prevention of excitotoxicity.
Searching for EAAT2 activators: a promising approach for the treatment of neurodegenerative diseases
Two approaches can be used to search for EAAT2 activators: transcriptional or translational activation.
β-lactam antibiotics, such as ceftriaxone, were identified as transcriptional activators of EAAT2. It may be possible to develop new derivatives of ceftriaxone with enhanced pharmacological and bioactivity properties.
Several series of novel small molecules that can specifically increase EAAT2 protein expression through translational activation have recently been identified. Further investigation into these small molecules is underway.
Acknowledgments
The authors are grateful to colleagues at Ohio State University and Brigham and Women’s Hospital who helped advance the EAAT2 activator project.
Key Terms
- Synaptic vesicle
Stores neurotransmitters to be released at synapses
- Synaptic cleft
Very small gap between presynaptic and postsynaptic cells through which neurotransmitters diffuse
- Ionotropic glutamate receptors
Form an ion channel pore that allows the flow of K+, Na+ and in some cases Ca2+ in response to glutamate binding
- Astrocytes
Type of glial cell in the CNS that have multiple functions including ionic concentration regulation in the intercellular space, uptake and/or breakdown of neurotransmitters, and blood–brain barrier formation
- Dementia
Loss of brain function that impairs memory, thinking, language, judgment, and behavior under certain diseases
- Motor neurons
Send impulses from the CNS to muscles, glands and other tissues
- Substantia nigra
Region of the brain structure located in the midbrain that is important in reward, addiction and movement
- Deep-brain stimulation
Treatment that involves surgical implantation of a brain pacemaker, capable of sending electrical impulses to specific parts of the brain
Footnotes
For reprint orders, please contact reprints@future-science.com
Financial & competing interests disclosure
The authors wish to thank the NIH/NINDS (Grants R01NS064275 and U01NS074601), the Thome Memorial Foundation and the Alzheimer’s association for their support. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as:
▪ of interest
▪▪ of considerable interest
- 1.Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65(1):1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- 2.Takeuchi H, Jin S, Wang J, et al. Tumor necrosis factor-alpha induces neurotoxicity via glutamate release from hemichannels of activated microglia in an autocrine manner. J Biol Chem. 2006;281(30):21362–21368. doi: 10.1074/jbc.M600504200. [DOI] [PubMed] [Google Scholar]
- 3.Wetherington J, Serrano G, Dingledine R. Astrocytes in the epileptic brain. Neuron. 2008;58(2):168–178. doi: 10.1016/j.neuron.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Choi DW. Ionic dependence of glutamate neurotoxicity. J Neurosci. 1987;7(2):369–379. doi: 10.1523/JNEUROSCI.07-02-00369.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yi JH, Hazell AS. Excitotoxic mechanisms and the role of astrocytic glutamate transporters in traumatic brain injury. Neurochem Int. 2006;48(5):394–403. doi: 10.1016/j.neuint.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 6.Hazell AS. Excitotoxic mechanisms in stroke: an update of concepts and treatment strategies. Neurochem Int. 2007;50(7–8):941–953. doi: 10.1016/j.neuint.2007.04.026. [DOI] [PubMed] [Google Scholar]
- 7.Acharya MM, Hattiangady B, Shetty AK. Progress in neuroprotective strategies for preventing epilepsy. Prog Neurobiol. 2008;84(4):363–404. doi: 10.1016/j.pneurobio.2007.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Van Den Bosch L, Van Damme P, Bogaert E, Robberecht W. The role of excitotoxicity in the pathogenesis of amyotrophic lateral sclerosis. Biochim Biophys Acta. 2006;1762(11–12):1068–1082. doi: 10.1016/j.bbadis.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 9.Koutsilieri E, Riederer P. Excitotoxicity and new antiglutamatergic strategies in Parkinson’s disease and Alzheimer’s disease. Parkinsonism Relat Disord. 2007;13(Suppl 3):S329–S331. doi: 10.1016/S1353-8020(08)70025-7. [DOI] [PubMed] [Google Scholar]
- 10.Shah RS, Lee HG, Xiongwei Z, Perry G, Smith MA, Castellani RJ. Current approaches in the treatment of Alzheimer’s disease. Biomed Pharmacother. 2008;62(4):199–207. doi: 10.1016/j.biopha.2008.02.005. [DOI] [PubMed] [Google Scholar]
- 11.Lipton SA. Paradigm shift in neuroprotection by NMDA receptor blockade: memantine and beyond. Nat Rev Drug Discov. 2006;5(2):160–170. doi: 10.1038/nrd1958. [DOI] [PubMed] [Google Scholar]
- 12.van Marum RJ. Update on the use of memantine in Alzheimer’s disease. Neuropsychiatr Dis Treat. 2009;5:237–247. doi: 10.2147/ndt.s4048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cheah BC, Vucic S, Krishnan AV, Kiernan MC. Riluzole, neuroprotection and amyotrophic lateral sclerosis. Curr Med Chem. 2010;17(18):1942–1199. doi: 10.2174/092986710791163939. [DOI] [PubMed] [Google Scholar]
- 14.Miller RG, Mitchell JD, Moore DH. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND) Cochr Data Syst Rev. 2012;3:CD001447. doi: 10.1002/14651858.CD001447. [DOI] [PubMed] [Google Scholar]
- 15.Storck T, Schulte S, Hofmann K, Stoffel W. Structure, expression, and functional analysis of a Na(+)-dependent glutamate/aspartate transporter from rat brain. Proc Natl Acad Sci USA. 1992;89(22):10955–10959. doi: 10.1073/pnas.89.22.10955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pines G, Danbolt NC, Bjoras M, et al. Cloning and expression of a rat brain L-glutamate transporter. Nature. 1992;360(6403):464–467. doi: 10.1038/360464a0. [DOI] [PubMed] [Google Scholar]
- 17.Kanai Y, Hediger MA. Primary structure and functional characterization of a high-affinity glutamate transporter. Nature. 1992;360(6403):467–471. doi: 10.1038/360467a0. [DOI] [PubMed] [Google Scholar]
- 18.Fairman WA, Vandenberg RJ, Arriza JL, Kavanaugh MP, Amara SG. An excitatory amino-acid transporter with properties of a ligand-gated chloride channel. Nature. 1995;375(6532):599–603. doi: 10.1038/375599a0. [DOI] [PubMed] [Google Scholar]
- 19.Arriza JL, Eliasof S, Kavanaugh MP, Amara SG. Excitatory amino acid transporter 5, a retinal glutamate transporter coupled to a chloride conductance. Proc Natl Acad Sci USA. 1997;94(8):4155–4160. doi: 10.1073/pnas.94.8.4155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rothstein JD, Martin L, Levey AI, et al. Localization of neuronal and glial glutamate transporters. Neuron. 1994;13(3):713–725. doi: 10.1016/0896-6273(94)90038-8. [DOI] [PubMed] [Google Scholar]
- 21.Liang J, Takeuchi H, Doi Y, et al. Excitatory amino acid transporter expression by astrocytes is neuroprotective against microglial excitotoxicity. Brain Res. 2008;1210:11–19. doi: 10.1016/j.brainres.2008.03.012. [DOI] [PubMed] [Google Scholar]
- 22.Chaudhry FA, Lehre KP, van Lookeren Campagne M, Ottersen OP, Danbolt NC, Storm-Mathisen J. Glutamate transporters in glial plasma membranes: highly differentiated localizations revealed by quantitative ultrastructural immunocytochemistry. Neuron. 1995;15(3):711–720. doi: 10.1016/0896-6273(95)90158-2. [DOI] [PubMed] [Google Scholar]
- 23.Rothstein JD, Dykes-Hoberg M, Pardo CA, et al. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. 1996;16(3):675–686. doi: 10.1016/s0896-6273(00)80086-0. [DOI] [PubMed] [Google Scholar]
- 24▪.Tanaka K, Watase K, Manabe T, et al. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science. 1997;276(5319):1699–1702. doi: 10.1126/science.276.5319.1699. First showed that a deficit in EAAT2 caused neurological problems in mice. [DOI] [PubMed] [Google Scholar]
- 25.Terry RD, Masliah E, Salmon DP, et al. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30(4):572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- 26.Greenamyre JT, Penney JB, Young AB, D’Amato CJ, Hicks SP, Shoulson I. Alterations in L-glutamate binding in Alzheimer’s and Huntington’s diseases. Science. 1985;227(4693):1496–1499. doi: 10.1126/science.2858129. [DOI] [PubMed] [Google Scholar]
- 27.Simpson MD, Royston MC, Deakin JF, Cross AJ, Mann DM, Slater P. Regional changes in [3H]D-aspartate and [3H]TCP binding sites in Alzheimer’s disease brains. Brain Res. 1988;462(1):76–82. doi: 10.1016/0006-8993(88)90587-2. [DOI] [PubMed] [Google Scholar]
- 28.Chalmers DT, Dewar D, Graham DI, Brooks DN, McCulloch J. Differential alterations of cortical glutamatergic binding sites in senile dementia of the Alzheimer type. Proc Natl Acad Sci USA. 1990;87(4):1352–1356. doi: 10.1073/pnas.87.4.1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Masliah E, Alford M, DeTeresa R, Mallory M, Hansen L. Deficient glutamate transport is associated with neurodegeneration in Alzheimer’s disease. Ann Neurol. 1996;40(5):759–766. doi: 10.1002/ana.410400512. [DOI] [PubMed] [Google Scholar]
- 30.Li S, Mallory M, Alford M, Tanaka S, Masliah E. Glutamate transporter alterations in Alzheimer disease are possibly associated with abnormal APP expression. J Neuropathol Exp Neurol. 1997;56(8):901–911. doi: 10.1097/00005072-199708000-00008. [DOI] [PubMed] [Google Scholar]
- 31.Jacob CP, Koutsilieri E, Bartl J, et al. Alterations in expression of glutamatergic transporters and receptors in sporadic Alzheimer’s disease. J Alzheimers Dis. 2007;11(1):97–116. doi: 10.3233/jad-2007-11113. [DOI] [PubMed] [Google Scholar]
- 32.Lauderback CM, Hackett JM, Huang FF, et al. The glial glutamate transporter, GLT-1, is oxidatively modified by 4-hydroxy-2-nonenal in the Alzheimer’s disease brain: the role of Abeta1–42. J Neurochem. 2001;78(2):413–416. doi: 10.1046/j.1471-4159.2001.00451.x. [DOI] [PubMed] [Google Scholar]
- 33.Butchbach ME, Tian G, Guo H, Lin CL. Association of excitatory amino acid transporters, especially EAAT2, with cholesterol-rich lipid raft microdomains: importance for excitatory amino acid transporter localization and function. J Biol Chem. 2004;279(33):34388–34396. doi: 10.1074/jbc.M403938200. [DOI] [PubMed] [Google Scholar]
- 34.Tian G, Kong Q, Lai L, Ray-Chaudhury A, Lin CL. Increased expression of cholesterol 24S-hydroxylase results in disruption of glial glutamate transporter EAAT2 association with lipid rafts: a potential role in Alzheimer’s disease. J Neurochem. 2010;113(4):978–989. doi: 10.1111/j.1471-4159.2010.06661.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McBean GJ, Roberts PJ. Neurotoxicity of L-glutamate and DL-threo-3-hydroxyaspartate in the rat striatum. J Neurochem. 1985;44(1):247–254. doi: 10.1111/j.1471-4159.1985.tb07137.x. [DOI] [PubMed] [Google Scholar]
- 36.Rothstein JD, Jin L, Dykes-Hoberg M, Kuncl RW. Chronic inhibition of glutamate uptake produces a model of slow neurotoxicity. Proc Natl Acad Sci USA. 1993;90(14):6591–6595. doi: 10.1073/pnas.90.14.6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Arias C, Arrieta I, Massieu L, Tapia R. Neuronal damage and MAP2 changes induced by the glutamate transport inhibitor dihydrokainate and by kainate in rat hippocampus in vivo. Exp Brain Res. 1997;116(3):467–476. doi: 10.1007/pl00005774. [DOI] [PubMed] [Google Scholar]
- 38.Rothstein JD. Current hypotheses for the underlying biology of amyotrophic lateral sclerosis. Ann Neurol. 2009;65(Suppl 1):S3–S9. doi: 10.1002/ana.21543. [DOI] [PubMed] [Google Scholar]
- 39.Boillee S, Vande Velde C, Cleveland DW. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron. 2006;52(1):39–59. doi: 10.1016/j.neuron.2006.09.018. [DOI] [PubMed] [Google Scholar]
- 40.Rothstein JD, Van Kammen M, Levey AI, Martin LJ, Kuncl RW. Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann Neurol. 1995;38(1):73–84. doi: 10.1002/ana.410380114. [DOI] [PubMed] [Google Scholar]
- 41.Bruijn LI, Becher MW, Lee MK, et al. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron. 1997;18(2):327–338. doi: 10.1016/s0896-6273(00)80272-x. [DOI] [PubMed] [Google Scholar]
- 42.Bendotti C, Tortarolo M, Suchak SK, et al. Transgenic SOD1 G93A mice develop reduced GLT-1 in spinal cord without alterations in cerebrospinal fluid glutamate levels. J Neurochem. 2001;79(4):737–746. doi: 10.1046/j.1471-4159.2001.00572.x. [DOI] [PubMed] [Google Scholar]
- 43.Howland DS, Liu J, She Y, et al. Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS) Proc Natl Acad Sci USA. 2002;99(3):1604–1609. doi: 10.1073/pnas.032539299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44▪.Guo H, Lai L, Butchbach ME, et al. Increased expression of the glial glutamate transporter EAAT2 modulates excitotoxicity and delays the onset but not the outcome of ALS in mice. Hum Mol Genet. 2003;12(19):2519–2532. doi: 10.1093/hmg/ddg267. First showed that increased EAAT2 has beneficial effects in an animal model of amyotrophic lateral sclerosis by transgenic mouse approach. [DOI] [PubMed] [Google Scholar]
- 45▪▪.Rothstein JD, Patel S, Regan MR, et al. Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature. 2005;433(7021):73–77. doi: 10.1038/nature03180. Identified the first EAAT2 transcriptional activator and demonstrated that increased EAAT2 is neuroprotective. [DOI] [PubMed] [Google Scholar]
- 46.Blandini F, Nappi G, Tassorelli C, Martignoni E. Functional changes of the basal ganglia circuitry in Parkinson’s disease. Prog Neurobiol. 2000;62(1):63–88. doi: 10.1016/s0301-0082(99)00067-2. [DOI] [PubMed] [Google Scholar]
- 47.Albin RL, Young AB, Penney JB. The functional anatomy of basal ganglia disorders. Trends Neurosci. 1989;12(10):366–375. doi: 10.1016/0166-2236(89)90074-x. [DOI] [PubMed] [Google Scholar]
- 48.Bezard E, Boraud T, Bioulac B, Gross CE. Involvement of the subthalamic nucleus in glutamatergic compensatory mechanisms. Eur J Neurosci. 1999;11(6):2167–2170. doi: 10.1046/j.1460-9568.1999.00627.x. [DOI] [PubMed] [Google Scholar]
- 49.Klockgether T, Turski L, Honore T, et al. The AMPA receptor antagonist NBQX has antiparkinsonian effects in monoamine-depleted rats and MPTP-treated monkeys. Ann Neurol. 1991;30(5):717–723. doi: 10.1002/ana.410300513. [DOI] [PubMed] [Google Scholar]
- 50.Greenamyre JT, Eller RV, Zhang Z, Ovadia A, Kurlan R, Gash DM. Antiparkinsonian effects of remacemide hydrochloride, a glutamate antagonist, in rodent and primate models of Parkinson’s disease. Ann Neurol. 1994;35(6):655–661. doi: 10.1002/ana.410350605. [DOI] [PubMed] [Google Scholar]
- 51.Starr MS. Antiparkinsonian actions of glutamate antagonists-- alone and with L-DOPA: a review of evidence and suggestions for possible mechanisms. J Neural Transm Park Dis Dement Sect. 1995;10(2–3):141–185. doi: 10.1007/BF02251229. [DOI] [PubMed] [Google Scholar]
- 52.Battaglia G, Busceti CL, Pontarelli F, et al. Protective role of group-II metabotropic glutamate receptors against nigro-striatal degeneration induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in mice. Neuropharmacology. 2003;45(2):155–166. doi: 10.1016/s0028-3908(03)00146-1. [DOI] [PubMed] [Google Scholar]
- 53.Corti C, Battaglia G, Molinaro G, et al. The use of knock-out mice unravels distinct roles for mGlu2 and mGlu3 metabotropic glutamate receptors in mechanisms of neurodegeneration/neuroprotection. J Neurosci. 2007;27(31):8297–8308. doi: 10.1523/JNEUROSCI.1889-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ossowska K, Konieczny J, Wolfarth S, Wieronska J, Pilc A. Blockade of the metabotropic glutamate receptor subtype 5 (mGluR5) produces antiparkinsonian-like effects in rats. Neuropharmacology. 2001;41(4):413–420. doi: 10.1016/s0028-3908(01)00083-1. [DOI] [PubMed] [Google Scholar]
- 55.Breysse N, Baunez C, Spooren W, Gasparini F, Amalric M. Chronic but not acute treatment with a metabotropic glutamate 5 receptor antagonist reverses the akinetic deficits in a rat model of parkinsonism. J Neurosci. 2002;22(13):5669–5678. doi: 10.1523/JNEUROSCI.22-13-05669.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Battaglia G, Busceti CL, Molinaro G, et al. Endogenous activation of mGlu5 metabotropic glutamate receptors contributes to the development of nigro-striatal damage induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in mice. J Neurosci. 2004;24(4):828–835. doi: 10.1523/JNEUROSCI.3831-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bergman H, Wichmann T, DeLong MR. Reversal of experimental parkinsonism by lesions of the subthalamic nucleus. Science. 1990;249(4975):1436–1438. doi: 10.1126/science.2402638. [DOI] [PubMed] [Google Scholar]
- 58.Limousin P, Krack P, Pollak P, et al. Electrical stimulation of the subthalamic nucleus in advanced Parkinson’s disease. N Engl J Med. 1998;339(16):1105–1111. doi: 10.1056/NEJM199810153391603. [DOI] [PubMed] [Google Scholar]
- 59.Wallace BA, Ashkan K, Heise CE, et al. Survival of midbrain dopaminergic cells after lesion or deep brain stimulation of the subthalamic nucleus in MPTP-treated monkeys. Brain. 2007;130(Pt 8):2129–2145. doi: 10.1093/brain/awm137. [DOI] [PubMed] [Google Scholar]
- 60.Chung EK, Chen LW, Chan YS, Yung KK. Downregulation of glial glutamate transporters after dopamine denervation in the striatum of 6-hydroxydopamine-lesioned rats. J Comp Neurol. 2008;511(4):421–437. doi: 10.1002/cne.21852. [DOI] [PubMed] [Google Scholar]
- 61.Holmer HK, Keyghobadi M, Moore C, Meshul CK. L-dopa-induced reversal in striatal glutamate following partial depletion of nigrostriatal dopamine with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Neuroscience. 2005;136(1):333–341. doi: 10.1016/j.neuroscience.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 62.Engel J., Jr A proposed diagnostic scheme for people with epileptic seizures and with epilepsy: report of the ILAE Task Force on Classification and Terminology. Epilepsia. 2001;42(6):796–803. doi: 10.1046/j.1528-1157.2001.10401.x. [DOI] [PubMed] [Google Scholar]
- 63.McNamara JO, Huang YZ, Leonard AS. Molecular signaling mechanisms underlying epileptogenesis. Sci STKE. 2006;2006(356):re12. doi: 10.1126/stke.3562006re12. [DOI] [PubMed] [Google Scholar]
- 64.Devinsky O. Diagnosis and treatment of temporal lobe epilepsy. Rev Neurol Dis. 2004;1(1):2–9. [PubMed] [Google Scholar]
- 65.Cavus I, Kasoff WS, Cassaday MP, et al. Extracellular metabolites in the cortex and hippocampus of epileptic patients. Ann Neurol. 2005;57(2):226–235. doi: 10.1002/ana.20380. [DOI] [PubMed] [Google Scholar]
- 66.During MJ, Spencer DD. Extracellular hippocampal glutamate and spontaneous seizure in the conscious human brain. Lancet. 1993;341(8861):1607–1610. doi: 10.1016/0140-6736(93)90754-5. [DOI] [PubMed] [Google Scholar]
- 67.Tian GF, Azmi H, Takano T, et al. An astrocytic basis of epilepsy. Nat Med. 2005;11(9):973–981. doi: 10.1038/nm1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Binder DK, Steinhauser C. Functional changes in astroglial cells in epilepsy. Glia. 2006;54(5):358–368. doi: 10.1002/glia.20394. [DOI] [PubMed] [Google Scholar]
- 69.Benarroch EE. Astrocyte-neuron interactions: implications for epilepsy. Neurology. 2009;73(16):1323–1327. doi: 10.1212/WNL.0b013e3181bd432d. [DOI] [PubMed] [Google Scholar]
- 70.Bjornsen LP, Eid T, Holmseth S, Danbolt NC, Spencer DD, de Lanerolle NC. Changes in glial glutamate transporters in human epileptogenic hippocampus: inadequate explanation for high extracellular glutamate during seizures. Neurobiol Dis. 2007;25(2):319–330. doi: 10.1016/j.nbd.2006.09.014. [DOI] [PubMed] [Google Scholar]
- 71.Sarac S, Afzal S, Broholm H, Madsen FF, Ploug T, Laursen H. Excitatory amino acid transporters EAAT-1 and EAAT-2 in temporal lobe and hippocampus in intractable temporal lobe epilepsy. APMIS. 2009;117(4):291–301. doi: 10.1111/j.1600-0463.2009.02443.x. [DOI] [PubMed] [Google Scholar]
- 72.Kong Q, Takahashi K, Schulte D, Stouffer N, Lin Y, Lin CL. Increased glial glutamate transporter EAAT2 expression reduces epileptogenic processes following pilocarpine-induced status epilepticus. Neurobiol Dis. 2012;47(2):145–154. doi: 10.1016/j.nbd.2012.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Compston A, Coles A. Multiple sclerosis. Lancet. 2008;372(9648):1502–1517. doi: 10.1016/S0140-6736(08)61620-7. [DOI] [PubMed] [Google Scholar]
- 74.Srinivasan R, Sailasuta N, Hurd R, Nelson S, Pelletier D. Evidence of elevated glutamate in multiple sclerosis using magnetic resonance spectroscopy at 3 T. Brain. 2005;128(Pt 5):1016–1025. doi: 10.1093/brain/awh467. [DOI] [PubMed] [Google Scholar]
- 75.Stover JF, Pleines UE, Morganti-Kossmann MC, Kossmann T, Lowitzsch K, Kempski OS. Neurotransmitters in cerebrospinal fluid reflect pathological activity. Eur J Clin Invest. 1997;27(12):1038–1043. doi: 10.1046/j.1365-2362.1997.2250774.x. [DOI] [PubMed] [Google Scholar]
- 76.Matute C, Domercq M, Sanchez-Gomez MV. Glutamate-mediated glial injury: mechanisms and clinical importance. Glia. 2006;53(2):212–224. doi: 10.1002/glia.20275. [DOI] [PubMed] [Google Scholar]
- 77.Smith T, Groom A, Zhu B, Turski L. Autoimmune encephalomyelitis ameliorated by AMPA antagonists. Nat Med. 2000;6(1):62–66. doi: 10.1038/71548. [DOI] [PubMed] [Google Scholar]
- 78.Wallstrom E, Diener P, Ljungdahl A, Khademi M, Nilsson CG, Olsson T. Memantine abrogates neurological deficits, but not CNS inflammation, in Lewis rat experimental autoimmune encephalomyelitis. J Neurol Sci. 1996;137(2):89–96. doi: 10.1016/0022-510x(95)00339-4. [DOI] [PubMed] [Google Scholar]
- 79.Bolton C, Paul C. MK-801 limits neurovascular dysfunction during experimental allergic encephalomyelitis. J Pharmacol Exp Ther. 1997;282(1):397–402. [PubMed] [Google Scholar]
- 80.Mitosek-Szewczyk K, Sulkowski G, Stelmasiak Z, Struzynska L. Expression of glutamate transporters GLT-1 and GLAST in different regions of rat brain during the course of experimental autoimmune encephalomyelitis. Neuroscience. 2008;155(1):45–52. doi: 10.1016/j.neuroscience.2008.05.025. [DOI] [PubMed] [Google Scholar]
- 81.Doyle KP, Simon RP, Stenzel-Poore MP. Mechanisms of ischemic brain damage. Neuropharmacology. 2008;55(3):310–318. doi: 10.1016/j.neuropharm.2008.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Benveniste H, Drejer J, Schousboe A, Diemer NH. Elevation of the extracellular concentrations of glutamate and aspartate in rat hippocampus during transient cerebral ischemia monitored by intracerebral microdialysis. J Neurochem. 1984;43(5):1369–1374. doi: 10.1111/j.1471-4159.1984.tb05396.x. [DOI] [PubMed] [Google Scholar]
- 83.Rossi DJ, Oshima T, Attwell D. Glutamate release in severe brain ischaemia is mainly by reversed uptake. Nature. 2000;403(6767):316–321. doi: 10.1038/35002090. [DOI] [PubMed] [Google Scholar]
- 84.Phillis JW, Ren J, O’Regan MH. Transporter reversal as a mechanism of glutamate release from the ischemic rat cerebral cortex: studies with DL-threo-beta-benzyloxyaspartate. Brain Res. 2000;868(1):105–112. doi: 10.1016/s0006-8993(00)02303-9. [DOI] [PubMed] [Google Scholar]
- 85.Rao VL, Dogan A, Bowen KK, Todd KG, Dempsey RJ. Antisense knockdown of the glial glutamate transporter GLT-1 exacerbates hippocampal neuronal damage following traumatic injury to rat brain. Eur J Neurosci. 2001;13(1):119–128. [PubMed] [Google Scholar]
- 86.Weller ML, Stone IM, Goss A, Rau T, Rova C, Poulsen DJ. Selective overexpression of excitatory amino acid transporter 2 (EAAT2) in astrocytes enhances neuroprotection from moderate but not severe hypoxia-ischemia. Neuroscience. 2008;155(4):1204–1211. doi: 10.1016/j.neuroscience.2008.05.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chu K, Lee ST, Sinn DI, et al. Pharmacological induction of ischemic tolerance by glutamate transporter-1 (EAAT2) upregulation. Stroke. 2007;38(1):177–182. doi: 10.1161/01.STR.0000252091.36912.65. [DOI] [PubMed] [Google Scholar]
- 88.Harvey BK, Airavaara M, Hinzman J, et al. Targeted over-expression of glutamate transporter 1 (GLT-1) reduces ischemic brain injury in a rat model of stroke. PLoS ONE. 2011;6(8):e22135. doi: 10.1371/journal.pone.0022135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Faden AI, Demediuk P, Panter SS, Vink R. The role of excitatory amino acids and NMDA receptors in traumatic brain injury. Science. 1989;244(4906):798–800. doi: 10.1126/science.2567056. [DOI] [PubMed] [Google Scholar]
- 90.Globus MY, Alonso O, Dietrich WD, Busto R, Ginsberg MD. Glutamate release and free radical production following brain injury: effects of posttraumatic hypothermia. J Neurochem. 1995;65(4):1704–1711. doi: 10.1046/j.1471-4159.1995.65041704.x. [DOI] [PubMed] [Google Scholar]
- 91.Bullock R, Zauner A, Woodward JJ, et al. Factors affecting excitatory amino acid release following severe human head injury. J Neurosurg. 1998;89(4):507–518. doi: 10.3171/jns.1998.89.4.0507. [DOI] [PubMed] [Google Scholar]
- 92.Koizumi H, Fujisawa H, Ito H, Maekawa T, Di X, Bullock R. Effects of mild hypothermia on cerebral blood flow-independent changes in cortical extracellular levels of amino acids following contusion trauma in the rat. Brain Res. 1997;747(2):304–312. doi: 10.1016/s0006-8993(96)01240-1. [DOI] [PubMed] [Google Scholar]
- 93.Yi JH, Hoover R, McIntosh TK, Hazell AS. Early, transient increase in complexin I and complexin II in the cerebral cortex following traumatic brain injury is attenuated by N-acetylcysteine. J Neurotrauma. 2006;23(1):86–96. doi: 10.1089/neu.2006.23.86. [DOI] [PubMed] [Google Scholar]
- 94.Yi JH, Pow DV, Hazell AS. Early loss of the glutamate transporter splice-variant GLT-1v in rat cerebral cortex following lateral fluid-percussion injury. Glia. 2005;49(1):121–133. doi: 10.1002/glia.20099. [DOI] [PubMed] [Google Scholar]
- 95.Zelenaia O, Schlag BD, Gochenauer GE, et al. Epidermal growth factor receptor agonists increase expression of glutamate transporter GLT-1 in astrocytes through pathways dependent on phosphatidylinositol 3-kinase and transcription factor NF-kappaB. Mol Pharmacol. 2000;57(4):667–678. doi: 10.1124/mol.57.4.667. [DOI] [PubMed] [Google Scholar]
- 96.Figiel M, Maucher T, Rozyczka J, Bayatti N, Engele J. Regulation of glial glutamate transporter expression by growth factors. Exp Neurol. 2003;183(1):124–135. doi: 10.1016/s0014-4886(03)00134-1. [DOI] [PubMed] [Google Scholar]
- 97▪.Tian G, Lai L, Guo H, et al. Translational control of glial glutamate transporter EAAT2 expression. J Biol Chem. 2007;282(3):1727–1737. doi: 10.1074/jbc.M609822200. First showed that EAAT2 expression is highly regulated at the translational level. [DOI] [PubMed] [Google Scholar]
- 98.Filosa A, Paixao S, Honsek SD, et al. Neuron-glia communication via EphA4/ephrin-A3 modulates LTP through glial glutamate transport. Nat Neurosci. 2009;12(10):1285–1292. doi: 10.1038/nn.2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yang Y, Gozen O, Watkins A, et al. Presynaptic regulation of astroglial excitatory neurotransmitter transporter GLT1. Neuron. 2009;61(6):880–894. doi: 10.1016/j.neuron.2009.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100▪.Su ZZ, Leszczyniecka M, Kang DC, et al. Insights into glutamate transport regulation in human astrocytes: cloning of the promoter for excitatory amino acid transporter 2 (EAAT2) Proc Natl Acad Sci USA. 2003;100(4):1955–1960. doi: 10.1073/pnas.0136555100. First showed the human EAAT2 promoter. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101▪▪.Colton CK, Kong Q, Lai L, et al. Identification of translational activators of glial glutamate transporter EAAT2 through cell-based high-throughput screening: an approach to prevent excitotoxicity. J Biomol Screen. 2010;15(6):653–662. doi: 10.1177/1087057110370998. First high-throughput screen in search for EAAT2 translational activators, which led to the identification of several series of novel small molecules. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Xing X, Chang LC, Kong Q, et al. Structure-activity relationship study of pyridazine derivatives as glutamate transporter EAAT2 activators. Bioorg Med Chem Lett. 2011;21(19):5774–5777. doi: 10.1016/j.bmcl.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]