

Abstract
Rheumatoid arthritis (RA) is a common human leukocyte antigen-associated disease. Most RA patients have a five-residue sequence motif called the shared epitope (SE) in the DRβ-chain of the HLA-DRB1 protein. The SE was found to activate nitric oxide (NO) production, suggesting a possible mechanism for RA development. The native conformation of the SE is presumed to be an α-helix, thus using cyclic peptides to stabilize this conformation may produce a potent SE mimetic which will have drug- like properties. We present the development of a backbone cyclic SE mimetic that activates NO production in the low nM range. Circular dichroism analysis revealed a conformational change from unstructured for the parent linear peptides to β– turn in the cyclic analogs. The most active cyclic analog is completely stable towards trypsin/chymotrypsin degradation while the linear 15-mer analogs completely degraded within 30 minutes. The outcome of this study is a potent cyclic peptide with drug-like properties that can be used as a template for drug development.
Keywords: Rheumatoid arthritis, Backbone cyclization, Peptidomimetics
Protein–protein interactions (PPIs)1 control many functions within the living cells, such as cell cycle, signal transduction and metabolism.2, 3α-helical interactions are abundant in PPIs and participate in key processes in many diseases,4 making these interactions targets for drug development. Helix mimetic molecules may have an antagonistic effect if the binding of the two proteins is essential for activity, such as in dimerization, or an agonistic effect if only the helix interaction itself is essential to induce the proper conformational change required for activity in the targeted protein.2 The main objective in developing helix-mimetic drugs is to generate a molecule that will specifically promote or inhibit interactions between α-helical regions and will have the appropriate pharmacological properties, such as metabolic stability, selectivity and oral bioavailability. Walenski et al. developed a hydrocarbon-stapled peptide that mimicked the α–helical structure of the BH3 domain of Bcl-2. This helix mimetic activated Bcl-2 mediated apoptosis pathways in leukemia cells in vivo.5 Moellering et al. targeted the NOTCH transcription factor complex and inhibited its assembly using α-helical stapled peptides derived from the MAML1 protein. Inhibition of NOTCH assembly induced apoptosis in T-ALL cells in vivo.6
Rheumatoid arthritis (RA) is one of the most common inflammatory diseases affecting both articular and extra articular tissues. Most RA patients have HLA-DRB1 alleles encoding a five-residue sequence motif that is commonly referred to as the shared epitope (SE), in residues 70–74 of the DRβ-chain.7 Immunogenetic analyses showed that residues 70, 71 and 74 are vital, while the identity of residues 72 and 73 is not significant. Thus, the (Arg/Gln)-(Arg/Lys)-X-X-Ala consensus motif was proposed as essential and sufficient to confer RA susceptibility.8 Further analyses showed that the identity of residue 70 is crucial for both protection against and susceptibility to RA. While Arg or Gln in position 70 are associated with increased RA susceptibility, Asp in that position confers protection against the disease.9–11 The native SE conformation in the DRβ-chain was predicted to be α-helical.12–14 However, the mechanism by which the SE affects disease susceptibility is unknown. Recently, a connection between the SE and the activation of nitric oxide (NO) signaling was found, suggesting a model for SE-related RA susceptibility.15–18 These studies indicated that the SE acts as a signaling ligand that bind and activates an aberrant NO- dependent signaling cascade through-cell surface calreticulin. The importance of the α-helical conformation to the potency of the SE was also observed,19 implying that stabilization of the α-helix conformation in short synthetic peptides bearing the SE motif can be beneficial to the development of potent SE-triggered signaling agonists and antagonists.
Short linear peptides lack important pharmacological properties needed to become drug-lead molecules, including metabolic stability, selectivity and bioavailability. They are usually in fast equilibrium among many conformations in solution with no single restricted conformation. Peptidomimetics are designed to retain the biological activity of their parent linear peptides, while conferring desirable pharmacological properties.20–23 Cyclization is a common strategy to confer drug-like properties upon peptides. The backbone cyclization methodology24 prepares cyclic peptides without utilizing the functional groups of the side chain residues. This feature is extremely important when all the functional groups in a peptide sequence are essential for biological activity.25–27 Helical structure in short peptide sequences is induced by a covalent bond for ring closure, which mimics the native hydrogen bond that stabilizes the helix. There is a wide variety of synthetic methods for creating these peptides, e.g., stapled peptides,5, 28 as well as a diversity of biological targets.29, 30
A SE mimetic agonist that activates NO production in the low nM range was developed. Three backbone cyclic peptide analogs containing the SE consensus motif were designed to induce a stable active conformation. The different potencies of the three analogs, which differ only in ring size, demonstrated the importance of conformational screening. Circular dichroism (CD) spectra revealed a slight conformational change from the parent linear peptides to the cyclic analogs. The most potent cyclic analog, c(HS4-4), is also completely stable towards enzymatic degradation while the linear peptides were degraded by trypsin/chymotrypsin within 30 minutes.
Helix mimetic cyclic analogs usually have bridges at positions i, i+4 or i, i+7.5, 6 The consensus SE motif Gln-Lys-X-X-Ala was incorporated into an i, i+4 backbone cyclic scaffold.31 A Trp residue was added to the amino terminus to aid in determining the concentrations of the cyclic analogs using UV spectroscopy.32 The synthesis and general structure of the backbone cyclic peptides are described in Scheme 1.
Scheme 1.

Synthesis of c(HSn-4) compounds. Conditions: a) 20% piperidine, DMF b) HBTU, DIPEA c) HATU, DIPEA d) Pd(PPh3)4 (0), PhSiH3 e) BTC, DIPEA f) TFA, TIPS, TDW
The characterization of the cyclic peptides is summarized in Table 1.
Table 1.
Characterization of the BC peptides
| Peptide Name | n | Bridge Size | Ring Size | Calcd MH+ | Observed MH+ (HRMS) | Purity HPLC (%) | TR HPLC* (min) |
|---|---|---|---|---|---|---|---|
| c(HS2-4) | 2 | 9 | 22 | 941.5428 | 941.5446 | >95 | 18.65 |
| c(HS4-4) | 4 | 11 | 24 | 969.5741 | 969.5744 | >95 | 18.68 |
| c(HS6-4) | 6 | 13 | 26 | 997.6054 | 997.6062 | >95 | 18.83 |
HPLC conditions.33
The conformational changes induced by cyclization and the variety of ring sizes were studied by CD of the cyclic peptides and two positive control 15-mers (65–79*0401, H-Lys-Asp-Leu-Leu-Glu-Gln-Lys-Arg-Ala-Ala-Val-Asp-Thr-Tyr-Cys-NH2, and 65- 79*0404, H-Lys-Asp-Leu-Leu-Glu-Gln-Arg-Arg-Ala-Ala-Val-Asp-Thr-Tyr-Cys- NH2).34 As predicted, the two linear 15-mer peptides had no defined structure and the CD spectra resembled a random coil (Figure 1a). CD spectra of the three cyclic analogs suggested a slight conformational change, as compared to the linear 15-mer peptides, however, no decisive conclusions could be drawn (Figure 1b). Although the bioactive pharmacophors of the peptide are assumed to acquire the needed helix conformation upon binding to their binding site, the restricted conformation of the short cyclic peptides compared with those of the longer linear ones may enhance activity.
Figure 1.
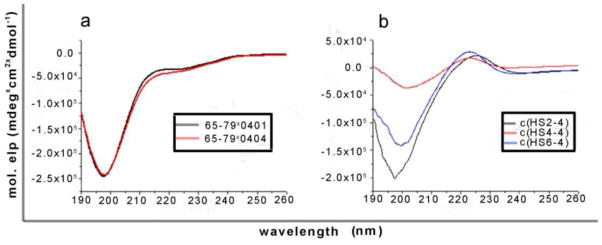
CD spectra of the cyclic and 15-mer linear peptides. a) Random coil CD spectra were observed for the two linear 15-mer peptides. b) A slightly different CD spectra were observed for the three cyclic analogs, as compared to the linear 15-mer peptides, suggesting a conformational change.
The ability of the cyclic analogs to activate NO production in fibroblasts was examined.35 The 15-mer peptides 65–79*0401 (H-Lys-Asp-Leu-Leu-Glu-Gln-Lys- Arg-Ala-Ala-Val-Asp-Thr-Tyr-Cys-NH2) and 65–79*0402 (H-Lys-Asp-Ile-Leu-Glu- Asp-Glu-Arg-Ala-Ala-Val-Asp-Thr-Tyr-Cys-NH2) were used as positive and negative controls, respectively. The optimal concentration for the positive control, 65–79*0401, was previously determined to be 50 μM.17–19 Figure 2 shows that the three cyclic analogs activated NO production significantly above the negative control levels. Furthermore, c(HS6-4) maintained the elevated NO production levels at concentrations five times lower than the positive control. The most potent analog by far was c(HS4-4), which maintained high NO production levels even in concentrations 50,000 times lower than 65–79*0401, and activated NO production even in the low nM concentrations.
Figure 2.

Dose-response experiments of NO production rate induced by cyclic analogs. Human M1 fibroblasts were incubated with different concentrations of the listed cyclic peptides, or with 50 μM of positive (65–79*0401) or negative (65- 79*0402) control peptides. NO production rates were determined using the fluorescent probe 4,5-diaminofluorescein diacetate (DAF-2DA).17–19 The results represent mean ± SEM from four replicate experiments.
These results demonstrate the importance of subtle changes in the conformation of the bioactive SE pharmacophors. The cyclic NO production activators are of paramount value to the ongoing efforts to resolve the mechanism by which the SE affects RA susceptibility and may also be used as lead scaffolds for developing potent SE-triggered signaling antagonists.
To assess the stability of the most potent cyclic analog, c(HS4-4), towards enzymatic degradation as compared to the linear 15-mers, 65–79*0401 and 65–79*0404, a trypsin/chymotrypsin stability assay was conducted.36 HPLC analysis was used to determine the percent of degradation (Figure 3), whereas MS analysis was used to identify the specific cleavage sites (Table 2). The cyclic analog, c(HS4-4) was completely stable towards trypsin/chymotrypsin degradation even after 4 hours of incubation with the proteases whereas the linear analogs, 65–79*0401 and 65–79*0404 were degraded completely after 30 minutes (Figure 3).
Figure 3.
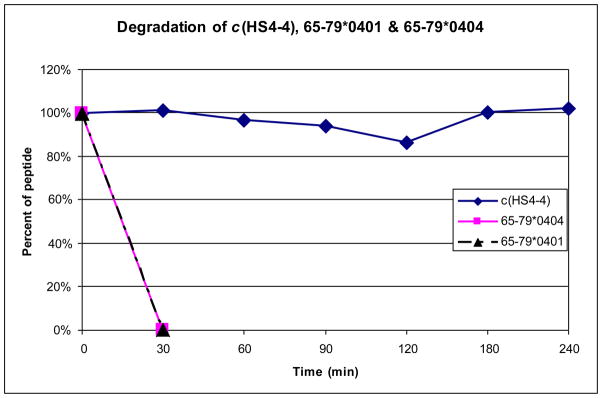
Comparison of the degradation rates of c(HS4-4), 65–79*0401 and 65- 79*0404 towards trypsin and chymotrypsin enzymatic cleavage.
Table 2.
Fragmentation of c(HS4-4), 65–79*0401 and 65–79*0404 after degradation by trypsin/chymotrypsin#
| Name | Deduced sequence of fragment | Observed MH+ |
|---|---|---|
| c(HS4-4) |
|
969.53 |
| 65–79*0401 | K-D-L-L-E-Q-K-R-A-A-V-D-T-Y-C | 1752.98 |
| Fragment 1 | K-D-L-L-E-Q-K-R-A-A-V-D-T-Y | 1649.93 |
| Fragment 2 | C | ## |
| Fragment 3 | K-D-L-L-E-Q-K-R | 1029.64 |
| Fragment 4 | A-A-V-D-T-Y | 638.51 |
| Fragment 5 | K-D-L-L-E-Q-K | 872.53 |
| Fragment 6 | R-A-A-V-D-T-Y | 795.43 |
| 65–79*0404 | K-D-L-L-E-Q-R-R-A-A-V-D-T-Y-C | 1778.84 |
| Fragment 1 | K-D-L-L-E-Q-R-R-A-A-V-D-T-Y | 1676.84 |
| Fragment 2 | C | ## |
| Fragment 3 | K-D-L-L-E-Q-R-R | 1057.23 |
| Fragment 4 | A-A-V-D-T-Y | 638.59 |
| Fragment 5 | K-D-L-L-E-Q-R | 901.71 |
| Fragment 6 | R-A-A-V-D-T-Y | 795.40 |
Fragments were identified by MS
Complementary fragment
The specific cleavage sites were determined using MS analysis to extract further information about the degradation of the peptides. Several cleavage sites were identified from the analyzed fragments of the linear 15-mer peptides, 65–79*0401 and 65–79*0404, containing the SE motif (residues 70–74) (Table 2). These cleavage sites were between Tyr78 and Cys79 (chymotrypsin), Arg72 and Ala73 (trypsin), and Arg71/Lys71 and Arg72 (trypsin). As two of the identified cleavage sites (after residue 71 and 72) are found within the SE segment and are conserved in the cyclic analog, we can conclude that the 15-mer peptides are good controls for comparison. Regarding the cyclic peptide, c(HS4-4), in accordance with the HPLC results, no fragments were observed in the MS analysis, suggesting that the restricted conformation induced by cyclization was not recognized by trypsin and chymotrypsin. The fact that cleavage was not observed at either of the cleavage sites within the conserved SE segment (after residue 71 and 72) in the cyclic analog strengthens the postulation that the use of backbone cyclization may confer enzymatic stability to peptides.
A SE mimetic agonist was designed. Three backbone cyclic peptide analogs, with varying ring sizes, were synthesized and evaluated as NO production activators. The different potencies of the three analogs demonstrate the importance of conformational screening in drug design. Although the CD spectra of the cyclic analogs were similar and did not form a regular secondary structure, it is safe to assume that conformational complementarity occurs upon binding to the binding site. Thus, the cyclic analog that enables the active pharmacophors to assume the correct binding conformation will be the most potent one. Stability studies revealed that the most potent backbone cyclic analog has superior properties compared to the linear 15-mer analogs. The enhanced potency along with the significant improved stability towards enzymatic degradation marks this peptide as a potential scaffold for the development of promising lead molecules for RA therapy.
Acknowledgments
We thank Timothy Samuel Haug from the Department of Chemical Engineering, University of Michigan for technical assistance. We thank Prof. Assaf Friedler and Dr. Deborah E. Shalev from the Hebrew University for helpful discussions.
References and Notes
- 1.The following abbreviations were used throughout the text. The abbreviations for amino acids are according to the IUPAC-IUB Commission of Biochemical Nomenclature, http://www.chem.qmul.ac.uk/iupac/AminoAcid/. ACN, acetonitrile; AGBU, Alloc glycine building units; Alloc, allyloxycarbonyl; BTC, bis(trichloromethyl)carbonate; CD, Circular dichroism; DCM, dichloromethane; DIPEA, diisopropylethylamine; Fmoc, 9-fluorenylmethyloxycarbonyl; HATU, (2-(7- Aza-1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate); HBTU, (2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate); HLA, human leukocyte antigen; MBHA, methylbenzhydrylamine; NMP, 1-methyl-2- pyrrolidinone; NO, nitric oxide; PPIs, Protein–protein interactions; RA, Rheumatoid arthritis; RP-HPLC, reverse phase high pressure liquid chromatography; SAR, structure activity relationship; SE, shared epitope; SPPS, solid phase peptide synthesis; TDW, tri-distilled water; TFA, trifluoroacetic acid.
- 2.Benyamini H, Friedler A. Future Med Chem. 2010;2:989. doi: 10.4155/fmc.10.196. [DOI] [PubMed] [Google Scholar]
- 3.Jones S, Thornton JM. Proc Natl Acad Sci U S A. 1996;93:13. doi: 10.1073/pnas.93.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guharoy M, Chakrabarti P. Bioinformatics. 2007;23:1909. doi: 10.1093/bioinformatics/btm274. [DOI] [PubMed] [Google Scholar]
- 5.Walensky LD, Kung AL, Escher I, Malia TJ, Barbuto S, Wright RD, Wagner G, Verdine GL, Korsmeyer SJ. Science. 2004;305:1466. doi: 10.1126/science.1099191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moellering RE, Cornejo M, Davis TN, Bianco CD, Aster JC, Blacklow SC, Kung AL, Gilliland DG, Verdine GL, Bradner JE. Nature. 2009;462:182. doi: 10.1038/nature08543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gregersen PK, Silver J, Winchester RJ. Arthritis Rheum. 1987;30:1205. doi: 10.1002/art.1780301102. [DOI] [PubMed] [Google Scholar]
- 8.Ou D, Mitchell LA, Tingle AJ. Hum Immunol. 1998;59:665. doi: 10.1016/s0198-8859(98)00067-6. [DOI] [PubMed] [Google Scholar]
- 9.Gourraud PA, Boyer JF, Barnetche T, Abbal M, Cambon-Thomsen A, Cantagrel A, Constantin A. Arthritis Rheum. 2006;54:593. doi: 10.1002/art.21630. [DOI] [PubMed] [Google Scholar]
- 10.van der Helm-van Mil AH, Huizinga TW, Schreuder GM, Breedveld FC, de Vries RR, Toes RE. Arthritis Rheum. 2005;52:2637. doi: 10.1002/art.21272. [DOI] [PubMed] [Google Scholar]
- 11.Ruiz-Morales JA, Vargas-Alarcon G, Flores-Villanueva PO, Villarreal-Garza C, Hernandez-Pacheco G, Yamamoto-Furusho JK, Rodriguez-Perez JM, Perez-Hernandez N, Rull M, Cardiel MH, Granados J. Hum Immunol. 2004;65:262. doi: 10.1016/j.humimm.2003.12.009. [DOI] [PubMed] [Google Scholar]
- 12.Winchester RJ, Gregersen PK. Springer Semin Immunopathol. 1988;10:119. doi: 10.1007/BF01857219. [DOI] [PubMed] [Google Scholar]
- 13.Kappes D, Strominger JL. Annu Rev Biochem. 1988;57:991. doi: 10.1146/annurev.bi.57.070188.005015. [DOI] [PubMed] [Google Scholar]
- 14.Shookster L, Matsuyama T, Burmester G, Winchester R. Hum Immunol. 1987;20:59. doi: 10.1016/0198-8859(87)90006-1. [DOI] [PubMed] [Google Scholar]
- 15.Holoshitz J, Ling S. Ann N Y Acad Sci. 2007;1110:73. doi: 10.1196/annals.1423.009. [DOI] [PubMed] [Google Scholar]
- 16.Holoshitz J, De Almeida DE, Ling S. Ann N Y Acad Sci. 2010;1209:91. doi: 10.1111/j.1749-6632.2010.05745.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ling S, Li Z, Borschukova O, Xiao L, Pumpens P, Holoshitz J. Arthritis Res Ther. 2007;9:R5. doi: 10.1186/ar2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ling S, Pi X, Holoshitz J. J Immunol. 2007;179:6359. doi: 10.4049/jimmunol.179.9.6359. [DOI] [PubMed] [Google Scholar]
- 19.Ling S, Lai A, Borschukova O, Pumpens P, Holoshitz J. Arthritis Rheum. 2006;54:3423. doi: 10.1002/art.22178. [DOI] [PubMed] [Google Scholar]
- 20.Ahn JM, Boyle NA, MacDonald MT, Janda KD. Mini Rev Med Chem. 2002;2:463. doi: 10.2174/1389557023405828. [DOI] [PubMed] [Google Scholar]
- 21.Gante J. Angewandte Chemie-International Edition. 1994;33:1699. [Google Scholar]
- 22.Naider F, Goodman M. In: Synthesis of Peptides and Peptidomimetics; Goodman M, Toniolo C, Moroder L, Felix A, editors. E22a. Thieme; Stuttgart New York: 2002. p. 1. [Google Scholar]
- 23.Vagner J, Qu HC, Hruby VJ. Curr Opin Chem Biol. 2008;12:292. doi: 10.1016/j.cbpa.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gilon C, Halle D, Chorev M, Selinger Z, Byk G. Biopolymers. 1991;31:745. doi: 10.1002/bip.360310619. [DOI] [PubMed] [Google Scholar]
- 25.Hruby VJ, al-Obeidi F, Kazmierski W. Biochem J. 1990;268:249. doi: 10.1042/bj2680249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Demmer O, Frank AO, Kessler H. In: Peptide and Protein Design for Biopharmaceutical Applications; KJ, editor. John Wiley and Sons Ltd; 2009. p. 133. [Google Scholar]
- 27.Ovadia O, Greenberg S, Laufer B, Gilon C, Hoffman A, Kessler H. Expert Opinion on Drug Discovery. 2010;5:655. doi: 10.1517/17460441.2010.493935. [DOI] [PubMed] [Google Scholar]
- 28.Stewart ML, Fire E, Keating AE, Walensky LD. Nature Chemical Biology. 2010;6:595. doi: 10.1038/nchembio.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Haridas V. European Journal of Organic Chemistry. 2009:5112. [Google Scholar]
- 30.Garner J, Harding MM. Organic & Biomolecular Chemistry. 2007;5:3577. doi: 10.1039/b710425a. [DOI] [PubMed] [Google Scholar]
- 31.All the peptides were synthesized using standard Fmoc SPPS procedures on Rink amide MBHA resin as the solid support. The urea backbone cyclic peptides, designated c(HSn-4), were synthesized according to the procedures described by Hurevich, et al. J Pept Sci. 2010;16:178. doi: 10.1002/psc.1218. using various AGBU, where n stands for the number of atoms in the N-alkyl chain on the glycine at position 2. The sequences of the linear 15 mer peptides are as follows: 65–79*0401: H-Lys-Asp-Leu-Leu-Glu- Gln-Lys-Arg-Ala-Ala-Val-Asp-Thr-Tyr-Cys-NH2 65–79*0402: H-Lys-Asp-Ile-Leu- Glu-Asp-Glu-Arg-Ala-Ala-Val-Asp-Thr-Tyr-Cys-NH2 65–79*0404: H-Lys-Asp- Leu-Leu-Glu-Gln-Arg-Arg-Ala-Ala-Val-Asp-Thr-Tyr-Cys-NH2.
- 32.Gill SC, von Hippel PH. Anal Biochem. 1989;182:319. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 33.All analytical HPLC were recorded at 220 nm at a flow of 1 ml/min on a RP- 18 column (5 μm 250 × 4.6 mm, 110 Å), eluents A (0.05% TFA in TDW) and B (0.05% TFA in ACN) were used in a linear gradient (95% A → 5% A in 35 min).
- 34.Samples of each peptide were prepared by dissolving a lyophilized peptide in TDW. Far-UV CD spectra were collected over 190–260 nm at room temperature using a J-810 spectropolarimeter (Jasco) in a 0.1 cm quartz cuvette for far-UV CD spectroscopy.
- 35.Human fibroblast M1 cells were plated at a density of 1 × 105 cells per well in 96-well plates the day prior to the Nitric Oxide assay. To determine the rate of NO production in fibroblast, cells were first loaded with 20 μM of the fluorescent NO probe 4,5-diaminofluorescein diacetate (DAF-2DA), incubated in the dark at 37°C for 1 hour and washed in 100 μL of DMEM/phenol red–free medium. The fluorescence level was recorded every 5 minutes over a period of 500 minutes, using a Fusion αHT system (PerkinElmer Life Sciences) at an excitation wavelength of 488 nm and emission wavelength of 515 nm. The NO production rate is expressed as the mean ± SEM fluorescence units per minute.
- 36.The trypsin stability assay was conducted as previously described by Tal-Gan, et al. Bioorg Med Chem. 2010;18:2976. doi: 10.1016/j.bmc.2010.02.031.. 400 μL of each peptide (1 mM) dissolved in 200 mM NH4HCO3 buffer solution (pH 8) were mixed with 1 μL of trypsin and chymotrypsin (porcine pancreas, Biological Industries Israel, Beit Haemek LTD) solution (2.5 mg/1 ml). The peptides were incubated at 37°C, 30 μL samples were taken every 30 min and mixed with 30 μL of 2% TFA and 30% ACN in water. Samples were analyzed by HPLC and by MALDI-TOF MS.