

Abstract
The onset of resistance to approved anti-AIDS drugs by HIV necessitates the search for novel inhibitors of HIV-1 reverse transcriptase (RT). Developing single molecular agents concurrently occupying the nucleoside and nonnucleoside binding sites in RT is an intriguing idea but the proof-of-concept has so far been elusive. As a first step, we describe molecular modeling to guide focused chemical syntheses of conjugates having nucleoside (d4T) and nonnucleoside (TIBO) moieties tethered by a flexible polyethylene glycol (PEG) linker. A triphosphate of d4T-6PEG-TIBO conjugate was successfully synthesized that is recognized as a substrate by HIV-1 RT and incorporated into a double-stranded DNA.
Keywords: HIV-1, Reverse Transcriptase, Bifunctional, NRTI, NNRTI, PEG
HIV reverse transcriptase (RT) plays a pivotal role in the viral life cycle, whose multiple functions include RNA-directed and DNA-directed DNA polymerization. HIV-1 RT inhibitors, critical components of the highly active anti-retroviral therapy (HAART) against AIDS, are designed to target either the active site or a nearby hydrophobic site of the viral polymerase, but not both. The nucleoside analog RT inhibitors (NRTIs) are prodrug nucleosides which are transported across cellular host membranes and phosphorylated to the metabolically active nucleotides. These nucleotides serve as substrates for RT and lacking a 3′ hydroxyl group, they serve to chain-terminate DNA synthesis. This chain termination and their competition with natural deoxynucleoside triphosphates (dNTPs) yield their clinical utility1. Structural evidence shows that the allosteric nonnucleoside RT inhibitors (NNRTIs) bind to an inducible pocket approximately 10 Å away from the polymerase site2,3, and steady-state kinetic studies suggest they are noncompetitive with respect to the binding of dNTP and oligonucleotides4–8. While NRTIs and NNRTIs are potent inhibitors of reverse transcription, resulting in an array of FDA-approved formulations, resistance at the level of RT proves to hinder their full utility–this phenomenon calls for new and improved antiviral inhibitors.
In light of viral resistance, RT has been targeted with using other strategies including dual NRTIs9, interference of RT and DNA binding10, among others. One approach that has previously received some attention is the concept of a “bifunctional inhibitor”. Pursuit of a “bifunctional inhibitor” for HIV-1 reverse transcriptase (a molecule which simultaneously targets both inhibitory pockets) has been both an intriguing and challenging endeavor, despite more than a decade of global research efforts11–15.
The genesis of the idea, in part, was inspired by kinetic observations of HIV-1 RT. Biochemical studies from our lab and others16–19 suggest that the natural dNTP and the NNRTI can simultaneously occupy their respective sites. Moreover, these studies also indicated that communication occurs between the active and the NNRTI binding sites since the inhibition of RT by NNRTIs is manifested through a remote effect on the chemical step of polymerization. At the molecular level, transient kinetic studies have shown that nonnucleoside inhibitors change the rate-limiting step of incorporation, resulting in chemistry as the slowest step16,17. This crosstalk between the two binding sites sets a hypothesis for designing a single molecular “bifunctional” inhibitor including an NRTI and NNRTI component tethered together to target both sites simultenaously. This innovative approach has been successful in yielding inhibitors of other enzyme systems, FKBP and stromelysin20,21.
The attraction of a bifunctional inhibitor against HIV-1 RT is many fold. First, combining two inhibitory moieties within one molecule can have synergistic effects that may improve potency. Second, the onset of resistance may decrease as a result of simultaneous mutations that must be produced at both binding pockets. Lastly, potential therapeutic agents based on the bifunctional inhibition designed specifically for HIV-1 RT would presumably not interfere with human polymerases such as the human mitochondrial DNA polymerase γ and thus result in fewer side effects. Unfortunately, in practice, the development of an HIV-1 RT bifunctional inhibitor has been extremely challenging11,13,14, 22–24 – attempts with d4T, AZT, and various NNRTI scaffolds have thus far not been successful to illustrate the bifunctional proof of concept. Renoud-Grappin et al. showed that a linkage to d4T did not reduce its antiviral inhibition, but further addition of the NNRTI decreased the antiviral activity 100-fold14. This loss of activity can be attributed to a lack of binding of the NNRTI, as this group of inhibitors showed activity against HIV-2 RT, for which no NNRTI pocket exists. Other compounds utilizing AZT and ddC as NRTI parents illustrated NRTI-linkage is detrimental at both the C5, N3 (of AZT) and C4 positions (of ddC)13, though the linkers chosen in this study may have influenced activity. Along with others, these two studies depict the complex structure-activity relationship that exists for putative bifunctional inhibitors, but a promising finding demonstrated that a linkage at the 5-position of d4T with a methylamino linker did not perturb activity of the NRTI in cell culture14. This contrasts with data from Gavriliu et al., which argues attachment of this methylamino linker indeed disrupts d4T’s antiviral activity in culture25. Comparison of these two studies yields significant differences in the linker terminus, further supporting the complexity in addressing linkage position and linker design for an RT bifunctional inhibitor. Work from our lab preparing a bifunctional inhibitor prodrug based on phenylethylthiazolylthiourea (PETT) compounds such as HI-236 as an NNRTI showed antiviral activity in cell culture however structural data with PETT-2 NNRTIs suggested the choice of position for linkage from the NNRTI was not optimal26,27.
In order to circumvent issues raised above and to fully understand the potential benefits of the bifunctional concept, several basic questions were addressed for the rational design. First is the choice of NRTI and the point of connection. To date, eight NRTIs have been approved for clinical use. The considerations in choosing one of these are two fold: (1) the chemical transformation to introduce properly substituted derivatives and (2) the in vivo transformation of the latter to its triphosphate by human kinases. N4-substitution of ddC (zalcitabine) has afforded active compounds against HIV replication, but the modifications made at this position substantially reduced potency relative to the parent compound ddC13. Substitution at both N-3 and C-5 of AZT (Retrovir) has lead to inactive antiviral compounds11. The substitutions at C-5 in d4T (stavudine, Figure 1) yield mixed results, but mostly result in inactive analogs14,25,28–30. Attempted modifications to AZT and d4T suggest the difficulty for human thymidine kinase (hTK) to phosphorylate 2′,3′-dideoxythymidine analogs, but there is evidence that C-5 substituted thymidine can be phosphorylated in vivo by thymidine kinase31–33. Guided by these previous studies, we chose d4T as our nucleoside because of the observed slight resistance in cell culture studies34–36, and at the enzymatic level, it is incorporated as efficiently as the natural substrate (dTTP) with both DNA/DNA and DNA/RNA primer-templates37.
Figure 1. Bifunctional Building Blocks.

Structures of d4T (1), 8-Cl-TIBO (2), and PEG (3). The 8-position of 8-Cl-TIBO is indicated with a (*).
Likewise, linkage to the NNRTI end should not interfere with the desired interactions between the inhibitor and the NNRTI pocket, namely the hydrogen bonding and aryl-aryl interactions. This is evident in the case of HEPT, an early generation NNRTI – substitution at N3 caused the loss of hydrogen bonding to the backbone carbonyl of lysine 101, diminishing its affinity for RT11, while derivatives of HEPT modified at position N1 preserved key interactions and activity38. The connection to N3 of TSAO-T also resulted in active compounds, presumably because of preserved hydrogen bonding between the 4-amino group and the backbone carbonyl of lysine 10111. At the time that initial compounds were developed for our study, only a few NNRTI structures were available and thus choosing an NNRTI depended on this limited structural data. Structures that were available gave insight into using 8-Cl-TIBO as our NNRTI of choice, since a plausible linkage could be attached39,40.
Design of a bifunctional HIV-1 RT inhibitor was guided by molecular modeling that benefitted from the availability of emerging x-ray crystallographic and biochemical data for RT. Based on the rational design, an iterative and systematic synthetic effort was undertaken to prepare, purify and characterize several generations of small molecule compounds. These compounds were then tested in biochemical and cellular assays to develop a bifunctional structure-activity relationship. With d4T and 8-Cl-TIBO as our initial building blocks to illustrate our “proof of concept,” here we present the first evidence of DNA incorporation of a rationally designed and fully synthesized bifunctional nucleoside triphosphate by HIV-1 RT.
Thymidine-PEG-TIBO Inhibitors
To begin our design, we chose polyethylene glycol9 as our linker (Figure 1), for its low toxicity in biological venues and for its amphiphilic nature, thus giving it favorable solubility in the cellular environment. We also chose to begin using a thymidine analog as our NRTI, to avoid potential synthetic pitfalls with other NRTIs. And at the time this study was initiated in our lab, structural evidence suggested that a bifunctional concept could be achieved with the NNRTI 8-Cl-TIBO39,40, with linkage stemming from the 8-position (Figure 1). To aid in the design of the inhibitor, modeling was performed to not only to verify that linkage could be achieved on position 8 of TIBO, but to also estimate the number of PEG units needed to bridge the two pockets. 8-Cl-TIBO was modeled into the ternary RT structure (solved with natural substrate dTTP)24, which suggested that the 8-position of TIBO was directed towards the dNTP pocket, and that approximately six PEG units could connect the NRTI to the NNRTI as shown in Figure 2.
Figure 2. Modeled Fit of dTTP-PEG-TIBO.
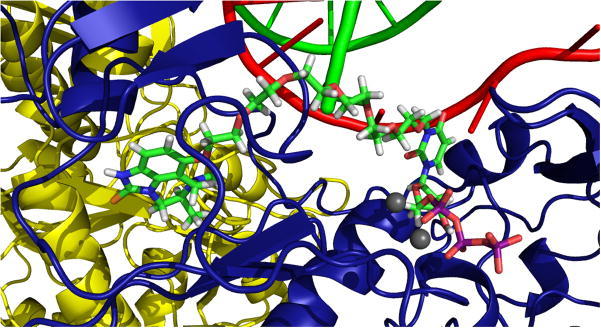
Using coordinates from the previously published ternary RT structure (RT, primer/template, and dTTP; pdb 1RTD)24 and the RT structure solved with 8-Cl TIBO (pdb 1HNV)40, the NNRTI pocket from 1HNV was aligned and superimposed onto 1RTD (see Experimental Section). In this hybrid modeled structure, the chlorine at position 8 of TIBO is clearly oriented towards the dTTP in the active site. The straight-line distance between 8-Cl of TIBO and 5-methyl of dTTP is 17.10 Å. Shown in blue and yellow are the p66 and p51 subunits of RT, respectively, and the green and red curved lines represent primer and template, respectively. The modeled bifunctional inhibitor is shown in ball and stick representation, and the grey spheres symbolize the active site magnesium ions.
Though the 5-position is a debatable site for derivatization (as stated earlier), linkage precedence guided the attachment of linker to this position12,14,33,41, as thymidine kinase can phosphorylate C-5 substituted pyrimidines and that d4T with long linkers remained active against HIV. We thus set out to synthesize the thymidine-6PEG-TIBO bifunctional inhibitor. As illustrated in Scheme 1, we chose a modular strategy for linker attachment to allow a variation in linker length and composition. Shown in Scheme 1 is the synthesis of the bifunctional molecule, where an ester linkage was introduced into the linker region for chemical feasibility. Molecules with four, five, or six PEG monomers were synthesized to develop a structure-activity relationship (SAR) for this group of inhibitors.
Scheme 1.

Synthesis of Thymidine-PEG-TIBO conjugates.
d4T-PEG-TIBO Inhibitors
We moved to synthetic efforts employing d4T as our NRTI moiety (Scheme 2). As mentioned earlier, d4T was chosen as our NRTI because of its similar kinetics of incorporation relative to natural thymidine and the low-level resistance developed against this NRTI. Modeling also suggested that we could pursue attachment of PEG to the 5-position of d4T, with the assumption that d4TTP bound in a similar manner to natural dTTP – a reasonable supposition based on the incorporation kinetics of d4TTP. An initial evaluation of antiviral activities of these compounds was carried out by examining their ability to inhibit HIV-1 replication in MT-2 cell culture. Table 1 shows the antiviral potency (EC50) and general cellular toxicity (IC50) for the bifunctional compounds 16a–c, containing 4, 5, and 6 PEG linkers. As illustrated in Table 1, these compounds showed some modest antiviral activity in the mid-micromolar range and the bifunctional molecule linked with four PEG units (16a) was most inhibitory, though the bifunctional inhibitors altogether fell short in illustrating a proof of concept. In trying to rationalize the poor activity a number of questions arise: (1) Was the nucleoside moiety in the bifunctional compounds phosphorylated as would be required for activity at the cellular level?; (2) was the ester functionality cleaved by cellular proteases to yield a cleaved inhibitor; and (3) was the linker length not optimal and/or did linker attachment perturb binding? As an initial step, we addressed the question of phosphorylation by synthesizing the protected monophosphate of d4T-4PEG-TIBO, with protecting groups utilized in stampidine, a prodrug formulation of d4T42. This approach did not yield an improvement in activity (data not shown). Synthesis of the corresponding triphosphate of d4T-4PEG-TIBO as a substrate was attempted to address the phosphorylation requirement for in vitro incorporation assays catalyzed by HIV-1 RT. However, a triphosphate product could not be synthesized (data not shown).
Scheme 2.

Synthesis of d4T-PEG-TIBO bifunctional RT inhibitors.
Table 1.
Activity of Potential Bifunctional Inhibitors in HIV-infected Cell Culture
| Compound | EC50 | IC50 |
|---|---|---|
| d4T | 2.2 μM | > 100 μM |
| 8-Cl-TIBO | 14 nM | > 100 nM |
| d4T-4PEG-TIBO (16a) | 45 μM | > 100 μM |
| d4T-5PEG-TIBO (16b) | 100 μM | > 100 μM |
| d4T-6PEG-TIBO (16c) | 120 μM | > 100 μM |
| N3-d4U-6PEG-TIBO (22) | 20 μM | 38 μM |
| TIBO-6PEG (2c) | 27 μM | > 100 μM |
Compounds were tested for their in vitro effect on viral replication. EC50 – antiviral potency; IC50 – cellular toxicity.
Though N3 substituted derivatives of d4T have been shown to be less active than d4T itself43, we attempted to synthesize a bifunctional inhibitor with N3 substitution (Scheme 3). Chemistry at this position was less problematic than C5 derivatization and a bifunctional inhibitor that displayed “proof of concept” might overcome the loss of activity that might be observed with N3 linkage. This includes the caveat that disrupted Watson-Crick base pairing may be a potential obstacle for activity with N3 substitution. The first compound of this series is shown in Figure 3A, where the PEG linker was directly attached to the N3 position of d4U without use of an ester moiety. This was deliberately planned through chemical synthesis to avoid ester cleavage in the cell as this may have been one problem associated with the lack of activity with the d4T-PEG-series. d4U was initially chosen for chemical feasibility and because the substitution now resides on N3, the linkage was lengthened by 2 PEG units. Estimation for PEG linker length was provided by modeling studies. In cell culture, this bifunctional compound, 22, did not illustrate a “proof of concept”, and we observed a minor increase in cell toxicity that could be generated by potential off-target interactions through the bifunctional inhibitor (Table 1). For nucleoside analogs, the first phosphorylation limits the rate of triphosphate formation44, and thus we again attempted to synthesize the prodrug version of N3-d4U-6PEG-TIBO (using stampidine formulation, see above). Again, this modification did not improve activity (data not shown) – suggesting that subsequent phosphorylations might also pose an obstacle to antiviral activity. Since our two initial prototypes were not showing significant activity, we asked if linkage at either the NRTI or NNRTI end might be affecting activity. The TIBO-6PEG fragment (Figure 3B) was tested in our cell culture assay. As shown in Table 1, the addition of linker significantly affects the activity of TIBO, decreasing activity by ~ 2000 fold. The perturbation upon TIBO’s activity certainly may account for our large hit on bifunctional activity.
Scheme 3.

Synthesis of N3-d4U-6PEG-TIBO.
Figure 3. Structures of the synthesized bifunctional inhibitors.

(A) Structure of N3-d4U-6PEG-TIBO. (B) Structure of TIBO-6PEG.
The MT-2 cell culture inhibition assay illustrated the pitfalls with our strategy, but it does not directly address potential interactions of inhibitors with RT. To focus on RT interactions, the triphosphate of N3-d4U-6PEG-TIBO (Figure 4A) was successfully synthesized and tested for incorporation with RT. Figure 4B shows a gel comparing the incorporation of the triphosphate of N3-d4U-6PEG-TIBO with that of natural dTTP. While the reaction using N3-d4U-6PEG-TIBO triphosphate showed a higher migrating product band, the migration appeared to be similar to that for a *D24-mer, which corresponds to a natural single deoxynucleotide incorporation band size (Figure 4B). This observation indicates the decomposition of the N3-d4U-6PEG-TIBO triphosphate into d4UTP serving as a substrate for RT, since the expected product mobility of the incorporated bifunctional inhibitor band size might be expected to be higher than the incorporated dTTP band size. We believe that the cleavage of the PEG linker along with the TIBO from the nucleotide part could occur due to the thermal instability of the bifunctional inhibitor either during the reaction time at 37°C or after the incorporation process when the samples were running on a denaturing gel. Because the temperature of the gel apparatus goes up to 55°C. Addition to this thermal denaturation, attaching the linker to the N3 position compels the nucleotide part to be in the unfavourable syn conformation that could impart additional strain on the inhibitor structure, and thus result in a higher propensity towards decomposition. This is supported by the observation that a very faint higher migrating band can be observed (Supplementary figure 1).
Figure 4. Incorporation of triphosphate of N3-d4U-6PEG-TIBO.
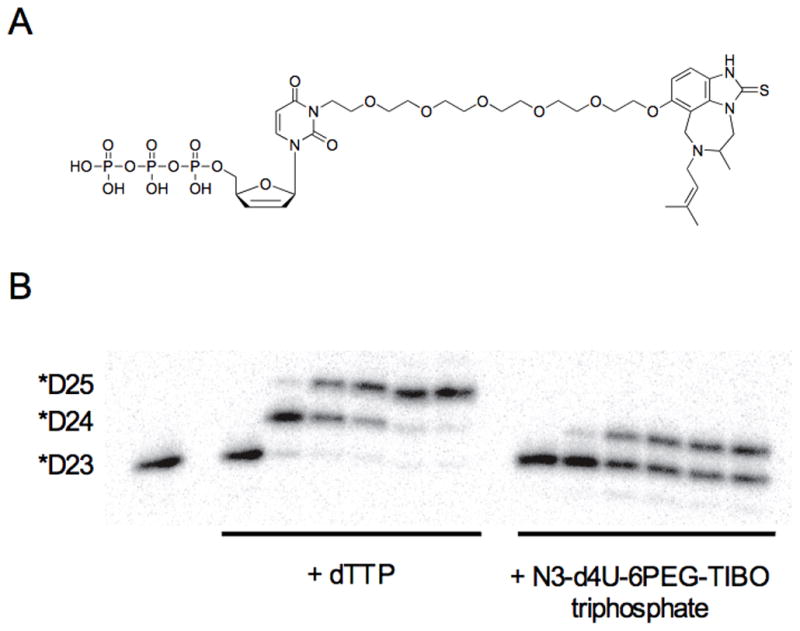
(A) Structure of triphosphate of N3-d4U-6PEG-TIBO (23). (B) Acrylamide gel showing incorporation of dTTP (100 μM) and triphosphate of N3-d4U-6PEG-TIBO (150 μM) over a time course under single turnover conditions (250 nM RT, 50 nM D23/D36 primer/template). Time points are t = 0, 30 sec, 5 min, 10 min, 30 min, and 60 min. Note the misincorporation of a second molecule of dTTP during this time course.
In the light of these findings, we synthesized a new bifunctional inhibitor (a 4PEG linker attached to the C5 position of d4T and a different NNRTI drug) in order to compare the incorporation efficiency with the TIBO bifunctional (Supplementary figure 2). The incorporation reaction of N3-d4U-6PEG-TIBO triphosphate at the room temperature resulted in a major denaturation band of the bifunctional product due to its intrinsic chemical and physical properties (Supplementary figure 1). However, the new bifunctional inhibitor (d4TTP-4PEG-TMC) was successfully incorporated by RT showing a much higher migration band corresponding to the size of the inhibitor with trace amounts of degredation product band likely owing to increased compound stability.
Implications for Future Research
Molecular modeling, as demonstrated in this study, should ecome an indispensable tool in the design of bifunctional RT inhibitors, so the probability of problems, such as wrong connection sites or inadequate linker length, can be minimized, and downstream synthetic efforts can be more focused. It should be appreciated, however, that the inhibitors designed from molecular modeling are “rough sketches”, not exact molecular “blueprints” to be taken literally. Rather, they should serve as the starting point from which structures can be refined based on experimental tests. For example, though structural and modeling studies suggested that the 8-position of 8-Cl-TIBO would be a favorable site for linkage, they should not preclude other positions, such as C-9, as feasible, even preferable, anchoring points. The ultimate support of the dual-binding of the bifunctional inhibitor relies on the availability of x-ray structural evidence of RT complexed with the inhibitor.
The effect of linker to the binding of the bifunctional inhibitor is underappreciated. More than an inert, weightless tether, the linker itself can significantly alter the way the bifunctional inhibitors bind to RT, and derail the intended interaction altogether. The 2000-fold decrease in activity of TIBO-6PEG vs. TIBO is one such example. On the other hand, the linker can be a source of positive contribution to binding. Creative use of diverse structures as linker that can bind to RT can add an unexplored dimension to bifunctional inhibitor design.
Synthetic difficulties remain a significant bottleneck in the preparation of the triphosphates of bifunctional inhibitor, as we encountered in numerous occurrences in our endeavors. Our successful synthesis of the triphosphate of N3-d4U-6PEG-TIBO and its small degree of incorporation by RT due to its thermal instability, however, illustrate a first step toward a successful RT bifunctional strategy.
Supplementary Material
Acknowledgments
We thank Stephen Hughes, Paul Boyer, and Andrea Ferris for the HIV-1 RTWT clone; Marina Udier-Blagovic, Theresa Lyons and William L. Jorgensen for their help in modeling studies. This work was supported by NIH GM49551 to K.S.A. and NIH R21GM060913 to K.A.P..
ABBREVIATIONS
- HIV-1
human immunodeficiency virus type 1
- RT
reverse transcriptase
- NRTIs
nucleoside RT inhibitors
- NNRTIs
nonnucleoside RT inhibitors
- dNTP
deoxynucleoside triphosphate
- dNMP
3′-deoxynucleoside monophosphate
- PPi
pyrophosphate
- AZT
β-D-(+)-3′-azido-3′-deoxythymidine
- d4T
β-D-(+)-2′,3′-didehydro-3′-deoxythymidine
- Kd
Dissociation constant
- WT
wild type
- TEAB
triethylammonium bicarbonate
- MOI
multiplicity of infection
- DMSO
dimethyl sulfoxide
- MTT
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- PEG
polyethylene glycol
- 8-Cl-TIBO
8-chloro tetrahydroimidazobenzodiazepinone
- SAR
structure-activity relationship
- MW
molecular weight
- TBDMS
tert-butyldimethylsilyl
- Tris
tris(hydroxymethyl)aminomethane
Footnotes
This work was supported by National Institutes of Health Grant 1R21GM060913-01A1 (to K.A.P.) and GM49551 (to K.S.A.)
Details on synthetic procedures, spectral characterization, molecular modeling, and biological assays are contained in Supporting Information.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Goody RS, Muller B, Restle T. Factors contributing to the inhibition of HIV reverse transcriptase by chain-terminating nucleotides in vitro and in vivo. FEBS Lett. 1991;291(1):1–5. doi: 10.1016/0014-5793(91)81089-q. [DOI] [PubMed] [Google Scholar]
- 2.Tantillo C, Ding J, Jacobo-Molina A, Nanni RG, Boyer PL, Hughes SH, Pauwels R, Andries K, Janssen PA, Arnold E. Locations of anti-AIDS drug binding sites and resistance mutations in the three-dimensional structure of HIV-1 reverse transcriptase. Implications for mechanisms of drug inhibition and resistance. J Mol Biol. 1994;243(3):369–87. doi: 10.1006/jmbi.1994.1665. [DOI] [PubMed] [Google Scholar]
- 3.Kohlstaedt LA, Wang J, Friedman JM, Rice PA, Steitz TA. Crystal structure at 3. 5 A resolution of HIV-1 reverse transcriptase complexed with an inhibitor. Science. 1992;256(5065):1783–90. doi: 10.1126/science.1377403. [DOI] [PubMed] [Google Scholar]
- 4.Merluzzi VJ, Hargrave KD, Labadia M, Grozinger K, Skoog M, Wu JC, Shih CK, Eckner K, Hattox S, Adams J, et al. Inhibition of HIV-1 replication by a nonnucleoside reverse transcriptase inhibitor. Science. 1990;250(4986):1411–3. doi: 10.1126/science.1701568. [DOI] [PubMed] [Google Scholar]
- 5.Carroll SS, Olsen DB, Bennett CD, Gotlib L, Graham DJ, Condra JH, Stern AM, Shafer JA, Kuo LC. Inhibition of HIV-1 reverse transcriptase by pyridinone derivatives. Potency, binding characteristics, and effect of template sequence. J Biol Chem. 1993;268(1):276–81. [PubMed] [Google Scholar]
- 6.Althaus IW, Chou JJ, Gonzales AJ, Deibel MR, Chou KC, Kezdy FJ, Romero DL, Aristoff PA, Tarpley WG, Reusser F. Steady-state kinetic studies with the non-nucleoside HIV-1 reverse transcriptase inhibitor U-87201E. J Biol Chem. 1993;268(9):6119–24. [PubMed] [Google Scholar]
- 7.Frank KB, Noll GJ, Connell EV, Sim IS. Kinetic interaction of human immunodeficiency virus type 1 reverse transcriptase with the antiviral tetrahydroimidazo[4,5,1-jk]-[1,4]-benzodiazepine-2-(1H)-thione compound, R82150. J Biol Chem. 1991;266(22):14232–6. [PubMed] [Google Scholar]
- 8.Debyser Z, Pauwels R, Andries K, Desmyter J, Kukla M, Janssen PA, De Clercq E. An antiviral target on reverse transcriptase of human immunodeficiency virus type 1 revealed by tetrahydroimidazo-[4,5,1-jk] [1,4]benzodiazepin-2 (1H)-one and -thione derivatives. Proc Natl Acad Sci U S A. 1991;88(4):1451–5. doi: 10.1073/pnas.88.4.1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peghini PA, Zahner R, Kuster H, Schott H, Schwendener RA. In vitro anti-human immunodeficiency virus and anti-hepatitis B virus activities and pharmacokinetic properties of heterodinucleoside phosphates containing AZT or ddC. Antivir Chem Chemother. 1998;9(2):117–26. doi: 10.1177/095632029800900203. [DOI] [PubMed] [Google Scholar]
- 10.Skillman AG, Maurer KW, Roe DC, Stauber MJ, Eargle D, Ewing TJ, Muscate A, Davioud-Charvet E, Medaglia MV, Fisher RJ, Arnold E, Gao HQ, Buckheit R, Boyer PL, Hughes SH, Kuntz ID, Kenyon GL. A novel mechanism for inhibition of HIV-1 reverse transcriptase. Bioorg Chem. 2002;30(6):443–58. doi: 10.1016/s0045-2068(02)00502-3. [DOI] [PubMed] [Google Scholar]
- 11.Velazquez S, Alvarez R, San-Felix A, Jimeno ML, De Clercq E, Balzarini J, Camarasa MJ. Synthesis and anti-HIV activity of [AZT]-[TSAO-T] and [AZT]-[HEPT] dimers as potential multifunctional inhibitors of HIV-1 reverse transcriptase. J Med Chem. 1995;38(10):1641–9. doi: 10.1021/jm00010a008. [DOI] [PubMed] [Google Scholar]
- 12.Ijichi K, Fujiwara M, Mori K, Morozumi M, Machida H, Shigeta S, Konno K, Yokota T, Baba M. Antiviral activities of nucleotide heterodimers against human immunodeficiency virus type 1 in vitro. Antiviral Res. 1996;31(1–2):115–20. doi: 10.1016/0166-3542(96)00945-x. [DOI] [PubMed] [Google Scholar]
- 13.Pontikis R, Dolle V, Guillaumel J, Dechaux E, Note R, Nguyen CH, Legraverend M, Bisagni E, Aubertin AM, Grierson DS, Monneret C. Synthesis and evaluation of “AZT-HEPT”, “AZT-pyridinone”, and “ddC-HEPT” conjugates as inhibitors of HIV reverse transcriptase. J Med Chem. 2000;43(10):1927–39. doi: 10.1021/jm991125l. [DOI] [PubMed] [Google Scholar]
- 14.Renoud-Grappin M, Fossey C, Fontaine G, Laduree D, Aubertin AM, Kirn A. Imidazo[1,5-b]pyridazine-d4T conjugates: synthesis and anti-human immunodeficiency virus evaluation. Antivir Chem Chemother. 1998;9(3):205–23. doi: 10.1177/095632029800900302. [DOI] [PubMed] [Google Scholar]
- 15.Nanni REJD, Jacobo-Molina A, Hughes SH, Arnold E. Review of HIV-1 reverse transcriptase three-dimensional structure: Implications for drug design. Perspectives in Drug Discovery and Design. 1993;1:129–150. [Google Scholar]
- 16.Rittinger K, Divita G, Goody RS. Human immunodeficiency virus reverse transcriptase substrate-induced conformational changes and the mechanism of inhibition by nonnucleoside inhibitors. Proc Natl Acad Sci U S A. 1995;92(17):8046–9. doi: 10.1073/pnas.92.17.8046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Spence RA, Kati WM, Anderson KS, Johnson KA. Mechanism of inhibition of HIV-1 reverse transcriptase by nonnucleoside inhibitors. Science. 1995;267(5200):988–93. doi: 10.1126/science.7532321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Basavapathruni A, Bailey CM, Anderson KS. Defining a molecular mechanism of synergy between nucleoside and nonnucleoside AIDS drugs. J Biol Chem. 2004;279(8):6221–6224. doi: 10.1074/jbc.C300523200. [DOI] [PubMed] [Google Scholar]
- 19.Basavapathruni A, Vingerhoets J, de Bethune MP, Chung R, Bailey CM, Kim J, Anderson KS. Modulation of human immunodeficiency virus type 1 synergistic inhibition by reverse transcriptase mutations. Biochemistry. 2006;45(23):7334–40. doi: 10.1021/bi052362v. [DOI] [PubMed] [Google Scholar]
- 20.Shuker SB, Hajduk PJ, Meadows RP, Fesik SW. Discovering high-affinity ligands for proteins: SAR by NMR. Science. 1996;274(5292):1531–4. doi: 10.1126/science.274.5292.1531. [DOI] [PubMed] [Google Scholar]
- 21.Hajduk PJ, Meadows RP, Fesik SW. Discovering high-affinity ligands for proteins. Science. 1997;278(5337):497, 499. doi: 10.1126/science.278.5337.497. [DOI] [PubMed] [Google Scholar]
- 22.Laduree D, Sugeac E, Fossey C, Schmidt S, Laumond G, Aubertin AM. Synthesis of certain heterodimers expected as HIV-1 reverse transcriptase inhibitors. Nucleosides, Nucleotides & Nucleic Acids. 2003;22(5–8):873–875. doi: 10.1081/NCN-120022675. [DOI] [PubMed] [Google Scholar]
- 23.Gavriliu D, Fossey C, Ciurea A, Delbederi Z, Sugeac E, Laduree D, Schmidt S, Laumond G, Aubertin AM. Synthesis and anti-HIV activity of [d4U]-[Trovirdine analogue] and [d4T]-[Trovirdine analogue] heterodimers as inhibitors of HIV-1 reverse transcriptase. Nucleosides Nucleotides Nucleic Acids. 2002;21(8–9):505–33. doi: 10.1081/NCN-120015066. [DOI] [PubMed] [Google Scholar]
- 24.Huang H, Chopra R, Verdine GL, Harrison SC. Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase: implications for drug resistance. Science. 1998;282(5394):1669–75. doi: 10.1126/science.282.5394.1669. [DOI] [PubMed] [Google Scholar]
- 25.Gavriliu D, Fossey C, Fontaine G, Benzaria S, Ciurea A, Delbederi Z, Lelong B, Laduree D, Aubertin AM, Kirn A. Synthesis and antiviral activity of C-5 substituted analogues of d4T bearing methylamino- or methyldiamino-linker arms. Nucleosides Nucleotides Nucleic Acids. 2000;19:1017–1031. doi: 10.1080/15257770008033040. [DOI] [PubMed] [Google Scholar]
- 26.Ren J, Diprose J, Warren J, Esnouf RM, Bird LE, Ikemizu S, Slater M, Milton J, Balzarini J, Stuart DI, Stammers DK. Phenylethylthiazolylthiourea (PETT) non-nucleoside inhibitors of HIV-1 and HIV-2 reverse transcriptases. Structural and biochemical analyses. J Biol Chem. 2000;275(8):5633–9. doi: 10.1074/jbc.275.8.5633. [DOI] [PubMed] [Google Scholar]
- 27.Younis Y, Hunter R, Muhanji CI, Hale I, Singh R, Bailey CM, Sullivan TJ, Anderson KS. [d4U]-spacer-[HI-236] double-drug inhibitors of HIV-1 reverse-transcriptase. Bioorg Med Chem. 2010;18(13):4661–73. doi: 10.1016/j.bmc.2010.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ciurea A, Fossey C, Benzaria S, Gavriliu D, Delbederi Z, Lelong B, Laduree D, Aubertin AM, Kirn A. Synthesis of 5-alkenylated D4T analogues via the Pd-catalyzed cross-coupling reaction. Nucleosides Nucleotides Nucleic Acids. 2001;20:1655–1670. doi: 10.1081/NCN-100105902. [DOI] [PubMed] [Google Scholar]
- 29.Delbederi Z, Fossey C, Fontaine G, Benzaria S, Gavriliu D, Ciurea A, Lelong B, Laduree D, Aubertin AM, Kirn A. Synthesis and antiviral activity of C-5 substituted beta-D- and beta-L-D4T analogues. Nucleosides Nucleotides Nucleic Acids. 2000;19:1441–1461. doi: 10.1080/15257770008033853. [DOI] [PubMed] [Google Scholar]
- 30.Laduree D, Fossey C, Renoud-Grappin M, Fontaine G, Camara F, Gavriliu D, Ciurea A, Aubertin AM, Kirn A. Synthesis of novel C-5 substituted d4T analogues bearing linker arms as potential anti-HIV agents. Nucleosides Nucleotides. 1999;18:883–884. doi: 10.1080/15257779908041592. [DOI] [PubMed] [Google Scholar]
- 31.Rong FG, Soloway AH, Ikeda S, Ives DH. Synthesis and Biochemical Activity of Hydrophilic Carborane-containing Pyrimidine Nucleosides as Potential Agents for DNA Incorporation and BNCT. Nucleosides & Nucleotides. 1997;16:379–401. [Google Scholar]
- 32.Soloway AH, Zhuo JC, Rong FG, Lunato AJ, Ives DH, Barth RF, Anisuzzaman AKM, Barth CD, Barnum BA. Identification, development, synthesis and evaluation of boron-containing nucleosides for neutron capture therapy. J Organometallic Chem. 1999;581:150–155. [Google Scholar]
- 33.Rong FG, Soloway AH, Ikeda S, Ives DH. Synthesis and biochemical activity of 5-tethered carborane-containing pyrimidine nucleosides as potential agents for DNA incorporation. Nucleosides & Nucleotides. 1995;14:1873–1887. [Google Scholar]
- 34.Mayers DL. Prevalence and incidence of resistance to zidovudine and other antiretroviral drugs. Am J Med. 1997;102(5B):70–5. doi: 10.1016/s0002-9343(97)00067-3. [DOI] [PubMed] [Google Scholar]
- 35.Lacey SF, Larder BA. Novel mutation (V75T) in human immunodeficiency virus type 1 reverse transcriptase confers resistance to 2′,3′-didehydro-2′,3′-dideoxythymidine in cell culture. Antimicrob Agents Chemother. 1994;38(6):1428–32. doi: 10.1128/aac.38.6.1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin PF, Samanta H, Rose RE, Patick AK, Trimble J, Bechtold CM, Revie DR, Khan NC, Federici ME, Li H, et al. Genotypic and phenotypic analysis of human immunodeficiency virus type 1 isolates from patients on prolonged stavudine therapy. J Infect Dis. 1994;170(5):1157–64. doi: 10.1093/infdis/170.5.1157. [DOI] [PubMed] [Google Scholar]
- 37.Vaccaro JA, Parnell KM, Terezakis SA, Anderson KS. Mechanism of inhibition of the human immunodeficiency virus type 1 reverse transcriptase by d4TTP: an equivalent incorporation efficiency relative to the natural substrate dTTP. Antimicrob Agents Chemother. 2000;44(1):217–21. doi: 10.1128/aac.44.1.217-221.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pontikis R, Benhida R, Aubertin AM, Grierson DS, Monneret C. Synthesis and anti-HIV activity of novel N-1 side chain-modified analogs of 1-[(2-hydroxyethoxy)methyl]-6-(phenylthio)thymine (HEPT) J Med Chem. 1997;40(12):1845–54. doi: 10.1021/jm960765a. [DOI] [PubMed] [Google Scholar]
- 39.Ding J, Das K, Moereels H, Koymans L, Andries K, Janssen PA, Hughes SH, Arnold E. Structure of HIV-1 RT/TIBO R 86183 complex reveals similarity in the binding of diverse nonnucleoside inhibitors. Nat Struct Biol. 1995;2(5):407–15. doi: 10.1038/nsb0595-407. [DOI] [PubMed] [Google Scholar]
- 40.Das K, Ding J, Hsiou Y, Clark AD, Jr, Moereels H, Koymans L, Andries K, Pauwels R, Janssen PA, Boyer PL, Clark P, Smith RH, Jr, Kroeger Smith MB, Michejda CJ, Hughes SH, Arnold E. Crystal structures of 8-Cl and 9-Cl TIBO complexed with wild-type HIV-1 RTand 8-Cl TIBO complexed with the Tyr181Cys HIV-1 RT drug-resistant mutant. J Mol Biol. 1996;264(5):1085–100. doi: 10.1006/jmbi.1996.0698. [DOI] [PubMed] [Google Scholar]
- 41.Rong FG, Soloway AH. Synthesis of 5-tethered carborane-containing pyrimidine nucleosides as potential agents for DNA incorporation. Nucleosides & Nucleotides. 1994;13:2021–2034. [Google Scholar]
- 42.Uckun FM, Pendergrass S, Venkatachalam TK, Qazi S, Richman D. Stampidine is a potent inhibitor of Zidovudine- and nucleoside analog reverse transcriptase inhibitor-resistant primary clinical human immunodeficiency virus type 1 isolates with thymidine analog mutations. Antimicrob Agents Chemother. 2002;46(11):3613–6. doi: 10.1128/AAC.46.11.3613-3616.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maillard M, Florent JC, Lemaitre M, Begassat F, Bugnicourt A, Ferrieux C, Rombi C, Pacaud ED, Thierry DE, Zerial A, Monneret C, Grierson DS. Preparation and anti-HIV activity of N-3 amino substituted thymidine nucleoside analogs. Bioorg Med Chem Lett. 1992;2:1469–1474. [Google Scholar]
- 44.Ho HT, Hitchcock MJ. Cellular pharmacology of 2′,3′-dideoxy-2′,3′-didehydrothymidine, a nucleoside analog active against human immunodeficiency virus. Antimicrob Agents Chemother. 1989;33(6):844–9. doi: 10.1128/aac.33.6.844. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.