

Abstract
In this Letter, we describe a novel approach for the general and enantioselective synthesis of a diverse array of small to large 1-azabicyclo[m.n.0]alkyl ring systems with an embedded olefin handle for further functionalization. The stereochemistry is established via a highly diastereoselective indium-mediated allylation of an Ellman sulfinimine in greater than 9:1 dr., which is readily separable by column chromatography to afford a single diastereomer. This methodology allows for the rapid preparation of 1-azabicyclo[m.n.0]alkane ring systems that are not readily accessible through any other chemistry in excellent overall yields and, for many systems, the only enantioselective preparation reported to date.
Keywords: azabicycle, olefin metathasis, enantioselective, allylation, alkaloid
Natural products and pharmaceutical compositions that possess azabicyclic ring systems 1–9 are very common; however, synthetic approaches to access these 1-azabicyclo[m.n.0]alkane systems (Fig. 1) are limited.1–8 In general, the existing strategies for the construction of azabicyclic ring systems rely on Staudinger-aza-Wittig approaches,9 7-exo-tet-cyclizations,10,11 cycloadditions ([5+2], [4+2] and [2+2+2]),12 ring-closing metathesis (RCM) strategies,13 Mitsunobu approaches14 and rearrangements (nitrone and intramolecular Schmidt rearrangements)15,16 For the larger azabicyclic ring systems, routes are very rare, and those reported lack stereocontrol.8,17
Figure 1.

Representative 1-azabicyclo[m.n.0]alkane ring systems, including their alternative IUPAC nomenclature, of interest in both natural product synthesis and drug discovery.
Towards the development of an enantioselective toolbox of synthetic routes to access these valuable azaheterocycles, we recently reported a novel six step approach for the rapid and enantioselective synthesis of azabicyclic systems such as 1–3 (Scheme 1).18 Here, an indium-mediated allylation of a chiral aldimine substrate 10, N-alkylation to afford 11, ring-closing metathesis (RCM) to provide 12, and finally a one-pot deprotection/acetal hydrolysis/reductive amination sequence to afford enantiopure azabicyclic ring systems 1–3.18 This methodology was then employed for the enantioselective total syntheses of (+)-grandisine D,18 cremastrine19 and amabiline.20
Scheme 1.
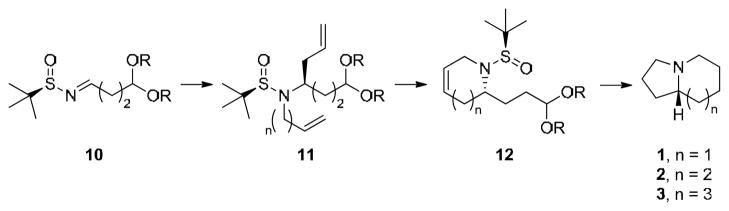
First generation rapid, enantioselective synthesis of azabicylic ring systems 1–3.
While this was a notable advance, we also wanted to develop a streamlined route to access the higher homologs 4–9 in an enantioselective manner. De Kimpe and co-workers recently demonstrated the asymmetric synthesis of 2-arylpyrrolidines 15 from γ-chloro N-(tert-butanesulfinyl)ketimines 13,11 and Brown and co-workers employed a related strategy for the total synthesis of (−)-tashiromine 17 and (−)-epilupinine 18 (Scheme 2, eq. 1).21
Scheme 2.

Application of γ–chloro N-(tert-butanesulfinyl)ketimines to access pyrroles and azabicyclic systems in high enantioselectivity.
Inspired by these results and our internal efforts, we envisioned a protocol that would subject various chloroalkyl N(tert-butanesulfinyl)aldehydes 19 to an asymmetric allylation reaction to provide 20 in high diastereomeric ratio (dr).18,22,23 Deprotection and alkylation of the pyrrolidine would provide azocines 21, substrates for a ring closing metathesis reaction24 to provide general, enantioselective access to 1-azabicyclo[m.n.0]alkane cores 1–9 with an embedded olefin as a handle for further functionalization (Scheme 3).
Scheme 3.
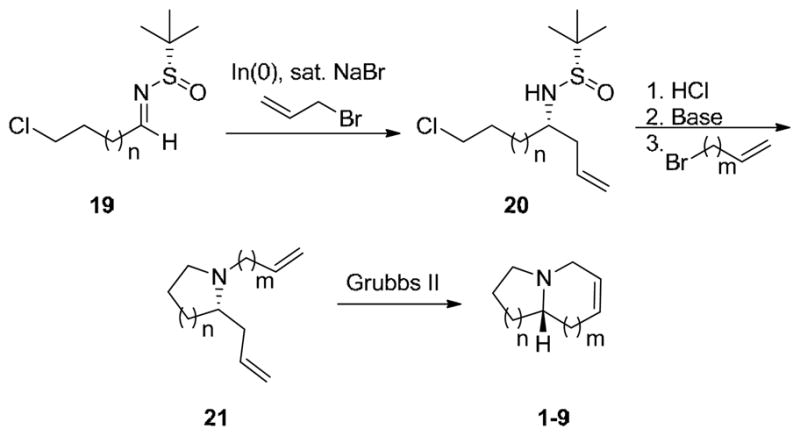
Envisioned route to employ chloroalkyl N-(tert-butanesulfinyl)aldehydes, an asymmetric allylation and a subsequent RCM to access diverse 1-azabicyclo[m.n.0]alkane cores in high enantioselectivity.
The requisite chloroalkyl N-(tert-butanesulfinyl)aldehydes 19 were easily prepared in 92–94% yields by condensing the corresponding chloroaldehydes 22 with the Ellman (S)-tert-butanesulfinamide 23 employing CuSO4 in DCM.25,26 A subsequent indium-mediated allylation reaction affords the anticipated (R)-anti-adducts 20 in >9:1 diastereoselectivity and up to 86% yield.18,22,23 After column chromatography, single diasteromers of analogs 20 resulted, which were carried forward. Following modification of the known protocols,11,21 acid-mediated deprotection and base-induced, microwave-assisted cyclization and alkyation with the required allyl, butenyl and pentenyl bromides smoothly afforded the chiral N-alkyl azocines 21 in 63–83% yields for the three step, one-pot reaction sequence (Scheme 4).27 To enable the one-pot sequence, a number of bases, solvents and temperatures were evaluated; however, K2CO3, NaI in DMF under microwave irradiation (120 °C, 15 min) proved to be general for all substrates, even the larger 7-azocine rings.
Scheme 4.

Enantioselective synthesis of N-alkyl azocines 24.
With all of the chiral N-alkyl azocines 21 in hand, we focused on the RCM to provide the 1-azabicyclo[m.n.0]alkane systems 1–9. Initial attempt following several known reaction conditions with Grubbs II18,20,24,28,29 failed to provide the desired unsaturated 1-azabicyclo[5.4.0]tridecane core of 7. A perusal of the literature regarding RCM methods with tertiary amines, suggested that ‘protection’ of the amine by in situ generation of ammonium salts enabled facile ring-closing.29–31 Thus, treatment of 21c1 with 1.1 equivalent of camphor sulfonic acid (CSA) in 0.05 M toluene, followed by the addition of 10 mol% of Grubbs II and microwave heating for 1 hour at 100 °C, provided the unsaturated 1-azabicyclo[5.4.0]tridecane core of 7 in 70% isolated yield (Scheme 5).27 An evaluation of additional acids led to the use of trifluoroacetic acid (TFA), which was equally effective (68% yield) and allowed for simpler purification.
Scheme 5.
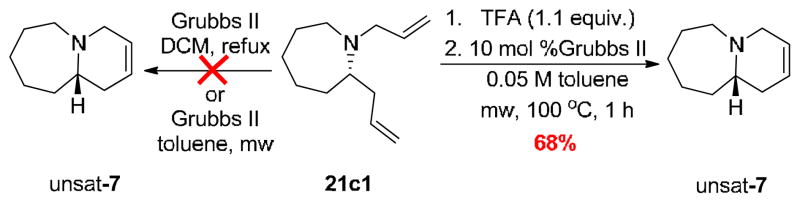
RCM approaches to access the 1-azabicyclo[m.n.0]alkane cores.
With a robust protocol in hand for the RCM, all of the chiral N-alkyl azocines 21 were converted, under these optimal conditions, into the desired unsaturated 1-azabicyclo[5.4.0]alkane cores 22 of 1–9 (Scheme 6). Yields for the RCM reaction averaged 70% for all the substrates 21, providing high yielding, enantioselective access to each of the key 1-azabicyclo[m.n.0]alkane systems 1–9 in short order (Fig. 2). Overall yields from the commercial aldehydes 22 ranged from 29 to 59%, and offer the synthetic and medicinal chemist a general route to access these important azabicyclic ring systems.
Scheme 6.
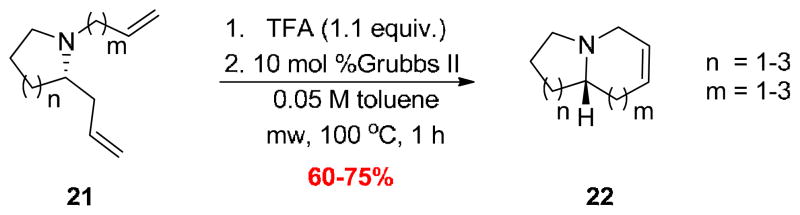
Optimal RCM conditions for the enantioselective synthesis of the unsaturated 1-azabicyclo[m.n.0]alkane cores 22 of 1–9.
Figure 2.

Mono-unsaturated 1-azabicyclo[m.n.0]alkane ring systems 22 synthesized that encompass all of the key azabicyclic ring systems 1–9. Yields are overall from commercial aldehydes 22, and ranged form 29–59%.
In summary, we have developed a novel approach for the general and enantioselective synthesis of a diverse array of small to large 1-azabicyclo[m.n.0]alkane ring systems with an embedded olefin handle for further functionalization. The stereochemistry is established via a highly diastereoselective indium-mediated allylation of an Ellman sulfinimine in greater than 9:1 dr., which is readily separable by column chromatography to afford a single diastereomer. This methodology allows for the rapid preparation of 1-azabicyclo[m.n.0]alkane ring systems that are not readily accessible through any other chemistry in excellent overall yields and, for many systems, the only enantioselective preparation reported to date.
Acknowledgments
The authors gratefully acknowledge funding from the Department of Pharmacology, Vanderbilt University Medical Center and the Warren Family & Foundation for funding the William K. Warren, Jr. Chair in Medicine
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Michael JP. Nat Prod Rep. 2005;22:603–626. doi: 10.1039/b413748p. [DOI] [PubMed] [Google Scholar]; b) Mitchinson A, Nadin A. J Chem Soc Perkin Trans. 2000;1:2862–2892. [Google Scholar]; c) Ohagan D. Nat Prod Rep. 1997;14:637–651. [Google Scholar]; d) Sakata K, Aoki K, Chang CF, Sakurai A, Tamura S, Murakoshi S. Agric Biol Chem. 1978;42:457–463. [Google Scholar]; e) Ye Y, Qin GW, Xu RS. Phytochemistry. 1994;37:1205–1208. [Google Scholar]; f) Shinozaki H, Ishida M. Brain Res. 1985;334:33–40. doi: 10.1016/0006-8993(85)90564-5. [DOI] [PubMed] [Google Scholar]; g) Pilli RA, De Oliveira MDCF. Nat Prod Rep. 2000;17:117–127. doi: 10.1039/a902437i. [DOI] [PubMed] [Google Scholar]; h) Pilli RA, Rosso GB, De Oliveira MDCF. Nat Prod Rep. 2010;27:1908–1937. doi: 10.1039/c005018k. [DOI] [PubMed] [Google Scholar]; i) Seger C, Mereiter K, Kaltenegger E, Pacher T, Greger H, Hofer O. Chem Biodiversity. 2004;1:265–279. doi: 10.1002/cbdv.200490023. [DOI] [PubMed] [Google Scholar]; j) Gregor H, Schinnerl J, Vajrodaya S, Brecker L, Hofer O. J Nat Prod. 2009;72:1708–1711. doi: 10.1021/np900294c. [DOI] [PubMed] [Google Scholar]
- 2.Morita H, Arisaka M, Yoshida N, Kobayashi J. J Org Chem. 2000;65:6241–6245. doi: 10.1021/jo000661e. [DOI] [PubMed] [Google Scholar]
- 3.a) Zou C, Li J, Lei H, Fu H, Lin W. J Chin Pharm Sci. 2000;9:113–115. [Google Scholar]; b) Williams DR, Shamim K, Khalida R, Reddy J, Amato GS, Shaw SM. Org Lett. 2003;5:3361–3364. doi: 10.1021/ol035368q. [DOI] [PubMed] [Google Scholar]
- 4.a) Kobayashi J, Watanabe D, Kawasaki N, Tsuda M. J Org Chem. 1997;62:9236–9239. [Google Scholar]; b) Nagata T, Nakagawa M, Nishida A. J Am Chem Soc. 2003;125:7484–7485. doi: 10.1021/ja034464j. [DOI] [PubMed] [Google Scholar]; c) Ono K, Nakagawa M, Nishida A. Angew Chem Int Ed. 2004;43:202–2023. doi: 10.1002/anie.200453673. [DOI] [PubMed] [Google Scholar]; d) Jakubec P, Cockfield DM, Dixon DJ. J Am Chem Soc. 2009;13:16632–16633. doi: 10.1021/ja908399s. [DOI] [PubMed] [Google Scholar]; e) Martin DBC, Vanderwal CD. Angew Chem Int Ed. 2010;49:2830–2832. doi: 10.1002/anie.201000045. [DOI] [PubMed] [Google Scholar]; f) Nilson MG, Funk RL. Org Lett. 2010;12:4912–4915. doi: 10.1021/ol102079z. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Cheng B, Wu F, Yang X, Zhou Y, Wan X, Zhai H. Chem Eur J. 2011;17:12569–12572. doi: 10.1002/chem.201102101. [DOI] [PubMed] [Google Scholar]
- 5.a) Johns SR, Lamberton JA, Sioumis AA. Chem Commun. 1968;21:1324–1325. [Google Scholar]; b) Johns SR, Lamberton JA, Sioumis AA. Aust J Chem. 1969;22:793–800. [Google Scholar]
- 6.Lin W, Xu R, Zhong Q. Huaxue Xuebao. 1991;49:927–931. [Google Scholar]
- 7.a) Valencia E, Fajardo V, Freyer AJ, Shamma M. Tetrahedron Lett. 1985;26:993–996. [Google Scholar]; b) Fang FG, Feigelson GB, Danishefsky SJ. Tetrahedron Lett. 1989;30:2743–2746. [Google Scholar]
- 8.For reviews on the synthesis of azabicyclic ring systems, see: Michael JP. Beilstein J Org Chem. 2007;3 doi: 10.1186/1860-5397-3-27.Hodgson DM, Winning LH. Org Biomol Chem. 2007;5:3071–3082. doi: 10.1039/b707566a.Toure BB, Hall DG. Chem Rev. 2009;109:4439–4486. doi: 10.1021/cr800296p.Michael JP. Nat Prod Rep. 2008;25:139–165. doi: 10.1039/b612166g.Pyne SG, Davis AS, Gates N, Hartley JP, Lindsay KB, Machan T, Tang M. Synlett. 2004;15:625–649.Enders D, Thiebes T. Pure & Appl Chem. 2001;73:573–578.Pili RA, Rosso GB, De Oliviera F, da Conceicao M. Nat Prod Rep. 2011;27:1908–1937. doi: 10.1039/c005018k.Aibes R, Figueredo M. Eu J Org Chem. 2009;15:2421–2435.Khim SK, Schultz AG. J Org Chem. 2004;69:7734–7736. doi: 10.1021/jo049083i.
- 9.a) Williams DR, Brown DL, Benbow JW. J Am Chem Soc. 1989;111:1923–1925. [Google Scholar]; b) Williams DR, Fromhold MG, Early JD. Org Lett. 2001;3:2721–2724. doi: 10.1021/ol016336a. [DOI] [PubMed] [Google Scholar]; c) Williams DR, Shamim K, Reddy JP, Amato GS, Shaw SM. Org Lett. 2003:3361–3364. doi: 10.1021/ol035368q. [DOI] [PubMed] [Google Scholar]
- 10.Kohno Y, Narasaka K. Bull Chem Soc Jpn. 1996;69:2063–2070. [Google Scholar]
- 11.a) Tehrani K, D’hooghe M, De Kimpe N. Tetrahedron. 2003;59:3099–3108. [Google Scholar]; b) Leemans E, Mangelinckx S, De Kimpe N. Chem Commun. 2010;46:3122–3124. doi: 10.1039/b925209f. [DOI] [PubMed] [Google Scholar]
- 12.a) Jacobi PA, Lee K. J Am Chem Soc. 1997;119:3409–3410. [Google Scholar]; b) Jacobi PA, Lee K. J Am Chem Soc. 2000;122:4295–4303. [Google Scholar]; c) Yu RT, Rovis T. J Am Chem Soc. 2006;128:12370–12371. doi: 10.1021/ja064868m. [DOI] [PubMed] [Google Scholar]; d) Roscini C, Cubbage KL, Berry M, Orr-Ewing AJ, Booker-Milburn KI. Angew Chem Int Ed. 2009;48:8716–8720. doi: 10.1002/anie.200904059. [DOI] [PubMed] [Google Scholar]
- 13.a) Alibes R, Figueredo M. J Org Chem. 2009;74:6199–6211. doi: 10.1021/jo901059n. [DOI] [PubMed] [Google Scholar]; b) Torssell S, Wanngren E, Somafi P. J Org Chem. 2007;72:4246–4249. doi: 10.1021/jo070498o. [DOI] [PubMed] [Google Scholar]; c) Sibi MP, Subramanian T. Synlett. 2004:1211–1214. [Google Scholar]; d) Olivo HF, Tovar-Miranda R, Barragan E. J Org Chem. 2006;71:3287–3290. doi: 10.1021/jo052364l. [DOI] [PubMed] [Google Scholar]; e) Hoye AT, Wipf P. Org Lett. 2011;13:2634–2637. doi: 10.1021/ol200743u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.De Kimpe N, Stanoeva E, Georgieva A, Keppens M, Kulinkovich O. Org Prep Proced Int. 1995;27:674–678. [Google Scholar]
- 15.Cid P, Closa M, De March P, Figueredo M, Font J, Sanfeliu E, Soria A. Eur J Org Chem. 2004:4215–4233. [Google Scholar]
- 16.Kapat A, Nyfeler E, Giuffredi GT, Renaud P. J Am Chem Soc. 2009;131:17746–17747. doi: 10.1021/ja908933s. [DOI] [PubMed] [Google Scholar]
- 17.Zeng Y, Smith BT, Hershberger J, Aubé J. J Org Chem. 2003;68:8065–8067. doi: 10.1021/jo035004b. [DOI] [PubMed] [Google Scholar]
- 18.Fadeyi O, Senter T, Hahn K, Lindsley C. Chem Eur J. 2012;18:5826–5831. doi: 10.1002/chem.201200629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hahn K, Fadeyi O, Cho P, Lindsley C. Tetrahedron Lett. 2012;53:3577–3580. doi: 10.1016/j.tetlet.2012.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Senter T, Fadeyi O, Lindsley C. Org Lett. 2012;14:1869–1871. doi: 10.1021/ol300466a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cutter A, Miller I, Keily J, Bellingham R, Light M, Brown R. Org Lett. 2011;13:3988–3991. doi: 10.1021/ol2015048. [DOI] [PubMed] [Google Scholar]
- 22.Schulte ML, Lindsley CW. Org Lett. 2011;13:5684–5687. doi: 10.1021/ol202415j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun XW, Liu M, Xu MH, Lin GQ. Org Lett. 2008;10:1259–1262. doi: 10.1021/ol8001514. [DOI] [PubMed] [Google Scholar]
- 24.Kuhn KM, Champange TM, Hong SH, Wei WH, Nickel A, Lee CW, Virgil SC, Grubbs RH, Pederson RL. Org Lett. 2010;125:984–987. doi: 10.1021/ol9029808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.a) Brinner KM, Ellman JA. Org Biomol Chem. 2005;3:2109–2113. doi: 10.1039/b502080h. [DOI] [PubMed] [Google Scholar]; b) Cogan DA, Ellman JA. J Am Chem Soc. 1999;121:268–269. [Google Scholar]; c) Tang TP, Ellman JA. J Org Chem. 2002;67:7819–7832. doi: 10.1021/jo025957u. [DOI] [PubMed] [Google Scholar]
- 26.Cogan DA, Liu G, Ellman JA. Tetrahedron. 1999;55:8883–8904. [Google Scholar]
-
27.Representative experimental:
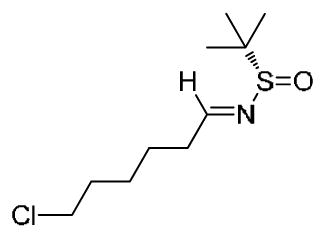 (S,E)-N-(6-chlorohexylidene)-2-methylpropane-2-sulfinamide 19c: To a solution of 6-chlorohexanal (7.37 g, 54.79 mmol) in DCM (219 ml) at ambient temperature was added CuSO4 (20.11 g, 126.02 mmol) and (S)-2-methylpropane-2-sulfinamide (7.64 g, 63.01 mmol) in a single batch. The reaction was stirred for 12 h, at which point the starting material was fully consumed by TLC analysis (4:1 Hex/EtOAc, rf = 0.35). The heterogeneous mixture was filtered through a silica pad and concentrated in vacuo to yield a viscous oil, which was purified by flash chromatography (4:1 Hex/EtOAc) to afford the desired product as a clear oil (12.34 g, 94%). [α]D20 = +157.9° (c = 1.5, MeOH). 1H NMR (400.1 MHz, CDCl3) δ (ppm): 8.05 (t, J = 4.5 Hz, 1H); 3.52 (t, J = 6.5 Hz, 2H); 2.53 (m, 2H); 1.79 (m, 2H); 1.66 (m, 2H); 1.51 (m, 2H); 1.18 (s, 9H). 13C NMR (100.6 MHz, CDCl3) δ (ppm): 169.26, 56.64, 44.82, 35.96, 32.37, 26.56, 24.78, 22.42. HRMS (TOF, ES+) C10H21NOSCl [M+H]+ calc’d 238.1032, found 238.1034.
(S,E)-N-(6-chlorohexylidene)-2-methylpropane-2-sulfinamide 19c: To a solution of 6-chlorohexanal (7.37 g, 54.79 mmol) in DCM (219 ml) at ambient temperature was added CuSO4 (20.11 g, 126.02 mmol) and (S)-2-methylpropane-2-sulfinamide (7.64 g, 63.01 mmol) in a single batch. The reaction was stirred for 12 h, at which point the starting material was fully consumed by TLC analysis (4:1 Hex/EtOAc, rf = 0.35). The heterogeneous mixture was filtered through a silica pad and concentrated in vacuo to yield a viscous oil, which was purified by flash chromatography (4:1 Hex/EtOAc) to afford the desired product as a clear oil (12.34 g, 94%). [α]D20 = +157.9° (c = 1.5, MeOH). 1H NMR (400.1 MHz, CDCl3) δ (ppm): 8.05 (t, J = 4.5 Hz, 1H); 3.52 (t, J = 6.5 Hz, 2H); 2.53 (m, 2H); 1.79 (m, 2H); 1.66 (m, 2H); 1.51 (m, 2H); 1.18 (s, 9H). 13C NMR (100.6 MHz, CDCl3) δ (ppm): 169.26, 56.64, 44.82, 35.96, 32.37, 26.56, 24.78, 22.42. HRMS (TOF, ES+) C10H21NOSCl [M+H]+ calc’d 238.1032, found 238.1034.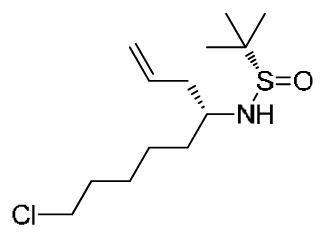 (S)-N-((R)-9-chloronon-1-en-4-yl)-2-methylpropane-2-sulfinamide 20c: NaBr (380 g) was dissolved in 841 mL of deionized H2O. To this fully dissolved saturated NaBr solution was added aldimine 19c (10.00g, 42.05 mmol) and indium powder (19.31 g, 168.2 mmol). The mixture was stirred vigorously for 5 min., then allyl bromide (20.35 g, 168.2 mmol) added in a single batch. Vigorous stirring was continued for 9 h, at which point the starting material was consumed by TLC analysis (1:1 Hex/EtOAc, rf = 0.34). The mixture was quenched with NaHCO3 and extracted x5 with EtOAc. The organic fractions were combined, washed with brine, dried over Na2SO4, and concentrated in vacuo to yield crude oil. Purification by flash chromatography (1:1 Hex/ EtOAc) afforded the desired product as a clear oil (10.10 g, 86%). [α]D20 = +25.7° (c = 1.5, MeOH). 1H NMR (400.1 MHz, CDCl3) δ (ppm): 5.77 (m, 1H);5.15 (m, 2H); 3.52 (t, J = 6.5Hz, 2H); 3.30 (sextet, J = 6.1 Hz, 1H); 3.21 (br d, J = 6.1 Hz, 1H); 2.35 (dp, J1 = 6.5 Hz, J2 = 10.4 Hz, 2H); 1.763 (quint., J = 6.5 Hz, 2H); 1.53-1.32 (m, 5H). 13C NMR (100.6 MHz, CDCl3) δ (ppm): 134.23, 119.14, 55.92, 54.83, 45.08, 40.56, 34.91, 32.60, 26.88, 24.92, 22.79. HRMS (TOF, ES+) C13H27NOSCl [M+H]+ calc’d 280.1502, found 280.1504.
(S)-N-((R)-9-chloronon-1-en-4-yl)-2-methylpropane-2-sulfinamide 20c: NaBr (380 g) was dissolved in 841 mL of deionized H2O. To this fully dissolved saturated NaBr solution was added aldimine 19c (10.00g, 42.05 mmol) and indium powder (19.31 g, 168.2 mmol). The mixture was stirred vigorously for 5 min., then allyl bromide (20.35 g, 168.2 mmol) added in a single batch. Vigorous stirring was continued for 9 h, at which point the starting material was consumed by TLC analysis (1:1 Hex/EtOAc, rf = 0.34). The mixture was quenched with NaHCO3 and extracted x5 with EtOAc. The organic fractions were combined, washed with brine, dried over Na2SO4, and concentrated in vacuo to yield crude oil. Purification by flash chromatography (1:1 Hex/ EtOAc) afforded the desired product as a clear oil (10.10 g, 86%). [α]D20 = +25.7° (c = 1.5, MeOH). 1H NMR (400.1 MHz, CDCl3) δ (ppm): 5.77 (m, 1H);5.15 (m, 2H); 3.52 (t, J = 6.5Hz, 2H); 3.30 (sextet, J = 6.1 Hz, 1H); 3.21 (br d, J = 6.1 Hz, 1H); 2.35 (dp, J1 = 6.5 Hz, J2 = 10.4 Hz, 2H); 1.763 (quint., J = 6.5 Hz, 2H); 1.53-1.32 (m, 5H). 13C NMR (100.6 MHz, CDCl3) δ (ppm): 134.23, 119.14, 55.92, 54.83, 45.08, 40.56, 34.91, 32.60, 26.88, 24.92, 22.79. HRMS (TOF, ES+) C13H27NOSCl [M+H]+ calc’d 280.1502, found 280.1504.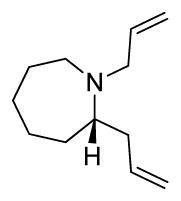 (S)-1,2-diallylazepane 21c1: A 4N solution of HCl / dioxanes (14.92 ml) was cooled to 0°C and slowly added to a microwave vial containing sulfinamide 20c (2.0 g, 7.15 mmol). Solution was then brought to ambient temperature and stirred for an additional 45 min, and concentrated in vacuo to afford the deprotected amine as the HCl salt in quantitative yield. The amine was dissolved in DMF (35.9 mL), then K2CO3 (1.98 g, 14.34 mmol) and NaI (1.18 g, 7.89 mmol) were added in a single batch. The vial was sealed and submitted to microwave irradiation at 120°C for 15 min. LC/MS analysis showed full consumption of starting material to the cyclized secondary amine. Allyl bromide (0.954 g, 7.89 mmol) and an additional equivalent of K2CO3 (0.99 g, 7.15 mmol) was then added to the pale yellow solution at ambient temperature, and stirring was continued for 4 h. Mixture was dissolved in 5% LiCl solution and extracted with Et2O (5 x 40 mL). Organic fractions were combined, washed with brine, dried over Na2SO4, and concentrated in vacuo to yield a pale yellow crude oil. Purification by flash chromatography (1:1 Hex/EtOAc) afforded the desired product as a clear oil (0.82g, 64%). [α]D20 = −5.0° (c = 0.9, MeOH). 1H NMR (400.1 MHz, CDCl3) δ (ppm): 5.82 (m, 2H); 5.19-4.93 (m, 4H); 3.22 (m, 2H); 2.85 (m, 1H); 2.70 (m, 2H); 2.26 (m, 1H); 2.06 (m, 1H); 1.75 (m, 1H); 1.66-1.38 (m, 7H). 13C NMR (100.6 MHz, CDCl3) δ (ppm): 137.98, 137.51, 115.99, 115.66, 62.45, 55.71, 49.83, 39.34, 32.65, 28.88, 27.53, 25.78. HRMS (TOF, ES+) C12H22N [M+H]+ calc’d 180.1752, found 180.1751.
(S)-1,2-diallylazepane 21c1: A 4N solution of HCl / dioxanes (14.92 ml) was cooled to 0°C and slowly added to a microwave vial containing sulfinamide 20c (2.0 g, 7.15 mmol). Solution was then brought to ambient temperature and stirred for an additional 45 min, and concentrated in vacuo to afford the deprotected amine as the HCl salt in quantitative yield. The amine was dissolved in DMF (35.9 mL), then K2CO3 (1.98 g, 14.34 mmol) and NaI (1.18 g, 7.89 mmol) were added in a single batch. The vial was sealed and submitted to microwave irradiation at 120°C for 15 min. LC/MS analysis showed full consumption of starting material to the cyclized secondary amine. Allyl bromide (0.954 g, 7.89 mmol) and an additional equivalent of K2CO3 (0.99 g, 7.15 mmol) was then added to the pale yellow solution at ambient temperature, and stirring was continued for 4 h. Mixture was dissolved in 5% LiCl solution and extracted with Et2O (5 x 40 mL). Organic fractions were combined, washed with brine, dried over Na2SO4, and concentrated in vacuo to yield a pale yellow crude oil. Purification by flash chromatography (1:1 Hex/EtOAc) afforded the desired product as a clear oil (0.82g, 64%). [α]D20 = −5.0° (c = 0.9, MeOH). 1H NMR (400.1 MHz, CDCl3) δ (ppm): 5.82 (m, 2H); 5.19-4.93 (m, 4H); 3.22 (m, 2H); 2.85 (m, 1H); 2.70 (m, 2H); 2.26 (m, 1H); 2.06 (m, 1H); 1.75 (m, 1H); 1.66-1.38 (m, 7H). 13C NMR (100.6 MHz, CDCl3) δ (ppm): 137.98, 137.51, 115.99, 115.66, 62.45, 55.71, 49.83, 39.34, 32.65, 28.88, 27.53, 25.78. HRMS (TOF, ES+) C12H22N [M+H]+ calc’d 180.1752, found 180.1751.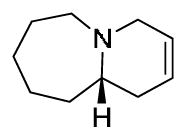 (S)-octahydropyrido[1,2-a]azepine unsat-7: To a solution of diene 21c1 (106 mg, 0.59 mmol) in toluene (11.8 mL) was added trifluoroacetic acid (71 mg, 0.62 mmol), and Grubbs 2nd generation catalyst (49 mg, 0.06 mmol). Solution was submitted to microwave irradiation for 1 h at 100°C. The toluene was removed in vacuo, and the crude residue purified to afford the desired product as the TFA salt (105 mg, 68%). [α]D20 = +45.2° (c = 1.25, MeOH). 1H NMR (400.1 MHz, CDCl3) δ (ppm): 5.88 (m, 1H); 5.64 (m, 1H); 3.94 (m, 1H); 3.57 (m, 2H); 3.18-3.04 (m, 2H); 2.73 (t, 1H); 2.25 (m, 1H); 2.24-1.92 (m, 3H); 1.89-1.72 (m, 3H); 1.58 (m, 2H). 13C NMR (100.6 MHz, CDCl3) δ (ppm): 126.83, 120.24, 63.64, 55.70, 54.32, 31.93, 30.59, 27.21, 26.00, 21.96. HRMS (TOF, ES+) C10H18N [M+H]+ calc’d 152.1439, found 152.1440.
(S)-octahydropyrido[1,2-a]azepine unsat-7: To a solution of diene 21c1 (106 mg, 0.59 mmol) in toluene (11.8 mL) was added trifluoroacetic acid (71 mg, 0.62 mmol), and Grubbs 2nd generation catalyst (49 mg, 0.06 mmol). Solution was submitted to microwave irradiation for 1 h at 100°C. The toluene was removed in vacuo, and the crude residue purified to afford the desired product as the TFA salt (105 mg, 68%). [α]D20 = +45.2° (c = 1.25, MeOH). 1H NMR (400.1 MHz, CDCl3) δ (ppm): 5.88 (m, 1H); 5.64 (m, 1H); 3.94 (m, 1H); 3.57 (m, 2H); 3.18-3.04 (m, 2H); 2.73 (t, 1H); 2.25 (m, 1H); 2.24-1.92 (m, 3H); 1.89-1.72 (m, 3H); 1.58 (m, 2H). 13C NMR (100.6 MHz, CDCl3) δ (ppm): 126.83, 120.24, 63.64, 55.70, 54.32, 31.93, 30.59, 27.21, 26.00, 21.96. HRMS (TOF, ES+) C10H18N [M+H]+ calc’d 152.1439, found 152.1440.
- 28.Suyama TL, Gerwick WH. Org Lett. 2006;8:4541–4543. doi: 10.1021/ol061736p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Merino P, Tejero T, Greco G, Marca E, Delso I, Gomez-SanJuan A, Matute R. HETEROCYCLES. 2012;84:75–100. [Google Scholar]
- 30.Woodward CP, Spiccia ND, Jackson WR, Robinson AJ. Chem Commun. 2011;47:779–781. doi: 10.1039/c0cc03716h. [DOI] [PubMed] [Google Scholar]
- 31.Verhelst SHL, Martinez BP, Timmer MSM, Lodder G, van der Marel GA, Overkleeft HS, van Boom JH. J Org Chem. 2003;68:9598–9603. doi: 10.1021/jo0350662. [DOI] [PubMed] [Google Scholar]