

Abstract
A concise and efficient synthetic approach to producing a novel non-cyclic nucleotide EPAC antagonist ESI-09 and its new analogs is reported. Key features of the synthesis include a mild and reliable one-pot procedure for an isoxazole synthon, as well as a modified one-pot protocol for the cyanomethyl ketone key intermediate. The synthesis requires inexpensive starting materials and only three linear steps for the completion in a total yield of 53%.
Keywords: EPAC antagonist, ESI-09, isoxazole, modified Kowalski's protocol, cyanomethyl ketone
Pancreatic ductal adenocarcinoma (PDA) is one of the leading causes of cancer death.1 Despite extensive research over the years in terms of therapeutic strategies, no effective treatment has come forward for this universally lethal disease.2 It is urgently needed to develop novel chemical probes and effective therapeutics that are based on new insights with better understanding of the molecular mechanism of pancreatic cancer. The exchange protein directly activated by cAMP (EPAC),3,4 especially EPAC1, is over-expressed in human PDA specimens, but the functional roles of this over-expression have never been investigated.5 Our team has previously reported the identification and characterization of a high throughput screening hit ESI-09 (1, chemical structure shown in Figure 1), a non-cyclic nucleotide small molecule that specifically inhibits EPAC1 and EPAC2.6,7 Using ESI-09 as a small molecular chemical probe, we demonstrated that EPAC1 plays an important role in pancreatic cancer cell migration and invasion, suggesting EPAC1 may represent an attractive target for novel therapeutic strategies for treating PDA.7
Figure 1.
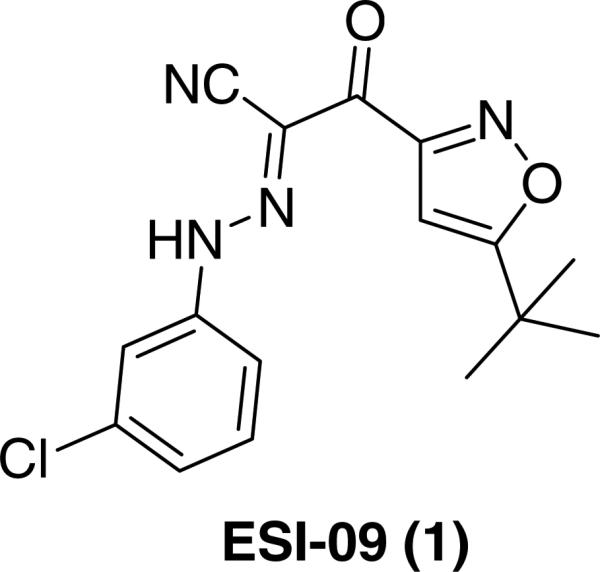
Structure of ESI-09 (1).
In our ongoing research to develop small molecules with high potency, specificity, and druglike properties based upon ESI-09, it is imperative to establish a practical and efficient method to readily access 1 suitable for a large scale as a reference compound as well as to further evaluate its pharmacological properties in vivo. Herein, we describe our optimized synthetic approaches to 1 starting from pinacolone in high overall yields.
Our retrosynthetic strategy is outlined in Scheme 1. Coupling of the key intermediate 8 with 3-chlorobenzenediazonium chloride 10 was expected to afford the desired product 1.8,9 Cyanomethyl derivative 8 could be formed from ethyl ester 6.10 Isoxazole 6 would be derived from the commercially available pinacolone 2 and oxalic acid diethyl ester 3.11
Scheme 1.

Retrosynthetic analysis of ESI-09 (1).
We first prepared the 3,5-disubstituted isoxazole 6 in two steps (Scheme 2). Starting from the commercially available and inexpensive pinacolone 2, oxalic acid diethyl ester 3 and NaH (60%), β-diketone 4 was readily prepared in 75% yield after purification. Further treatment of β-diketone 4 with hydroxylamine hydrochloride for about 2 h at room temperature afforded the intermediate oxime 5. Upon stirring at 75 °C for another 4 h, followed by simple extraction procedures and short column silica gel chromatography, the desired product 6 was obtained in 90% yield. Despite the good yields for preparation of isoxazole 6, this two-step procedure required two column purification steps. Based on the reagents which were used in this procedure, we reasoned that we could use hydroxylamine hydrochloride directly without further purification of β-diketone 4. In this event, treatment of the reaction solution of β-diketone 4 with the solution of hydroxylamine hydrochloride in ethanol afforded the desired product 6 after refluxing at 75 °C for 24 h. To our delight, we found that only a single clear spot was detectable by TLC. After short column silica gel chromatography, isoxazole 6 was obtained in 87% yield.
Scheme 2.

One-pot synthesis of 5-tert-butylisoxazole-3-carboxylic acid ethyl ester 6.
Next, we followed Kowalski's protocol12 to synthesize the key intermediate 7, which was carried out at -78 °C in the presence of 2.0 equiv of LDA and 2.0 equiv of dibromomethane (Scheme 3). Based upon our findings, the reaction was incomplete after 1 h, and the product was a complex reaction mixture of several components. After purification with silica gel column, the desired bromomethyl ketone 7 was only obtained in 6% yield and we also isolated the trace amount of the dibromo by-product 11 (Table 1, entry 1). We then investigated the different bases for this reaction and the results showed that MeLi was the best base (entry 3), but the yield was still low (only 25% yield). It was noteworthy that this modified condition (entry 3) resulted in a clear reaction while approximately 50% of the starting material was recovered and the by-product 11 was also isolated in 25% yield. To improve the yield of this reaction, we increased the amount of MeLi and extended the reaction time from 1 h to 2 h (entry 4 to 6). To our delight, the reaction was also clear and the desired product 7 was obtained in 73% yield by using 4.0 equiv of MeLi (entry 6). Considering the mechanism of this reaction,12 we reasoned that 1.2 equiv of dibromomethane would be enough for this reaction. As expected, the desired product 7 was obtained in 84% yield under an optimized condition by using 1.2 equiv of dibromomethane and 3.0 equiv of MeLi (entry 7).
Scheme 3.
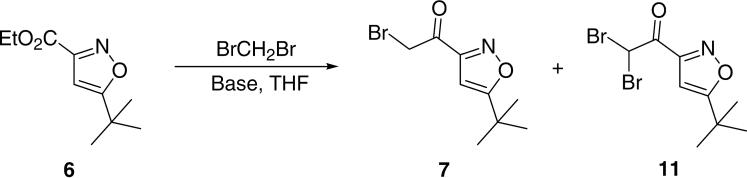
Synthesis of 2-bromo-1-(5-tert-butylisoxazol-3-yl)ethanone 7.
Table 1.
Optimization of Reactions Conditions of Bromomethyl Ketone 7a
| entry | base (equiv) | BrCH2Br (equiv) | time | products (yieldb) |
|---|---|---|---|---|
| 1 | LDA (2.0) | 2.0 | 1 h | 7 (6%), 11 (trace) |
| 2 | n-BuLi (2.0) | 2.0 | 1 h | 7 (11%), 11 (trace) |
| 3 | MeLi (2.0) | 2.0 | 1 h | 7 (25%), 11 (25%) |
| 4 | MeLi (2.5) | 2.0 | 2 h | 7 (57%), 11 (5%) |
| 5 | MeLi (3.0) | 2.0 | 2 h | 7 (65%), 11 (3%) |
| 6 | MeLi (4.0) | 2.0 | 2 h | 7 (73%), 11 (trace) |
| 7 | MeLi (3.0) | 1.2 | 2 h | 7 (84%), 11 (trace) |
The concentration was 0.25 M in THF.
Isolated yield.
With bromomethyl ketone 7 in hand, we used trimethylsilyl cyanide (TMSCN) as cyanide source to covert 7 into cyanomethyl derivative 8.13 In the presence of tetrabutylammonium fluoride (TBAF), cyanomethyl ketone 8 was smoothly generated in the solvent of CH3CN at room temperature for about 3 h (Scheme 4). This reaction was clear with only a small polar spot observed via TLC. However, it was noteworthy that cyanomethyl ketone 8 could decompose upon purification by standard column chromatography (unstable in silica gel column or neutral Al2O3 column) and therefore, we used this intermediate directly for the next step without further purification.
Scheme 4.
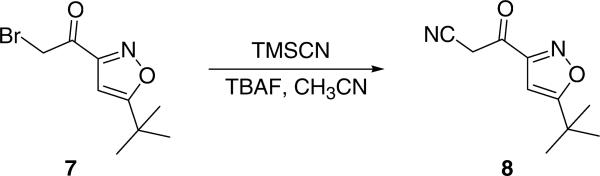
Synthesis of 3-(5-tert-butylisoxazol-3-yl)-3-oxo-propionitrile 8.
Aryldiazonium salt 10 was prepared from 3-chloroaniline 9 by using sodium nitrite in the presence of hydrochloric acid at 0 °C for 2 min. Direct coupling of the crude cyanomethyl ketone 8 with aryldiazonium chloride 10 afforded the desired product 1 in 51% yield in two steps (Scheme 5). The chemical structure of 1 was further confirmed by single-crystal X-ray structural analysis.14
Scheme 5.

Synthesis of 3-(5-tert-butylisoxazol-3-yl)-2-[(3-chlorophenyl)-hydrazono]-3-oxo-propionitrile 1.
Despite the synthesis of ESI-09 in moderate yields (an overall yield of 37% in four steps), we attempted to shorten the synthetic route by means of exploring one-pot procedure for converting isoxazole 6 to cyanomethyl ketone 8 according to our modified Kowalski's protocol for bromomethyl ketone (Table 1, entry 7). Replacing CH3CN with dibromomethane, we found that 6 was converted to 8 in high yield by using 2.0 equiv MeLi as the base (Scheme 6).15 Consistent with experimental observations from the synthesis of bromomethyl ketone 7, it was noted that MeLi is the optimal base for preparation of ketonitrile 8 (less than 50% yields by using n-BuLi or LDA as the base under the same reaction condition). Using the same subsequent coupling reaction, desired product 116 was obtained in 61% yield in two steps from isoxazole 6. To further explore the synthetic potential for generating ESI-09 analogs using this optimized approach, a series of new derivatives 12a-d (Scheme 7) have been synthesized in good yields (62-76%, two steps).
Scheme 6.
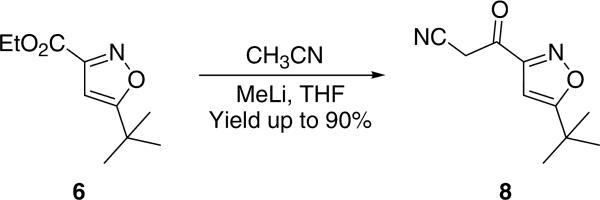
Synthesis of 3-(5-tert-butylisoxazol-3-yl)-3-oxo-propionitrile 8 from 6.
Scheme 7.

Synthesis of ESI-09 analogs 12a-d.
In conclusion, we have developed a straightforward method to synthesize 1 starting from pinacolone in an excellent overall yield of 53% in three steps. The efficient approach included an one-pot procedure for the isoxazole synthon, followed by a modified protocol for the key cyanomethyl ketone and the subsequent coupling step. The described synthetic strategies and optimized reaction conditions may have general applications in the convenient preparation of various isoxazole and cyanomethyl ketone building blocks. Moreover, the established concise and efficient synthesis of the novel EPAC antagonist ESI-09 will facilitate its further in vitro and in vivo pharmacological evaluations, as well as the ongoing synthesis of new analogs for the structure-activity relationship study.
Supplementary Material
Acknowledgments
This work was supported by grants P30DA028821, R21MH093844 (JZ), R01GM066170, R21NS066510 (XC), and R01GM106218 (XC & JZ) from the National Institute of Health, R. A. Welch Foundation Chemistry and Biology Collaborative Grant from Gulf Coast Consortia (GCC) for Chemical Genomics, Sealy and Smith Foundation grant (to the Sealy Center for Structural Biology and Molecular Biophysics), John Sealy Memorial Endowment Fund, and the Center for Addiction Research (CAR) at the University of Texas Medical Branch.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors declare no competing financial interest.
Supplementary data
Supplementary data associated with this article can be found, in the online version, at doi:xxx.
References
- 1.Hariharan D, Saied A, Kocher HM. HPB (Oxford) 2008;10:58. doi: 10.1080/13651820701883148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.a Hezel AF, Kimmelman AC, Stanger BZ, Bardeesy N, Depinho RA. Genes. Dev. 2006;20:1218. doi: 10.1101/gad.1415606. [DOI] [PubMed] [Google Scholar]; b Hidalgo M. N. Engl. J. Med. 2010;362:1605. doi: 10.1056/NEJMra0901557. [DOI] [PubMed] [Google Scholar]
- 3.a de Rooij J, Zwartkruis FJ, Verheijen MH, Cool RH, Nijman SM, Wittinghofer A, Bos JL. Nature. 1998;396:474. doi: 10.1038/24884. [DOI] [PubMed] [Google Scholar]; b Kawasaki H, Springett GM, Mochizuki N, Toki S, Nakaya M, Matsuda M, Housman DE, Graybiel AM. Science. 1998;282:2275. doi: 10.1126/science.282.5397.2275. [DOI] [PubMed] [Google Scholar]
- 4.a Holz GG, Chepurny OG, Schwede F. Cell. Signal. 2008;20:10. doi: 10.1016/j.cellsig.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Gloerich M, Bos JL. Annu. Rev. Pharmacol. Toxicol. 2010;50:355. doi: 10.1146/annurev.pharmtox.010909.105714. [DOI] [PubMed] [Google Scholar]; c Grandoch M, Roscioni SS, Schmidt M. Br. J. Pharmacol. 2010;159:265. doi: 10.1111/j.1476-5381.2009.00458.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Breckler M, Berthouze M, Laurent AC, Crozatier B, Morel E, Lezoualc'h F. Cell. Signal. 2011;23:1257. doi: 10.1016/j.cellsig.2011.03.007. [DOI] [PubMed] [Google Scholar]
- 5.Lorenz R, Aleksic T, Wagner M, Adler G, Weber CK. Pancreas. 2008;37:102. doi: 10.1097/MPA.0b013e318160748f. [DOI] [PubMed] [Google Scholar]
- 6.a Tsalkova T, Mei FC, Cheng X. PLos One. 2012;7:e30441. doi: 10.1371/journal.pone.0030441. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Tsalkova T, Mei FC, Li S, Chepurny OG, Leech CA, Liu T, Holz GG, Woods VL, Jr., Cheng X. Proc. Natl. Acad. Sci. U.S.A. 2012;109:18613. doi: 10.1073/pnas.1210209109. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Chen H, Tsalkova T, Mei FC, Hu Y, Cheng X, Zhou J. Bioorg. Med. Chem. Lett. 2012;22:4038. doi: 10.1016/j.bmcl.2012.04.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Almahariq M, Tsalkova T, Mei FC, Chen H, Zhou J, Sastry SK, Schwede F, Cheng X. Mol. Pharmacol. 2013;83:122. doi: 10.1124/mol.112.080689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a Abdel-Motaleb RM, Makhloof AA, Ibrahim HM, Elnagdi MH. J. Heterocycl. Chem. 2006;43:931. [Google Scholar]; b Aziz SI, Anwar HF, Fleita DH, Elnagdi MH. J. Heterocycl. Chem. 2007;44:725. [Google Scholar]
- 9.a Hassaneen HME. Synth. Commun. 2007;37:3579. [Google Scholar]; b Behbehani H, Ibrahim HM, Makhseed S, Mahmoud H. Eur. J. Med. Chem. 2011;46:1813. doi: 10.1016/j.ejmech.2011.02.040. [DOI] [PubMed] [Google Scholar]
- 10.a Arseniyadis S, Kyler KS, Watt DS. Org. React. (N.Y.) 1984;31:1. [Google Scholar]; b Katritzky AR, Abdel-Fattah AAA, Wang M. J. Org. Chem. 2003;68:4932. doi: 10.1021/jo026796x. [DOI] [PubMed] [Google Scholar]; c Ji Y, Trenkle WC, Vowles JV. Org. Lett. 2006;8:1161. doi: 10.1021/ol053164z. [DOI] [PubMed] [Google Scholar]
- 11.a Pinho e Melo TMVD. Curr. Org. Chem. 2005;9:925. [Google Scholar]; b Lepage F, Tombret F, Cuvier G, Marivain A, Gillardin JM. Eur. J. Med. Chem. 1992;27:581. [Google Scholar]
- 12.Kowalski CJ, Haque MS. J. Org. Chem. 1985;50:5140. [Google Scholar]
- 13.Soli ED, Manoso AS, Patterson MC, DeShong P, Favor DA, Hirschmann R, Smith AB., III J. Org. Chem. 1999;64:3171. doi: 10.1021/jo982302d. [DOI] [PubMed] [Google Scholar]
- 14.The crystallographic data have been deposited in full at the Cambridge Crystallographic Data Centre, reference CCDC 913321, accessible at http://www.ccdc.cam.ac.uk.
- 15.1H and 13C NMR spectra (see spectra of crude product in the Supporting Information) showed that the crude residue was only contaminated with some solvent impurities and suitable for direct use in the next step.
- 16.One-pot synthesis of 5-tert-butylisoxazole-3-carboxylic acid ethyl ester (6). To a solution of pinacolone (5.0 g, 50.0 mmol) in THF (100 mL) was added NaH (60%) (2.2 g, 55.0 mmol) at 0 °C. The reaction mixture was stirred at r.t. for about 30 min. Diethyl oxalate (7.3 g, 50.0 mmol) was added at 0 °C and the mixture was then stirred at r.t. overnight. To the resulting solution, hydroxylamine hydrochloride (3.8 g, 55.0 mmol) in ethanol (100 mL) was added dropwise. The mixture was heated at reflux for 16 h. After this time the sodium chloride was removed by filtration and the filtrate was concentrated under reduced pressure. The residue was purified by short column chromatography on silica gel eluting with hexane/ethyl acetate (4/1) to provide the title compound as a colorless oil (8.57 g, 87%). 1H NMR (600 MHz, CDCl3) δ 6.37 (s, 1H), 4.43 (q, 2H, J = 7.2 Hz), 1.41 (t, 3H, J = 7.2 Hz), 1.37 (s, 9H). 13C NMR (150 MHz, CDCl3) δ 183.3, 160.5, 156.2, 99.3, 62.1, 33.1, 28.9, 14.3. HRMS (ESI) calcd for C10H16NO3 198.1125 (M + H)+, found 198.1131.2-Bromo-1-(5-tert-butylisoxazol-3-yl)ethanone (7). To a solution of 5-tert-butylisoxazole-3-carboxylic acid ethyl ester (0.5 g, 2.5 mmol) in anhydrous tetrahydrofuran (10 mL) was added dibromomethane (0.52 g, 3.0 mmol) under nitrogen. The mixture was cooled to -78 °C and 1.6 M methyllithium in diethyl ether (4.7 mL, 7.5 mmol) was added dropwise. The solution was stirred at -78 °C for 40 min and then quenched with acetic acid (0.45 g, 7.5 mmol). The mixture was warmed to 0 °C and poured onto ice/water (20 mL) and extracted with ethyl acetate (50 mL). The organic layer was dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by short column chromatography on silica gel eluting with hexane/ethyl acetate (4/1) to provide the title compound as a colorless oil (524 mg, 84%). 1H NMR (600 MHz, CDCl3) δ 6.40 (s, 1H), 4.57 (s, 2H), 1.38 (s, 9H). 13C NMR (150 MHz, CDCl3) δ 185.7, 183.9, 159.7, 97.7, 33.2, 31.7, 28.9. HRMS (ESI) calcd for C9H13BrNO2 246.0124 (M + H)+, found 246.0131.2,2-Dibromo-1-(5-tert-butylisoxazol-3-yl)ethanone (11). Colorless oil. 1H NMR (600 MHz, CDCl3) δ 6.40 (s, 1H), 4.90 (s, 1H), 1.38 (s, 9H). 13C NMR (150 MHz, CDCl3) δ 193.8, 183.6, 159.2, 97.2, 66.7, 33.1, 28.9. HRMS (ESI) calcd for C9H12Br2NO2 323.9229 (M + H)+, found 323.9320.3-(5-tert-Butylisoxazol-3-yl)-3-oxo-propionitrile (8). To a solution of 2-bromo-1-(5-tert-butylisoxazol-3-yl)ethanone (0.62 g, 2.5 mmol) in 10 mL of acetonitrile was added trimethylsilyl cyanide (0.25 g, 2.5 mmol), followed by 2.5 mL (2.5 mmol) of tetrabutylammonium fluoride solution (1 M in THF). The reaction mixture was stirred at r.t. for 0.5 h, and then diluted with brine (10 mL). The product was extracted with ethyl acetate (50 mL). The organic layer was dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was obtained as a yellow oil and used directly for next step without further purification. It is noteworthy that cyanomethyl ketone 8 was unstable in silica gel column or neutral Al2O3 column. The pure compound for characterization was purified by preparative plate chromatography (silica gel, hexane/ethyl acetate 2:1) to obtain 8 as a yellow oil. 1H NMR (600 MHz, CDCl3) δ 6.42 (s, 1H), 4.18 (s, 2H), 1.38 (s, 9H). 13C NMR (150 MHz, CDCl3) δ 184.6, 182.0, 160.0, 112.9, 97.3, 33.2, 29.9, 28.8. HRMS (ESI) calcd for C10H13N2O2 193.0972 (M + H)+, found 193.1020.One-pot synthesis of 3-(5-tert-butylisoxazol-3-yl)-3-oxo-propionitrile (8) from 6. To a solution of CH3CN (0.41 g, 10.0 mmol) in anhydrous THF (5 mL) was added 1.6 M methyllithium in diethyl ether (3.1 mL, 5.0 mmol) at -78 °C under nitrogen. The mixture was stirred at -78 °C for 0.5 h, and 5-tert-butylisoxazole-3-carboxylic acid ethyl ester (0.5 g, 2.5 mmol) in THF (5 mL) was then added dropwise. The solution was stirred at -78 °C for 1 h and then quenched with acetic acid (0.3 g, 5.0 mmol). The mixture was warmed to 0 °C and poured onto ice/water (10 mL) and extracted with ethyl acetate (50 mL). The organic lay was dried over Na2SO4, filtered and concentrated under reduced pressure. The crude residue (490 mg) was obtained as a yellow oil and directly used for next step without further purification.3-(5-tert-Butylisoxazol-3-yl)-2-[(3-chlorophenyl)-hydrazono]-3-oxo-propionitrile (1). To a solution of 3-chloroaniline (30 mg, 0.24 mmol) in H2O (1 mL cooled to -5°C) was added 0.24 mL of 1 N HCl (aq.). To the resulting acidic aniline solution, 1 mL solution of sodium nitrite (16 mg, 0.24 mmol) in H2O was added dropwise to generate the aryldiazonium salt solution. To the aryldiazonium salt solution was added sodium acetate (33 mg, 0.4 mmol), followed by 1 mL solution of crude 3-(5-tert-butylisoxazol-3-yl)-3-oxo-propionitrile (38 mg, 0.2 mmol) in ethanol. The reaction mixture was stirred at 0 °C for 5 min, and then poured onto H2O (2 mL) and extracted with ethyl acetate (20 mL). The organic layer was dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by short column chromatography on silica gel eluting with hexane/ethyl acetate (2/1) to provide the desired product ESI-09 (40 mg, 61%, two steps from 6) as a yellow solid (mp 146-147 °C). HPLC purity 99.6% (tR = 21.72 min). 1H NMR (600 MHz, DMSO-d6) δ 12.70 (bs, 1H), 7.44-7.47 (m, 3H), 7.25-7.26 (m, 1H), 6.70 (s, 1H), 1.39 (s, 9H). 13C NMR (150 MHz, DMSO-d6) δ 181.1, 179.4, 160.1, 143.6, 134.0, 131.2, 125.1, 116.2, 115.8, 113.4, 110.5, 100.4, 32.5, 28.5. HRMS (ESI) calcd for C16H16ClN4O2 331.0956 (M + H)+, found 331.0969.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.