

Abstract
Multiple Sclerosis (MS) is a neurodegenerative autoimmune disorder caused by chronic inflammation and demyelination within the central nervous system (CNS). Clinical studies in MS patients have demonstrated efficacy with B cell targeted therapies such as anti-CD20. However, the exact role that B cells play in the disease process is unclear. Activation Induced cytidine deaminase (AID) is an essential enzyme for the processes of antibody affinity maturation and isotype switching. To evaluate the impact of affinity maturation and isotype switching, we have interrogated the effect of AID-deficiency in an animal model of MS. Here, we show that the severity of experimental autoimmune encephalomyelitis (EAE) induced by the extracellular domain of human myelin oligodendrocyte glycoprotein (MOG1-125) is significantly reduced in Aicda deficient mice, which, unlike wild-type mice, lack serum IgG to myelin associated antigens. MOG specific T cell responses are comparable between wild-type and Aicda knockout mice suggesting an active role for antigen experienced B cells. Thus affinity maturation and/or class switching are critical processes in the pathogenesis of EAE.
Keywords: AID, EAE, MS, affinity maturation, Isotype switching
Introduction
MS is a chronic demyelinating disease in which the myelin of the CNS is the target of an autoimmune process [1]. B cells may play an important role in the pathogenesis of several human autoimmune diseases including MS [reviewed in [2]]. B cells are efficient antigen presenting cells (APCs) that can activate and provide T cell help to mount effective immune responses [3]. B cells can also produce cytokines to modulate the inflammatory response [4,5]. Additionally, autoantibodies can result in immune-mediated tissue destruction in experimental models [6–13]. The oligoclonal bands identified from cerebrospinal fluid of MS patients are composed of immunoglobulins and recent studies have suggested that their presence may be a biomarker for prognosis and/or subtypes of MS [14,15].
Supporting this notion, B cells found in the CNS of MS patients have been shown to be clonally related, exhibit a plasmablast phenotype and have undergone affinity maturation, implicating antigen driven B cell responses in MS [16]. IgG specific for myelin oligodendrocyte glycoprotein (MOG) have been demonstrated in MS patients [17,18]. In an attempt to block these various effector functions, therapies aimed at modulating B cells and immune responses are in various stages of preclinical and clinical research [reviewed in [2]]. Rituximab, a monoclonal antibody that selectively depletes CD20 expressing B lymphocytes, is approved for rheumatoid arthritis (RA) and can reduce MS symptoms [19].
In B cells, activation induced cytidine deaminase (AID) is essential for isotype switching and affinity maturation of immunoglobulins [20]. Through somatic hypermutation (SHM), AID introduces single point mutations at high frequency into the variable regions of the rearranged Ig heavy and light chains to generate high affinity antibodies [21]. Through class switch recombination (CSR), the Cm heavy chain constant region is exchanged for Ca, Cg or Ce to produce IgA, IgG or IgE. Each class of Ab has a different effector function increasing the versatility of the Abs made by a B cell [21]. Increased expression of AID has been observed in inflammatory diseases including RA [22] allergic rhinitis [23], Sjogren's syndrome [24], as well as several B cell lymphomas [25].
The role of AID in the pathogenesis of autoimmune diseases has been documented in experimental models. Inactivation of the Aicda gene in the MRL/lpr mouse model of systemic lupus significantly enhances survival [26,27]. BXD2 mice, which are also autoimmune prone, over-express AID, produce pathogenic auto-antibodies and develop severe arthritis and glomerulonephritis [28] all of which can be suppressed by transgenic expression of a dominant negative AID [29].
In this study, we aimed to specifically test the role of AID in the pathogenesis of recombinant human myelin oligodendrocyte glycoprotein (rhMOG) EAE [30]. Our results demonstrate that in the absence of AID, rhMOG-EAE is profoundly attenuated suggesting that AID-dependent events such as affinity maturation and isotype switching are critical processes involved in the EAE pathogenesis. Accordingly, we show that MOG specific, high affinity IgG are abundant in WT mice with EAE and that serum IgG1 from these mice bind to brain tissue, whereas such antibodies are below the limit of detection in Aicda deficient mice.
Materials and Methods
Animals
All animals used in this study were housed and maintained at Genentech in accordance with American Association of Laboratory Animal Care guidelines. All experimental studies were conducted under protocols (#12–1009 and subletters) approved by the Institutional Animal Care and Use Committee of Genentech Lab Animal Research in an AAALACi-accredited facility in accordance with the Guide for the Care and Use of Laboratory Animals and applicable laws and regulations. B cell deficient (uMT KO) animals were purchased from Jackson Laboratories (Bar Harbor, ME; colony #002288) along with control WT animals (colony #000664).
Generation of Aicda deficient mice
The construct for targeting the C57BL/6 Aicda locus in ES cells was made using a combination of recombineering as well as standard molecular cloning techniques [31,32]. Briefly, a 8755 bp fragment (assembly NCBI37/mm9, chr6:122,508,416-122,517,170) from a mouse BAC (RP23-470E2) was first retrieved into plasmid pBlight-TK [31]. Second a 940 bp loxP-em7-kanamycin-loxP cassette was inserted upstream of exon 3 between position chr6:122,510,801 and 122,510,802. Correctly targeted plasmid was transformed into arabinose-induced SW106 cells [33,34] to remove kanamycin and leave behind a single loxP site. Finally, an frt-PGK-em7-Neo-BGHpA-frt-loxP cassette was inserted downstream of exon 3 between position chr6:122,511,674 and 122,511,675, resulting in the Aicda targeting targeting vector. The final vector was confirmed by DNA sequencing.
The Aicda vector was linearized with NotI, C57BL/6 ES cells were targeted using standard methods (G418 positive and gancyclovir negative selection), and positive clones were identified using PCR and TaqMan® analysis. Correctly targeted ES cells were transfected with a Cre plasmid to remove Neo and create the Aicda KO allele. Aicda KO ES cells were then injected into blastocysts using standard techniques, and germline transmission was obtained after crossing resulting chimaeras with C57BL/6 females.
Production of human MOG
Recombinant human MOG was expressed and purified as previously described [35,36]. Briefly, the protein was expressed in inclusion bodies in Escherichia coli and processed under denaturing conditions. The resulting material was purified and refolded on-column using Ni-NTA-superflow resin (Qiagen, Carlsbad, CA). Endotoxin was removed from the protein by incorporating a 0.1% Triton X-114 wash step prior to applying the glutathione redox buffers for refolding. The eluted protein was then dialyzed into 100 mM sodium phosphate buffer pH8.0. Purity of the obtained preparation was verified by SDS-PAGE and the protein's molecular weight was confirmed by mass spectrometry.
EAE induction and clinical scoring
EAE was induced in 8–12-week-old female animals using recombinant human myelin oligodendrocyte glycoprotein (MOG) [35] and complete freunds adjuvant as previously described. Briefly, animals were injected subcutaneously on the back with a 200 microliter emulsification of 50–100 micrograms of recombinant human MOG 1–125 in incomplete freund's adjuvant supplemented with 8 mg/ml mycobacterium tuberculosis (Difco Laboratories, Detroit, MI). The final dose of mycobacterium tuberculosis per mouse was 800 ug. Animals were also injected with 200 ng pertussis toxin immediately after immunization and again 48 hours later. Clinical scoring of disease was performed 3 times per week starting at day 9. Animals were assessed based on a 5-point system: 0 = no clinical disease, 1 = loss of tail tone only, 2 = mild monoparesis or paraparesis, 3 = severe paraparesis, 4 = paraplegia and/or quadraparesis, and 5 = moribund or death.
Histopathology
Brain and spines were fixed in 10% neutral buffered formalin. Tissues were decalcified, trimmed and processed to 4 μm sections stained with H+E. Microscopic examination was performed on 4 coronal sections of brain from each animal. Microscopic examination of spine, including spinal cord at three levels (cervical, thoracic and lumbar) was performed on 2 to 4 transverse sections from each level for each animal. Lesions were scored according to severity on a scale of 0 (no lesions) to 3 (severe). Average lesion severity across all sections examined was compared across groups and graphed using Aabel software v3.0.6.
Detection of brain tissue binding IgG in EAE derived sera by immunofluorescence
A brain sample collected from a SCID mouse (to avoid interference with endogenous immunoglobulin) was frozen in OCT (Tissue-Tek, Cat#4583, Torrance, CA) [37]. Sections were cut on a cryotome and briefly fixed in cold acetone. Sections were probed with sera collected at terminal sacrifice of mice induced with MOG to produce EAE. Mice represented Aicda.wt, Aicda.ko, uMT.wt and uMT.ko genotypes. Normal mouse serum at 1:20 was used as a control. Sera were diluted 1:20 in PBS and incubated for 60 minutes at room temperature. After washing with PBS, binding of IgG1 or IgM on brain tissue was detected using IgG1 and IgM specific secondary antibodies (Invitrogen, A21121 and A21042; 5 ug/ml) conjugated to Alexa Fluor 488. Slides were imaged in a region rich in myelinized white matter (brain stem at the cerebellar peduncle) using identical acquisition conditions. Images were examined for presence or absence of white-matter associated fluorescent signal without image modification.
Flow cytometry
Single cell suspensions were prepared from draining lymph nodes (DLNs). Cells were stained with FITC-conjugated anti-CD44, PE-conjugated anti-CD4, PerCp conjugated anti-CD8 and APC-conjugated anti-CD62L (BD Biosciences, San Jose, CA). Cell acquisition was performed on a FACScalibur (Becton Dickinson, Mountain View, CA) and data was analyzed with Flowjo software (Ashland, OR). Isotype switching analysis was performed as described previously [38].
In vitro cytokine assay
Ten days after immunization, draining lymph node cells were isolated and cultured in 96-well flat-bottom plates at a concentration of 5 × 105 cells/well in complete RPMI 1640 medium that contained 10% heat-inactivated FCS, 1 mM glutamine, 1% penicillin-streptavidin, 1 mM nonessential amino acids, and 5 × 10− 5 M 2-ME with various concentrations of recombinant human MOG protein (0, 2, 10 and 50 ug/ml) or MOG35–55 peptide (0, 1, 5, and 25 ug/ml). Supernatants were collected 4 days after culture for cytokine detection by luminex (BioRad, Hercules, CA).
Detection of antibodies by ELISA
Serum samples were collected before and 28 days after immunization. For detection of anti-rMOG and anti-MOG35–55 peptide autoantibodies, ELISA plates (Dynex Technologies, Chantilly, VA) were coated with rMOG (5 ìg/ml) or MOG35–55 peptide (100 ug/ml) in PBS. Plates were washed with PBS/0.05% Tween-20 and blocked with PBS/0.1% BSA. Serum samples were diluted at various concentrations and bound antibodies were detected with AP-conjugated goat anti-mouse IgG and AP-conjugated goat anti-mouse IgM (Southern Biotechnology Associates, Birmingham, AL) using 1-step PNPP substrate (Thermo Scientific, Rockford, IL). The OD was measured at 405 nm by a SpectraMax 250 Microplate Reader (Molecular Devices, Menlo Park, CA). Experimental values were normalized to mixed day-28 sera of WT immunized mice for IgG antibodies and to mixed day 28 sera of Aicda KO immunized mice for IgM antibodies (arbitrarily defined as 100 U).
Anti-rhMOG-specific IgM assay
For measurement of anti-rhMOG-specific IgM in mouse serum, 96-well microtiter ELISA plates were coated overnight at 4°C with various rhMOG concentrations in a carbonate buffer. Plates were washed, blocked with 200 μL ELISA assay diluent with BSA for 1–2 hours at room temperature and washed. Normalized 0.1 mg/ml of total IgM from diluted mouse serum were added and incubated for 2 hours at room temperature. The plates were washed three times and incubated with HRP-conjugated goat anti-mouse IgM-specific antibody for 30 min.
After washing three times, bound enzyme was detected with the same steps as described below. For measurement of total mouse IgM in serum, 96-well microtiter ELISA plates (Greiner, Germany) were coated overnight at 4°C with 2 μg/ml Goat F(ab')2 anti-mouse Ig (SouthernBiotech) in a carbonate buffer (pH 9.6). Plates were washed, blocked with 200 μL ELISA assay diluent with BSA for 1–2 hours at room temperature and washed. Diluted mouse serum samples or mouse IgM standards (BD BioSciences) were added and incubated for 2 hours at room temperature. Total mouse IgM levels were quantitated using HRP-conjugated Goat anti-mouse IgM-specific antibody (Invitrogen) for 30 min. After washing three times, bound enzyme was detected by addition of 100 μL/well TMB substrate (BioFX Laboratories) for 5 minutes. The reactions were stopped by adding 100 μL/well of stop reagent (BioFX Laboratories) and detection of color at A 650 nm.
Surface plasmon resonance
Surface plasmon resonance (SPR) was run on a Biacore 3000 (GE healthcare, Piscataway, NJ) at 25°C using recombinant MOG (described previously) amine-coupled to a Biacore CM5 sensor chip at 2,500 response units (RU) and a reference flow cell with human IgG amine-coupled at 5,000 RUs. Injection flow rates were 5 ul/min and sensors were regenerated using 10 mM glycine pH 2.0 for 30 seconds at a flow rate of 30 ul/min. Serum samples were used diluted 1:100 in HBS-P buffer (0.01 M Hepes, pH 7.4, 0.15 M NaCl, 0.005% Surfactant P20). Anti-mouse IgM (Southern Biotech, cat# 1140–01), anti-mouse IgG2a (Southern Biotech, cat# 1080–01), and anti-mouse IgG1 (Southern Biotech, cat# 1070–01) were used at 12.5 ug/ml diluted in HBS-P buffer. For the blocking study, recombinant MOG (2 uM) was incubated with 1:100 diluted serum (Day 28, WT) for 1 hr before injection over the sensor.
Results
Generation and characterization of Aicda-deficient mice
The targeting strategy used to generate Aicda-deficient mice in C57BL/6 background is depicted in Figure 1A. The deletion of exon-3 removes the majority of the catalytic domain of the Aicda gene and renders the transcript out of frame. To assess the effect on class switch recombination (CSR), splenocytes were stimulated with a combination of LPS and IL-4 for 4 days, followed by FACS analysis for isotype usage. This regimen promotes class switching from μ to γ1. Wild-type (WT) splenic B cells were 25% IgG1+ whereas AID-deficient splenocytes yielded negligible IgG1+B cells (Figure 1B). Thus the deletion of exon-3 completely abrogates isotype switching in mouse B cells as expected [20].
Figure 1. .

Generation and functional characterization of Aicda deficient mice. A) Gene targeting strategy used to delete the catalytic domain (exon-3) of Aicda locus. The structure of the AID domains is shown in top. The location of the homology arms in the targeting vector is indicated. The schematic of targeting construct used to delete the catalytic domain of AID (exon-3) is shown in the middle. The solid triangles represent Cre recombination sites and the Neomycin cassette was subsequently removed by expression of Cre-recombinase. Representative WT or KO PCR products using 3′ or 5′ arm analysis at the Aicda locus is shown in the right panel. The size of Aicda KO PCR product is indicated and corresponds to the modified locus. Notice that one primer is located in the Neomycin cassette and therefore only detects the targeted Aicda locus. B) FACS analysis of LPS/IL4 stimulated WT or AID KO B cells in vitro. Percentage of IgM or IgG1 are indicated in each quadrant. B220-FITC is used as a B-cell marker.
Aicda-deficient mice show decreased susceptibility to rhMOG-induced EAE
Full or partial length recombinant antigens produced in E. coli present an opportunity to involve B cells in the EAE disease model. These antigens have three dimensional structures and have been shown to be bound by antibodies in their native conformation [39–41]. Strikingly, uMT-deficient animals are resistant to disease induction with recombinant MOG protein (1–125) but susceptible to MOG peptide 35–55 induced EAE [42,43]. To determine whether or not AID plays a role in this resistance to rhMOG induced EAE, we immunized a cohort of animals containing wild-type and uMT-deficient mice (Jackson Labs) and Aicda wild-type and Aicda-deficient mice (bred at Genentech).
In these experiments, the incidence of disease was significantly reduced in animals deficient for either uMT (25% of the animals become sick) or AID (10%) as compared to wild-type controls (100%; Figure 2, Table 1). Detailed histopathology analysis of EAE CNS lesions revealed that both Aicda-deficient and uMT-deficient animals are nearly devoid of CNS lesions compared to the two WT cohort controls (Figure 2C-G). EAE lesions such as myelinopathy and gliosis were present in the CNS in WT mice, whereas none of the Aicda KO histology sections revealed such severity. Together these data suggest that AID is a critical component of the development of EAE.
Figure 2. .

Aicda-deficient (Aicda KO) and uMT-deficient (uMT KO) are resistant to rhMOG induced EAE. Development of EAE in Aicda KO and uMT KO animals (A) and average daily clinical score (B; p < 0.0001 by Oneway Dunnet's test for Aicda KO as compared to Aicda WT and p < 0.0001 for uMT KO and compared to uMT WT). Immunohistochemistry of spinal cord, lumbar. D, F: Aicda.wt (1–1630). E, G Aicda KO (2–1613). Mild superficial myelinopathy characterized by polymicrocavitation of myelin (arrowheads) and sparse leptomeningeal collections of mononuclear cells (arrows) are consistent with mild, subacute to chronic EAE. Corresponding lesions are absent in a representative section from an Aicda KO animal. Bar = 200 μm D, E); 50 μm F, G). WM = white matter; GM = gray matter. Boundary between gray and white matter outlined with dashed line.
Table 1. .
Incidence and disease induction in uMT- or AID- deficient mice.
| Genotype | Dose (ug) | Incidence | Day of onset (Ave ± SEM)* | Peak MSOD (Ave ± SEM)* |
|---|---|---|---|---|
| uMT+/+ | 100 | 100 (8/8) | 11.1 ± 1.1 | 3.4 ± 0.2 |
| uMT − / − | 100 | 25 (2/8) | 20** | 3** |
| AID+/+ | 100 | 100 (9/9) | 11.3 ± 0.6 | 3.7 ± 0.2 |
| AID − / − | 100 | 10 (1/10) | 16.5** | 4** |
*Calculated only for those animals that received a disease score greater than or equal to 1.
**SEM not shown as number of animals in calculation is less than 3.
Aicda KO mice do not show a defect in MOG-specific T cell response
Helper T cells and IL-17 play an important role in the development of EAE [44]. To determine whether the T cell arm is compromised in Aicda deficient mice, we examined T cell activation and effector function in the draining lymph nodes (dLN) of rhMOG-immunized animals. Draining LN cells were isolated from mice 10 days after immunization with rhMOG. We did not observe any appreciable difference in the cellularity of WT or Aicda -/- dLNs (Figure 3A, 41.1 ± 3.3 vs 44.3 ± 8.9 millions, p = 0.59). The relative frequency of CD4+(15.6 ± 1.6 vs 19.6 ± 3.2, p = 0.21) and CD8+T cells (14.1 ± 1.3 vs 14.0 ± 2.3, p = 0.92) was comparable between groups (Figure 4B top panels).
Figure 3. .

Similar state of T cell activation in draining lymph nodes and post antigenic stimulation. A, Total cell numbers in two DLNs of WT and Aicda KO mice. B, T cell activation is normal in Aicda KO mice. DLN cells were stained with CD4, CD8, CD44 and CD62L and analyzed by flow cytometry. C) Aicda KO mice do not show defect in the MOG-specific T cell IL-17 production. WT and Aicda KO mice were immunized with MOG and 10 days later, draining lymph node cells from mice were isolated and cultured with various concentrations of rMOG or MOG35–55 peptide for 4 days and subsequently the supernatants were collected and analyzed by Luminex.
Figure 4. .

Characterization of anti-MOG antibodies using ELISA at day zero and 28. A) IgG anti-rhMOG, B) IgM anti-rhMOG, C) IgG anti-MOG peptide 35–55, D) IgM anti-MOG peptide 35–55. Mixed day 28 sera of WT or KO immunized mice were used as standard (arbitrarily defined as 100 U) for IgG or IgM antibodies, respectively. E) IgM binding to rhMOG coated at different concentrations. Sera from 8 mice were pooled for each of the following sample sets: Day 0 WT, Day 0 KO, Day 28 WT, and Day 28 KO. Pooled mice sera IgM Abs normalized at 0.1 mg/ml in the assay. Anti-mouse IgM was used as detection to exclusively measure the binding of IgM antibodies in serum.
Activated T cells up-regulate CD44 expression and down-regulate CD62L expression. To compare the activation states of CD4+T cells, we stained CD4+cells with CD44 and CD62L activation markers. We found a comparable population of CD44hiCD62Llo (21.2 ± 2.0 vs 20.9 ± 1.4 millions, p = 0.81) and CD44hiCD62Lhi populations (11.8 ± 0.8 vs 10.3 ± 2.0 millions, p = 0.31) in WT and Aicda KO mice (Figure 4B lower panels). These studies suggest that the activation of CD4+T cells is not affected in Aicda KO mice. To further test the function of T cells, dLN cells were re-stimulated with various doses of rhMOG or MOG35–55 peptide ex vivo. As shown in Figure 3C, dLN cells from both WT and Aicda KO mice produce substantial amounts of IL-17 in a dose-dependent manner to either rhMOG (left panel) or MOG35–55 peptide (right panel) stimulation. In total, these data suggest that the resistance to disease induction observed in Aicda-deficient animals is not due to changes in T cell activation or effector function.
High affinity IgG anti-MOG auto-antibodies are only abundant in WT animals
AID is essential for both affinity maturation and isotype switching. We measured the level of MOG-specific IgG or IgM auto-antibodies in WT and Aicda-deficient mice. We found that WT mice produced a substantial amount of IgG against rhMOG (Figure 4A) and MOGp35–55 (Figure 4C) 28 days after immunization. As expected, the levels of IgG against rhMOG and MOGp35–55 in Aicda-deficient mice were below the limit of detection as these mice are unable to undergo class switch recombination [20]. In contrast, WT mice produced only a small amount of IgM against rhMOG (Figure 4B) or MOGp35–55 (Figure 4D), whereas Aicda-deficient mice interestingly produced more IgM antibodies against rhMOG and MOGp35–55.
We then determined how well WT or IgM anti-MOG antibodies bind to rhMOG at limiting concentrations to gain further insights into the affinity and abundance of these antibodies. Our results confirmed the ELISA study that WT mice produced only a small amount of IgM Abs against rhMOG whereas Aicda KO have substantially higher amount of anti-MOG antibodies (Figure 4E). It is possible that a large fraction of rhMOG-specific antibodies from WT mice might have class-switched to IgG isotype, whereas this process is defective in Aicda-deficient mice resulting in expansion of MOG specific germline encoded antibodies which might also have higher affinity. Although the total IgM Abs in mouse serum has been normalized, we cannot rule out that the starting anti-MOG specific antibodies are the same across different groups. Our data suggests that IgM anti-MOG antibodies in WT mice do not appear to be a major source of pathogenic antibodies since similar abs are even more represented in the Aicda KO group.
To better document the specificity, binding properties and the isotype of the anti-MOG autoantibodies, surface plasmon resonance (SPR) analysis was used. SPR uses a continuous flow technology to monitor the progress of biomolecular interactions in real time. An rhMOG specific binding response was detected in WT immunized animals and this signal could be inhibited with soluble rhMOG protein (Figure 5A). The majority of this binding was mediated by IgG1 class of anti-MOG antibodies (Figure 5B). Thus IgG1 anti-MOG high affinity auto-antibodies are highly abundant in WT mice that might be the major source of pathogenic antibodies.
Figure 5. .

Surface plasmon resonance (SPR) analysis of serum antibody binding to MOG. A. SPR sensorgrams of serum samples binding to sensor immobilized MOG. Serum from 8 mice were pooled for each of the following sample sets: Day 0 WT, Day 0 KO, Day 28 WT, and Day 28 KO. Start and stop injection times are indicated by arrows. Day 28 WT serum showed the highest binding response, and the majority of this binding signal could be competed with soluble MOG (2 uM) preincubated with the serum sample before injection. B. Detection of the major binding isotype in Day 28 WT serum. Subsequent injections of three anti-isotype antibodies were used to probe the MOG bound component. Start and stop injection times are indicated by arrows for each sample (Day 28 WT serum, anti-mIgM, anti-mIgG2a and anti-mIgG1). The data indicate that the majority of the binding signal is specific to IgG1. No background binding of anti-mIgG1 to MOG alone was detected (data not shown).
Figure 6. .
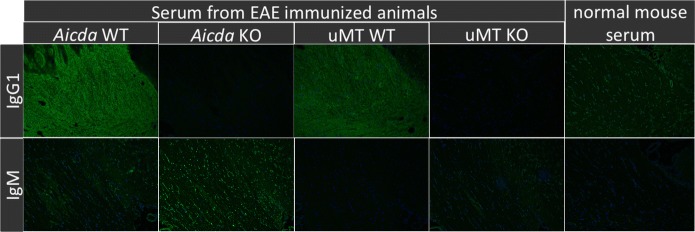
Indirect fluorescence for binding of serum antibody to normal brain tissue. Serum from EAE affected WT mice show diffuse, strong white matter associated signal consistent with the presence of MOG specific IgG1. By contrast, corresponding signal is absent using serum from MOG induced KO mice, or healthy controls. There is no difference of signal intensity or distribution for IgM binding to brain, regardless of MOG induction or genotype. Representative images are shown.
Myelin-specific IgG1 and not IgM autoantibodies strongly bind to myelin tissue in CNS
To better understand the in vivo properties of the IgG1 or IgM autoantibodies observed, we asked whether or not these antibodies could recognize and bind CNS tissues [12,37]. We performed indirect immunofluorescence assays using SCID brain tissue (to avoid the interference with endogenous immunoglobulin). Intriguingly, sera derived from EAE affected Aicda wt or uMT wt mice produced strong signal in the white matter when probed with anti-IgG1, indicating the presence of isotype switched autoantibodies targeting white matter myelin in these mice (Figure 6). By contrast, white matter signal was virtually absent in Aicda.ko or uMT.ko mice, reflecting the absence of IgG, including myelin-specific autoantibodies, in these animals.
Normal mouse serum (i.e., derived from healthy, not EAE induced mice) yielded weak background signal, clearly different from the signal achieved with EAE derived serum. When examined for binding of IgM, brain sections probed with sera from MOG induced mice had similar signal distribution and low-level intensity regardless of genotype and comparable to normal mouse serum, indicating that IgM do not contribute to the pathogenesis of EAE in this model. This data suggests that the high affinity IgG1 anti-myelin antibodies play an important role in EAE and the absence of these high affinity IgG1 anti-MOG antibodies in Aicda deficient mice may contribute to the resistance in disease induction as supported by other studies [9,12,13,45].
Discussion
We report that Aicda-deficient mice are protected against rhMOG-induced EAE in a manner similar to B cell-deficient (uMT) mice, supporting the hypothesis that high affinity isotype switched B cells and/or antibodies play a critical role in the EAE pathogenesis. uMT mice have a block in early B cell development and lack mature B cells due to a targeted disruption of the transmembrane IgM heavy chain [46]. In contrast, naïve B cells are present in the Aicda-deficient mice; AID-deficient B cells are only defective in SHM and CSR [20]. Given the SHM/CSR specific defects in the Aicda -deficient mice, our results have broad implications for the role of B cells in autoimmunity and therapeutic interventions aimed at different subsets of B cells.
Antigen specific mature B cells are important modulators of the immune response. First, B cells are highly sensitive APC compared to other professional APCs due to selective recognition of antigen through the B cell receptor and may be the first APCs to capture and present self-antigens to T cells [3]. Second, activated B cells also produce cytokines and membrane associated molecules that provide cognate help to modulate the inflammatory response [4,5]. Finally, secreted auto-antibodies may mediate tissue damage [6–13]. High affinity B cell receptor or auto-antibodies that are isotype switched may impact all aspects of B cell functions outlined above; however, our data is consistent with the hypothesis that the resistance in Aicda-deficient animals may be mediated primarily by the lack of high affinity pathogenic auto-antibodies.
Our in vitro T cell functional studies suggest that the protection observed in Aicda-deficient animals is not due to a defect in T cell activation or effector function. Thus there is not a defect in T cell priming in the Aicda deficient animals. In this context, the BCR-mediated antigen recognition or processing does not appear to profoundly affect T cell function however further studies in different anatomical locations are needed to rule this possibility out. For example, access of neoantigen is limited in the CNS and autoantibodies could facilitate B cell APC function in CNS via opsonization of the neoantigens [47].
The pathogenic role of autoantibodies in rhMOG induced EAE has been established [9,12,13,45]. These auto-antibodies may enhance demyelination [6–9,12,13,45], affect trafficking of encephalitogenic T cells into the CNS [45,47] or impact immune regulation [4,48]. Although IgM anti-MOG antibodies are detected in WT and Aicda KO mice, these antibodies in both groups do not appear to be of high affinity based on our MOG antigen binding studies and immunofluorescence on brain tissue. Our competition binding measurements show that a major portion of the high affinity anti-MOG antibodies is IgG1 that are completely absent in the Aicda KO mice.
The WT IgG1 antibodies are generated from antigen experienced activated B cells and therefore usually selected as high affinity antibodies. Accordingly, we show that white matter binding IgG1 can be demonstrated by indirect fluorescence in the serum of EAE affected WT animals, whereas such antibodies are not present in normal mouse serum or serum of MOG immunized Aicda KO mice. This is consistent with rhMOG induced EAE where autoantibodies can exacerbate disease [9,45].
An interesting observation from this study is that IgM anti-MOG antibodies are even more abundant in Aicda KO mice than WT. The exact role of the unmutated pool of antibodies is unclear, but others have proposed that unmutated germ-line encoded IgM antibodies might play a protective role by clearing apoptotic cells and/or antigens from injury sites to minimize inflammation or autoimmunity [49–55]. It is important to note that in addition to its use in immune deficiencies, intravenous immunoglobulin therapy is used in the treatment of several inflammatory conditions, including Kawasaki disease, dermatomyositis and antineutrophil cytoplasmic antibody (ANCA)-positive vasculitis [56].
In experimental models, the therapeutic benefit of pooled intravenous IgM for immune-mediated inflammatory diseases such as experimental autoimmune uveitis, experimental multiple sclerosis and experimental myas thenia gravis has been established [55]. In addition, transgenic mice engineered to produce high titers of auto-antibodies against MOG have an exacerbated form of EAE [45,57] and MS patients have a higher prevalence of IgG switched, but not IgM isotype, anti-MOG antibodies than healthy controls [17]. Additional studies are needed to address how precisely B cell function is altered in Aicda KO mice in the context of the autoimmunity experimental models.
The contribution of B cells in EAE model has been controversial and part of this is due to the nature of the antigen that is used to invoke the autoimmune response. Properly folded recombinant human MOG assumes a three dimensional conformation and contains diverse epitopes that may facilitate B cell specific activation [58]. In contrast, MOG peptide 35–55 mimics a linear epitope that can directly bind to MHC class II on any APC to induce EAE [59]. Correspondingly, B cells do not appear to play a major role in induction or maintenance of MOG peptide 35–55 induced EAE, as both B cell deficient (uMT) [60] and Aicda-deficient mice were equally susceptible in this model [61]. Therefore the selection of the appropriate EAE model is important when interrogating the contribution of various cell types such as B or T lymphocytes.
Although considerable work needs to be performed to better understand the mechanisms by which B cells influence autoimmune diseases, clinical data is suggesting an important role for B cells in human patients (reviewed in [2,62]). The elucidation of how AID dependent processes contribute to MS pathogenesis ultimately will help with the design of better treatments for MS and other inflammatory diseases by investing on druggable pathways or molecules that modulate relevant B cell functions.
Acknowledgement
We thank Robert Schwingendorf, Ben Grellman and Vida Asghari for genotyping of animals and managing the mice colonies.
Declaration of interest: All the authors are the employees of Genentech, a wholly owned subsidiary of Roche. The authors declare no competing financial interests. The authors are responsible for the content and the writing of this paper.
References
- 1.Sospedra M., Martin R. Immunology of multiple sclerosis. Annual Rev. Immunol. 2005;23:683–747. doi: 10.1146/annurev.immunol.23.021704.115707. [DOI] [PubMed] [Google Scholar]
- 2.Townsend M. J., Monroe J. G., Chan A. C. B-cell targeted therapies in human autoimmune diseases: An updated perspective. Immunol. Rev. 2010;237:264–283. doi: 10.1111/j.1600-065X.2010.00945.x. [DOI] [PubMed] [Google Scholar]
- 3.Lanzavecchia A. Receptor-mediated antigen uptake and its effect on antigen presentation to class II-restricted T lymphocytes. Ann. Rev. Immunol. 1990;8:773–793. doi: 10.1146/annurev.iy.08.040190.004013. [DOI] [PubMed] [Google Scholar]
- 4.Duddy M. E., Alter A., Bar-Or A. Distinct profiles of human B cell effector cytokines: A role in immune regulation? J. Immunol. 2004;172:3422–3427. doi: 10.4049/jimmunol.172.6.3422. [DOI] [PubMed] [Google Scholar]
- 5.Matsushita T., Yanaba K., Bouaziz J. D., Fujimoto M., Tedder T. F. Regulatory B cells inhibit EAE initiation in mice while other B cells promote disease progression. J. Clin. Invest. 2008;118:3420–3430. doi: 10.1172/JCI36030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Genain C. P., Nguyen M. H., Letvin N. L., et al. Antibody facilitation of multiple sclerosis-like lesions in a nonhuman primate. J. Clin. Invest. 1995;96:2966–2974. doi: 10.1172/JCI118368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Linington C., Bradl M., Lassmann H., Brunner C., Vass K. Augmentation of demyelination in rat acute allergic encephalomyelitis by circulating mouse monoclonal antibodies directed against a myelin/oligodendrocyte glycoprotein. Amer. J. Pathol. 1988;130:443–454. [PMC free article] [PubMed] [Google Scholar]
- 8.Kerlero de Rosbo N., Honegger P., Lassmann H., Matthieu J. M. Demyelination induced in aggregating brain cell cultures by a monoclonal antibody against myelin/oligodendrocyte glycoprotein. J. Neurochem. 1990;55:583–587. doi: 10.1111/j.1471-4159.1990.tb04173.x. [DOI] [PubMed] [Google Scholar]
- 9.Lyons J. A., Ramsbottom M. J., Cross A. H. Critical role of antigen-specific antibody in experimental autoimmune encephalomyelitis induced by recombinant myelin oligodendrocyte glycoprotein. Euro. J. Immunol. 2002;32:1905–1913. doi: 10.1002/1521-4141(200207)32:7<1905::AID-IMMU1905>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 10.Marta C. B., Oliver A. R., Sweet R. A., Pfeiffer S. E., Ruddle N. H. Pathogenic myelin oligodendrocyte glycoprotein antibodies recognize glycosylated epitopes and perturb oligodendrocyte physiology. Proc. Natl. Acad. Sci. USA. 2005;102:13992–13997. doi: 10.1073/pnas.0504979102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mason L. J., Ravirajan C. T., Latchman D. S., Isenberg D. A. A human anti-dsDNA monoclonal antibody caused hyaline thrombi formation in kidneys of ‘leaky’ SCID mice. Clin. Exper. Immunol. 2001;126:137–142. doi: 10.1046/j.1365-2249.2001.01651.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuerten S., Pauly R., Rottlaender A., et al. Myelin-reactive antibodies mediate the pathology of MBP-PLP fusion protein MP4-induced EAE. Clin. Immunol. 2011;140:54–62. doi: 10.1016/j.clim.2011.03.009. [DOI] [PubMed] [Google Scholar]
- 13.Oliver A. R., Lyon G. M., Ruddle N. H. Rat and human myelin oligodendrocyte glycoproteins induce experimental autoimmune encephalomyelitis by different mechanisms in C57BL/6 mice. J. Immunol. 2003;171:462–468. doi: 10.4049/jimmunol.171.1.462. [DOI] [PubMed] [Google Scholar]
- 14.Avasarala J. R., Cross A. H., Trotter J. L. Oligoclonal band number as a marker for prognosis in multiple sclerosis. Arch. Neurol. 2001;58:2044–2045. doi: 10.1001/archneur.58.12.2044. [DOI] [PubMed] [Google Scholar]
- 15.Joseph F. G., Hirst C. L., Pickersgill T. P., et al. CSF oligoclonal band status informs prognosis in multiple sclerosis: a case control study of 100 patients. J. Neurol. Neurosurg. Psych. 2009;80:292–296. doi: 10.1136/jnnp.2008.150896. [DOI] [PubMed] [Google Scholar]
- 16.Owens G. P., Bennett J. L., Lassmann H., et al. Antibodies produced by clonally expanded plasma cells in multiple sclerosis cerebrospinal fluid. Ann. Neurol. 2009;65:639–649. doi: 10.1002/ana.21641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou D., Srivastava R., Nessler S., et al. Identification of a pathogenic antibody response to native myelin oligodendrocyte glycoprotein in multiple sclerosis. Proc. Natl. Acad. Sci. USA. 2006;103:19057–19062. doi: 10.1073/pnas.0607242103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.O'Connor K. C., Appel H., Bregoli L., et al. Antibodies from inflamed central nervous system tissue recognize myelin oligodendrocyte glycoprotein. J. Immunol. 2005;175:1974–1982. doi: 10.4049/jimmunol.175.3.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hauser S. L., Waubant E., Arnold D. L., et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. New Engl. J. Med. 2008;358:676–688. doi: 10.1056/NEJMoa0706383. [DOI] [PubMed] [Google Scholar]
- 20.Muramatsu M., Kinoshita K., Fagarasan S., et al. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102:553–563. doi: 10.1016/s0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- 21.Hackney J. A., Misaghi S., Senger K., et al. DNA targets of AID evolutionary link between antibody somatic hypermutation and class switch recombination. Adv. Immunol. 2009;101:163–189. doi: 10.1016/S0065-2776(08)01005-5. [DOI] [PubMed] [Google Scholar]
- 22.Humby F., Bombardieri M., Manzo A., et al. Ectopic lymphoid structures support ongoing production of class-switched autoantibodies in rheumatoid synovium. PLoS Med. 2009;6:e1. doi: 10.1371/journal.pmed.0060001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Payne S. C., Chen P. G., Borish L. Local class switching in nonallergic rhinitis. Curr. Opin. Otolaryngol. Head Neck Surg. 2011;19:193–198. doi: 10.1097/MOO.0b013e328345005c. [DOI] [PubMed] [Google Scholar]
- 24.Le Pottier L., Devauchelle V., Fautrel A., et al. Ectopic germinal centers are rare in Sjogren's syndrome salivary glands and do not exclude autoreactive B cells. J. Immunol. 2009;182:3540–3547. doi: 10.4049/jimmunol.0803588. [DOI] [PubMed] [Google Scholar]
- 25.Gu X., Shivarov V., Strout M. P. The role of activation-induced cytidine deaminase in lymphomagenesis. Curr. Opin. Hematol. 2012;19:292–298. doi: 10.1097/MOH.0b013e328353da3a. [DOI] [PubMed] [Google Scholar]
- 26.Jiang C., Foley J., Clayton N., et al. Abrogation of lupus nephritis in activation-induced deaminase-deficient MRL/lpr mice. J. Immunol. 2007;178:7422–7431. doi: 10.4049/jimmunol.178.11.7422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jiang C., Zhao M. L., Diaz M. Activation-induced deaminase heterozygous MRL/lpr mice are delayed in the production of high-affinity pathogenic antibodies and in the development of lupus nephritis. Immunology. 2009;126:102–113. doi: 10.1111/j.1365-2567.2008.02882.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hsu H. C., Yang P., Wang J., et al. Interleukin 17-producing T helper cells and interleukin 17 orchestrate autoreactive germinal center development in autoimmune BXD2 mice. Nature Immunol. 2008;9:166–175. doi: 10.1038/ni1552. [DOI] [PubMed] [Google Scholar]
- 29.Hsu H. C., Wu Y., Yang P., et al. Overexpression of activation-induced cytidine deaminase in B cells is associated with production of highly pathogenic autoantibodies. J. Immunol. 2007;178:5357–5365. doi: 10.4049/jimmunol.178.8.5357. [DOI] [PubMed] [Google Scholar]
- 30.Weissert R. Experimental Autoimmune Encephalomyelitis—Models, Disease Biology and Experimental Therapy. InTech. 2012 [Google Scholar]
- 31.Warming S., Rachel R. A., Jenkins N. A., Copeland N. G. Zfp423 is required for normal cerebellar development. Molecular and cellular biology. 2006;26:6913–6922. doi: 10.1128/MCB.02255-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu P., Jenkins N. A., Copeland N. G. A highly efficient recombineering-based method for generating conditional knockout mutations. Genome Res. 2003;13:476–484. doi: 10.1101/gr.749203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee E. C., Yu D., Martinez de Velasco J., et al. A highly efficient Escherichia coli-based chromosome engineering system adapted for recombinogenic targeting and subcloning of BAC DNA. Genomics. 2001;73:56–65. doi: 10.1006/geno.2000.6451. [DOI] [PubMed] [Google Scholar]
- 34.Warming S., Costantino N., Court D. L., Jenkins N. A., Copeland N. G. Simple and highly efficient BAC recombineering using galK selection. Nucl. Acids Res. 2005;33:e36. doi: 10.1093/nar/gni035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bettadapura J., Menon K. K., Moritz S., Liu J., Bernard C. C. Expression, purification, and encephalitogenicity of recombinant human myelin oligodendrocyte glycoprotein. J. Neurochem. 1998;70:1593–1599. doi: 10.1046/j.1471-4159.1998.70041593.x. [DOI] [PubMed] [Google Scholar]
- 36.Linares D., Echevarria I., Mana P. Single-step purification and refolding of recombinant mouse and human myelin oligodendrocyte glycoprotein and induction of EAE in mice. Protein Express. Purif. 2004;34:249–256. doi: 10.1016/j.pep.2003.11.016. [DOI] [PubMed] [Google Scholar]
- 37.DeVoss J., Hou Y., Johannes K., et al. Spontaneous autoimmunity prevented by thymic expression of a single self-antigen. J. Exper. Med. 2006;203:2727–2735. doi: 10.1084/jem.20061864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Misaghi S., Garris C. S., Sun Y., et al. Increased targeting of donor switch region and IgE in Sγ1-deficient B cells. J. Immunol. 2010;185:166–173. doi: 10.4049/jimmunol.1000515. [DOI] [PubMed] [Google Scholar]
- 39.Stefferl A., Brehm U., Storch M., et al. Myelin oligodendrocyte glycoprotein induces experimental autoimmune encephalomyelitis in the “resistant” Brown Norway rat: disease susceptibility is determined by MHC and MHC-linked effects on the B cell response. J. Immunol. 1999;163:40–49. [PubMed] [Google Scholar]
- 40.Clements C. S., Reid H. H., Beddoe T., et al. The crystal structure of myelin oligodendrocyte glycoprotein, a key autoantigen in multiple sclerosis. Proc. Natl. Acad. Sci. USA. 2003;100:11059–11064. doi: 10.1073/pnas.1833158100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Breithaupt C., Schubart A., Zander H., et al. Structural insights into the antigenicity of myelin oligodendrocyte glycoprotein. Proc. Natl. Acad. Sci. USA. 2003;100:9446–9451. doi: 10.1073/pnas.1133443100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lyons J. A., San M., Happ M. P., Cross A. H. B cells are critical to induction of experimental allergic encephalomyelitis by protein but not by a short encephalitogenic peptide. Euro. J. Immunol. 1999;29:3432–3439. doi: 10.1002/(SICI)1521-4141(199911)29:11<3432::AID-IMMU3432>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 43.Svensson L., Abdul-Majid K. B., Bauer J., et al. A comparative analysis of B cell-mediated myelin oligodendrocyte glycoprotein-experimental autoimmune encephalomyelitis pathogenesis in B cell-deficient mice reveals an effect on demyelination. Euro. J. Immunol. 2002;32:1939–1946. doi: 10.1002/1521-4141(200207)32:7<1939::AID-IMMU1939>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 44.Komiyama Y., Nakae S., Matsuki T., et al. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J. Immunol. 2006;177:566–573. doi: 10.4049/jimmunol.177.1.566. [DOI] [PubMed] [Google Scholar]
- 45.Litzenburger T., Fassler R., Bauer J., et al. B lymphocytes producing demyelinating autoantibodies: development and function in gene-targeted transgenic mice. J. Exper. Med. 1998;188:169–180. doi: 10.1084/jem.188.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kitamura D., Roes J., Kuhn R., Rajewsky K. A B cell-deficient mouse by targeted disruption of the membrane exon of the immunoglobulin mu chain gene. Nature. 1991;350:423–426. doi: 10.1038/350423a0. [DOI] [PubMed] [Google Scholar]
- 47.Trotter J., DeJong L. J., Smith M. E. Opsonization with antimyelin antibody increases the uptake and intracellular metabolism of myelin in inflammatory macrophages. J. Neurochem. 1986;47:779–789. doi: 10.1111/j.1471-4159.1986.tb00679.x. [DOI] [PubMed] [Google Scholar]
- 48.Matsushita S., Suzuki K., Murayama M., et al. Serotonin transporter regulatory region polymorphism is associated with anorexia nervosa. American journal of medical genetics. Pt. B, Neuropsych. Genet. 2004;128B:114–117. doi: 10.1002/ajmg.b.30022. [DOI] [PubMed] [Google Scholar]
- 49.Gaipl U. S., Kuhn A., Sheriff A., et al. Clearance of apoptotic cells in human SLE. Curr. Direct. Autoimmun. 2006;9:173–187. doi: 10.1159/000090781. [DOI] [PubMed] [Google Scholar]
- 50.Ehrenstein M. R., Cook H. T., Neuberger M. S. Deficiency in serum immunoglobulin (Ig)M predisposes to development of IgG autoantibodies. J. Exper. Med. 2000;191:1253–1258. doi: 10.1084/jem.191.7.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ogden C. A., Kowalewski R., Peng Y., Montenegro V., Elkon K. B. IGM is required for efficient complement mediated phagocytosis of apoptotic cells in vivo. Autoimmunity. 2005;38:259–264. doi: 10.1080/08916930500124452. [DOI] [PubMed] [Google Scholar]
- 52.Binder C. J., Silverman G. J. Natural antibodies and the autoimmunity of atherosclerosis. Springer Semin. Immunopathol. 2005;26:385–404. doi: 10.1007/s00281-004-0185-z. [DOI] [PubMed] [Google Scholar]
- 53.Werwitzke S., Trick D., Kamino K., et al. Inhibition of lupus disease by anti-double-stranded DNA antibodies of the IgM isotype in the (NZB × NZW)F1 mouse. Arthritis Rheumat. 2005;52:3629–3638. doi: 10.1002/art.21379. [DOI] [PubMed] [Google Scholar]
- 54.Boes M., Schmidt T., Linkemann K., et al. Accelerated development of IgG autoantibodies and autoimmune disease in the absence of secreted IgM. Proc. Natl. Acad. Sci. USA. 2000;97:1184–1189. doi: 10.1073/pnas.97.3.1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hurez V., Kazatchkine M. D., Vassilev T., et al. Pooled normal human polyspecific IgM contains neutralizing anti-idiotypes to IgG autoantibodies of autoimmune patients and protects from experimental autoimmune disease. Blood. 1997;90:4004–4013. [PubMed] [Google Scholar]
- 56.Bayry J., Negi V. S., Kaveri S. V. Intravenous immunoglobulin therapy in rheumatic diseases. Nature Rev. Rheumatol. 2011;7:349–359. doi: 10.1038/nrrheum.2011.61. [DOI] [PubMed] [Google Scholar]
- 57.Du C., Sriram S. Increased severity of experimental allergic encephalomyelitis in lyn-/- mice in the absence of elevated proinflammatory cytokine response in the central nervous system. J. Immunol. 2002;168:3105–3112. doi: 10.4049/jimmunol.168.6.3105. [DOI] [PubMed] [Google Scholar]
- 58.de Graaf K. L., Albert M., Weissert R. Autoantigen conformation influences both B- and T-cell responses and encephalitogenicity. J. Biol. Chem. 2012;287:17206–17213. doi: 10.1074/jbc.M111.304246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Slavin A. J., Soos J. M., Stuve O., et al. Requirement for endocytic antigen processing and influence of invariant chain and H-2M deficiencies in CNS autoimmunity. J. Clin. Invest. 2001;108:1133–1139. doi: 10.1172/JCI13360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hjelmstrom P., Juedes A. E., Fjell J., Ruddle N. H. B-cell-deficient mice develop experimental allergic encephalomyelitis with demyelination after myelin oligodendrocyte glycoprotein sensitization. J. Immunol. 1998;161:4480–4483. [PubMed] [Google Scholar]
- 61.Sekiguchi Y., Ichikawa M., Takamoto M., et al. Antibodies to myelin oligodendrocyte glycoprotein are not involved in the severity of chronic non-remitting experimental autoimmune encephalomyelitis. Immunol. Lett. 2009;122:145–149. doi: 10.1016/j.imlet.2008.08.009. [DOI] [PubMed] [Google Scholar]
- 62.Martin F., Chan A. C. B cell immunobiology in disease: evolving concepts from the clinic. Ann. Rev. Immunol. 2006;24:467–496. doi: 10.1146/annurev.immunol.24.021605.090517. [DOI] [PubMed] [Google Scholar]