

Abstract
Schwannomas are tumors formed by proliferation of dedifferentiated Schwann cells. Patients with neurofibromatosis 2 (NF2) and schwannomatosis develop multiple schwannomas in peripheral and cranial nerves. Although benign, these tumors can cause extreme pain and compromise sensory/motor functions, including hearing and vision. At present, surgical resection is the main treatment modality, but it can be problematic because of tumor inaccessibility and risk of nerve damage. We have explored gene therapy for schwannomas, using a model in which immortalized human NF2 schwannoma cells expressing a fluorescent protein and luciferase are implanted in the sciatic nerve of nude mice. Direct injection of an adeno-associated virus (AAV) serotype 1 vector encoding caspase-1 (ICE) under the Schwann-cell specific promoter, P0, leads to regression of these tumors with essentially no vector-mediated neuropathology, and no changes in sensory or motor function. In a related NF2 xenograft model designed to cause measurable pain behavior, the same gene therapy leads to tumor regression and concordant resolution of tumor-associated pain. This AAV1-P0-ICE vector holds promise for clinical treatment of schwannomas by direct intratumoral injection to achieve reduction in tumor size and normalization of neuronal function.
Prabhakar and colleagues demonstrate that direct injection of an adeno-associated virus (AAV) serotype 1 vector encoding caspase-1 leads to regression of Schwann cell tumors in a mouse model. Importantly, this approach does not cause vector-mediated neuropathology or changes in sensory motor function.
Introduction
Multiple schwannomas in peripheral and cranial nerves are the hallmark of neurofibromatosis 2 (NF2) and schwannomatosis, two forms of neurofibromatoses, classified as neurocutaneous syndromes, with incidences of about 1 in 32,000 and 1 in 1,000,000, respectively (Antinheimo et al., 2000; Baser et al., 2006). Schwannomas are benign tumors composed of neoplastic dedifferentiated Schwann cells. Although typically nonmalignant and slow growing, these tumors can have devastating consequences for patients. Schwannomas in NF2 are frequently associated with neurological deficits, such as paresthesias, weakness, or hearing loss, and similar tumors in schwannomatosis often cause excruciating pain (Huang et al., 2004; Lu-Emerson and Plotkin, 2009). Some schwannomas become very large, causing compression of adjacent organs or structures, and can lead to paralysis or death due to progressive spinal cord or brainstem compression. The standard of care for patients with NF2 and schwannomatosis is surgical resection or radiosurgery of symptomatic tumors to reduce tumor size. Unlike in the case of sporadic schwannomas, in which typically only a single tumor is present and surgery is generally an efficacious treatment strategy as long as the lesion is accessible for resection (Lu-Emerson and Plotkin, 2009), in schwannomatosis and NF2, which present with multiple tumors, resection is confounded by both the inaccessibility of many tumors and by risk of nerve damage, including major motor dysfunction, significant sensory loss (including deafness in the case of NF2 vestibular schwannomas), and neuropathic pain. Thus, for most individuals there is substantial morbidity associated with schwannomas in both NF2 and schwannomatosis, as well as with the current therapies. This suffering and debility, in combination with the paucity of therapeutic options, makes the treatment of schwannomas a major unmet medical need.
The underlying molecular abnormality in NF2 is a germline mutation of the NF2 gene. Somatic loss of the normal remaining NF2 allele in Schwann cells leads to deregulated growth of neoplastic Schwann cells with schwannoma formation (Rouleau et al., 1993). The timing of loss of the second wild-type allele may occur during development, as Schwann cells move out along axons and begin myelination, or in response to injury when Schwann cells dedifferentiate and commence proliferation (Jessen and Mirsky, 2005; McClatchey and Giovannini, 2005). In schwannomatosis some patients have a germline mutation in the SMARCB/IN11 gene with a second hit in Schwann cells leading to schwannoma formation (Hulsebos et al., 2007); in addition, the majority of these tumors harbor additional mutations in the NF2 gene (Jacoby et al., 1997; Hadfield et al., 2008).
In initial treatment paradigms, we used a mouse schwannoma model in which an immortalized cell line derived from human NF2 schwannoma cells (HEI-193) (Hung et al., 2002) was genetically modified to express firefly luciferase (Fluc) and the fluorescent protein, mCherry (designated HEI-193FC), and implanted subcutaneously in nude mice. Using this model, we demonstrated that intratumoral injection of a herpes simplex virus-1 (HSV-1) amplicon vector expressing caspase-1 (interleukin-β-converting enzyme; ICE) under the Schwann cell-specific P0 promoter led to tumor regression (Prabhakar et al., 2010). This gene therapy approach had two limitations; the subcutaneous location used was not clinically relevant and the HSV amplicon vector employed has never been used in clinical trials.
In the present study we used a more biologically relevant tumor model in which these human HEI-193FC schwannoma cells are implanted into the sciatic nerve of nude mice (Saydam et al., 2011). We explored the use of recombinant adeno-associated viral (AAV) vectors, which are well established as safe in clinical trials, and found that among the serotypes tested by direct injection into the mouse sciatic nerve (AAV1, AAVrh8, AAVrh10), AAV1 proved to be the most efficient in transducing Schwann cells (M.S.E. and G.J.B., unpublished data). Here, we demonstrate that direct injection of an AAV1-P0-ICE vector into intrasciatic distal schwannomas both prevented tumor development and led to regression of well-established tumors, as assessed by in vivo bioluminescence imaging and correlative histopathology. Tumors injected with a control vector, AAV1-P0-GFP, continued to grow. In a proximal sciatic nerve implantation model, designed to cause painlike behavior as assessed by plantar von Frey withdrawal threshold (i.e., mechanical sensitization), intratumoral AAV1-P0-ICE injection alleviated pain sensitivity in parallel with regression of tumors. Neuropathological evaluation of Epon and paraffin sections of sciatic nerves of both nude and immunocompetent mice injected with AAV1-P0-ICE showed no evidence of inflammation or axonal degeneration, and only rare macrophages, consistent with the absence of behavioral changes in pain sensitivity or motor skills in these vector-injected animals.
Materials and Methods
Cell culture
The HEI-193 human schwannoma cell line (from D.J. Lim, House Ear Institute, Los Angeles, CA) was established from a schwannoma in a patient with NF2, immortalized with human papillomavirus E6/E7 genes (Hung et al., 2002) and maintained in Dulbecco's modified Eagle's medium (DMEM) with 2 μM forskolin (Calbiochem, San Diego, CA), recombinant glial growth factor (14 ng/ml; Sigma-Aldrich, St. Louis, MO), and G418 disulfate salt (50 μg/ml; Sigma-Aldrich). To obtain the HEI-193FC cell line, HEI-193 cells were infected with lentivirus encoding Fluc and mCherry (Prabhakar et al., 2010). Human neuroblastoma cell line SH-SY5Y (American Type Culture Collection [ATCC], Manassas, VA) was grown in DMEM–F12 (1:1) (GIBCO-BRL, Rockville, MD). HEK-293T human embryonic kidney cells (from M. Calos, Stanford University, Stanford, CA) were grown in DMEM. For all cell types, growth media were supplemented with 10% fetal bovine serum (FBS; Sigma-Aldrich) and 1% penicillin–streptomycin (Cellgro, Herndon, VA) and cells were maintained at 37°C in a humidified atmosphere of 5% CO2 and 95% air.
AAV vector design and packaging
AAV vector plasmids dsAAV-P0-ICE and dsAAV-P0-GFP were derived from the plasmid dsAAV-CBA-GFP-BGHpA (Sena-Esteves laboratory). This plasmid carries two AAV2 inverted terminal repeat (ITR) elements, one wild type and one in which the terminal resolution site was deleted, as described (McCarty et al., 2003), generating a vector that is packaged as a double-stranded molecule. The dsAAV-P0-ICE plasmid was generated by replacing the CBA-GFP cassette in the parent plasmid with the PCR-amplified rat P0 promoter (1.1 kb; for full details of the P0 promoter see Brown and Lemke, 1997) and cDNA, using the plasmid HSV-P0-ICE (Prabhakar et al., 2010) as a template and the following primers: P0-1, AAAGGTACCacgagcattctcgaactctccaaa; P0-2, AAAACTAGTtctgcagaattcgatatcaagcttgg; mCaspase-1.1, aaaactagtgccaccatggctgtgagggcaaagaggaaG; mCaspase-1.2, AAAGCGGCCGCttaatgtcccgggaagaggtagAAA. The dsAAV-P0-GFP plasmid was generated by replacing the chicken β-actin (CBA) promoter in the parent plasmid with the PCR-amplified rat P0 promoter. Both AAV vectors carry the bovine growth hormone polyadenylation signal. The identity of all PCR-amplified sequences was confirmed by sequencing.
AAV1 serotype vectors were produced by transient cotransfection of 293T cells by calcium phosphate precipitation of vector plasmid (dsAAV-P0-ICE or dsAAV-P0-GFP), adenoviral helper plasmid pFΔ6, and a plasmid encoding the AAV1 cap gene (pXR1), as previously described (Broekman et al., 2006). Briefly, AAV vectors were purified by iodixanol gradient centrifugation followed by column chromatography with HiTrap Q anion-exchange columns (GE Healthcare, Piscataway, NJ). The virus-containing fractions were concentrated with Centricon 100-kDa molecular weight cutoff (MWCO) centrifugal devices (Millipore, Billerica, MA) and the titer (genome copies [GC]/ml) was determined by real-time PCR amplification with primers and probe specific for the bovine growth hormone polyadenylation signal.
Luminescence cell viability assay
The CellTiter-Glo luminescent cell viability assay (Promega, Madison, WI) was used to determine the number of viable cells in culture on the basis of quantitation of ATP levels, an indicator of metabolically active cells. The system detects as few as 15 cells per well 10 min after adding reagent and mixing, resulting in cell lysis and the generation of a luminescent signal proportional to the amount of ATP present. Ten thousand HEI-193, 293T, or SH-SY5Y cells were plated into the wells of 96-well plates and infected in triplicate with the AAV1-P0-ICE or AAV1-P0-GFP vector at a multiplicity of infection (MOI) of 10,000 GC/cell to achieve >90% infection. After 72 hr, medium was aspirated off and cells were incubated in 100 μl of fresh medium containing 25 μl of CellTiter-Glo reagent for 10 min at room temperature. The luminescence was measured with a luminometer (Dynex Technologies, Chantilly, VA).
Animals
All animal experimentation was approved by and conducted under the oversight of the Massachusetts General Hospital (Boston, MA) Institutional Animal Care and Use Committee. Animals, nu/nu and C57BL/6 mice, were kept on a 12:12 light-to-dark cycle with ad libitum access to food and water. Animals were checked daily to evaluate health.
Generation of tumors and vector injection
Sciatic nerve schwannomas were generated by direct injection of schwannoma cells into the left sciatic nerve of isoflurane-anesthetized mice, as described (Saydam et al., 2011). Specifically, cells were implanted approximately 4 mm distal to the sciatic notch at a point midway between the sciatic notch and the trifurcation of the sciatic nerve into the common peroneal, tibial, and sural branches. For all experiments other than the experiment shown in Fig. 7, HEI-193FC cells were trypsinized and rinsed, and 30,000 cells in a volume of 1 μl of culture medium were injected into the distal sciatic nerve of athymic nude mice (nu/nu, 5-week-old females; National Cancer Institute [NCI]), using a glass micropipette and a gas-powered microinjector (IM-300; Narishige, Tokyo, Japan). For the data shown in Fig. 7, a similar implantation procedure was performed, differing only in that cells (60,000 in 1 μl) were implanted more proximally in the sciatic nerve. Tumor growth was monitored by in vivo bioluminescence imaging at weekly intervals, using the CMIR Image program (an image display and analysis suite developed in the Interactive Data Language [IDL; Research Systems/Exelis Visual Information Solutions, Boulder, CO]) as described (Prabhakar et al., 2010). Briefly, mice were injected intraperitoneally with the Fluc substrate d-luciferin, and, 5 min later, signal was acquired with a high-efficiency IVIS Spectrum (Caliper Life Sciences, Hopkinton, MA) with an XGI-8 gas anesthesia system (Caliper Life Sciences) for Fig. 3A and a cryogenically cooled, high-efficiency charge-coupled device (CCD) camera system (Roper Scientific, Trenton, NJ) for Fig. 3B.
FIG. 7.

GFP expression after injection of AAV1-P0-GFP or AAV1-CBA-GFP into the sciatic nerve of young (postnatal day 21 [P21]) and adult (14 week) C57BL/6 mice. Naive sciatic nerves of immune-competent (C57BL/6) mice (n=3/age group/vector, 12 total) were injected with AAV1-P0-GFP or AAV1-CBA-GFP vector (1013 GC in 1 μl) in P21 and 14-week-old mice. After 10 days, the (A) sciatic nerves (both longitudinal and transverse sections shown) and (B) dorsal root ganglia (DRG) were collected, frozen serial sections were obtained, and GFP immunofluorescence was determined. Representative images are shown. Original magnification: ×10.
FIG. 3.

Monitoring schwannoma volumes in vivo by bioluminescence imaging. Thirty thousand HEI-193FC cells were implanted into the distal sciatic nerve of nude mice and tumor volume was monitored at weekly intervals by in vivo bioluminescence imaging. At the time points indicated [arrows; weeks 4 and 5 (A) and weeks 5 and 6 (B)] after tumors were established, they were injected twice (i.e., two separate injections 1 week apart) with either AAV1-P0-ICE or AAV1-P0-GFP vector (1×1010 GC in 1 μl per injection). Graphs in (A) and (B) were obtained with different bioluminescence imaging systems, and thus the luciferase signal (ordinate data) is different in scale. Data points are shown as mean values±SEM (n=6 per group for each experiment). *Significant difference (by post-hoc analysis) between AAV1-P0-ICE and control groups at p<0.002 (A) and p<0.001 (B). In both (A) and (B), one in the control group out of six tumors regressed spontaneously, and all animals were taken into consideration in data analysis.
After 4 weeks, tumors that grew progressively were treated with AAV1-P0-ICE or AAV1-P0-GFP. The schwannomas were injected twice, 1 week apart, with 1010 vector GC in 1 μl, using either AAV1-P0-ICE or AAV1-P0-GFP (over approximately 10 sec), under direct visualization as described for injection of tumor cells (Saydam et al., 2011), targeting the enlarged part of the nerve where tumor cells were implanted. Volumetric changes in tumors were tracked by in vivo bioluminescence imaging out to 18–19 weeks.
Histology
After treatment with vectors, the animals were killed with isoflurane (3%) followed by decapitation. Sciatic nerves were taken out and fixed in 10% formalin for hematoxylin and eosin (H&E) staining (Messerli et al., 2002). The sciatic nerve was removed; one-third of each of these biopsies was fixed in glutaraldehyde for Epon embedding, one-third was fixed in 10% buffered formalin for paraffin embedding, and the remaining third was fresh-frozen at −80°C for PCR analysis of biodistribution of vector. Myelin integrity of sciatic nerves injected with AAV1-P0-ICE vector was assessed in toluidine blue-stained ultrathin sections prepared as follows: nerves were placed into a modified Karnovsky fixative (2.5% glutaraldehyde, 2.0% paraformaldehyde, and 0.025 calcium chloride in 0.1 M sodium cacodylate buffer), pH 7.4, and fixed overnight at 4°C. Further processing was done with a Leica Lynx automatic tissue processor. Briefly, the nerves were postfixed in aqueous 1.3% osmium tetroxide, dehydrated in graded ethanol solutions, en bloc stained with 3.0% uranyl acetate in the 70% ethanol step, infiltrated with epoxy and propylene oxide mixtures, and embedded in pure epoxy. The nerves were flat embedded (to maintain orientation) and allowed to polymerize overnight at 60°C. One-micron sections were cut with a DuPont-Sorvall MT-1 ultramicrotome and glass knives. The sections were then stained on a warm hotplate with a solution of 0.5% toluidine blue in 0.5% sodium borate, rinsed with water, dried, and coverslipped (Eichler et al., 2009). The formalin-fixed tissue was processed and embedded in paraffin. Sections were stained with H&E in accordance with routine protocols.
Behavioral analysis
The mice were tested by the von Frey method for pain/mechanical sensitivity and by rotarod for gross motor function according to published methods (Agarwal et al., 2007; Kirschbaum et al., 2009). Both nu/nu and C57BL/6 mice were used for the behavioral experiments. One microliter (1013 GC/ml) of the vector AAV1-P0-ICE, 1 μl of AAV1-P0-GFP, or 1 μl of phosphate-buffered saline (PBS) was injected into the sciatic nerve twice, with injections spaced 1 week apart (n=12 per group). All animals were allowed to habituate to the behavioral apparatus for 1 week before testing for baseline. Three baseline measurements on three separate days preceded the first injection. Mice were then tested the day after each injection and twice per week for 8 weeks.
Mechanical sensitivity of the hind paw was measured by determining withdrawal thresholds assessed with von Frey filaments employed to determine mechanical sensitivity of the plantar surface of both hind paws (only data for the hind paw ipsilateral to the experimental schwannoma is shown). A set of 20 von Frey filaments (North Coast Medical, Gilroy, CA), generating bending forces ranging from 0.008 to 300 grams, were used. Mice were “rested” in the test cages for 30 min before establishment of withdrawal threshold. All tests were performed on the right (control) and left (injected) hind paws. The middle plantar surface of the paw was stimulated with a series of ascending force von Frey monofilaments. The 50% threshold for each paw withdrawal was calculated, as previously described (Chen et al., 2009).
A rotating rod apparatus (Columbus Instruments, Columbus, OH) was used to assess motor performance. Mice were placed on the elevated accelerating rod beginning at 1 rpm/min for two trials per day twice per week. Each trial lasted 3 min, during which time the rotating rod underwent a linear acceleration from 1 to 40 rpm. Animals were scored for their latency (in seconds) to fall in each trial, and the average of two trials is reported. Animals rested a minimum of 10 min between trials to avoid fatigue (Chen et al., 2009).
Data analysis
All data are presented as group averages±SEM. The baseline value for all tests before injection used the average of all measurements before injection. Data were analyzed with SPSS 19 (IBM, Armonk, NY). Repeated-measure analysis of variance (ANOVA) was used when data were collected in multiple trials (Harris et al., 2012). p<0.05 was accepted as significant.
Genomic DNA extraction and real-time PCR: biodistribution
At necropsy, tissues were harvested, snap frozen in liquid nitrogen, and stored at −80°C until genomic DNA was extracted. Genomic DNA (gDNA) was isolated from the spinal cord, ipsilateral and contralateral dorsal root ganglia (DRG), and ipsilateral and contralateral sciatic nerves, using a DNeasy blood and tissue kit (Qiagen, Valencia, CA) according to the manufacturer's instructions. gDNA concentrations were determined with a biophotometer (Eppendorf, Hamburg, Germany). AAV GCs in the gDNA were quantified by real-time PCR with an ABI 7900 HT sequence detection system (Applied Biosystems, Foster City, CA), according to the manufacturer's instructions, and results were analyzed with the SDS 2.3 software. Briefly, primer pairs and probe were designed to the bovine growth hormone (BGH) poly(A) region of the scAAV1-P0-GFP vector, using ABI Primer Express software version 2.0. A standard curve was created, using scAAV1-P0-GFP plasmid DNA containing the BGH poly(A) region. The PCRs contained a total volume of 100 μl and were run under the following conditions: 50°C for 2 min, 95°C for 10 min, and 45 cycles of 95°C for 15 sec and 60°C for 1 min.
DNA samples were assayed in triplicate. To control for false negatives due to endogenous tissue-related PCR inhibition, the third replicate was spiked with plasmid DNA at a ratio of 100 copies/μg gDNA. If this replicate was greater than 40 copies/μg gDNA then the results were considered acceptable. If a sample contained ≥100 copies/μg gDNA it was considered positive for vector genomes. If a sample contained fewer than 100 copies/μg gDNA it was considered negative for vector genomes. If less than 1 μg of gDNA was analyzed, the vector copy number reported was normalized per microgram of gDNA and the plasmid spike-in was reduced to maintain the ratio of 100 copies/μg gDNA.
Results
Effects of delivery of AAV1-P0-ICE vectors to schwannomas
To assess the ability of AAV1 vectors to kill schwannoma cells, we generated AAV1 vectors encoding ICE (Juan et al., 1996) or GFP under the control of the rat P0 promoter (shown for AAV1-P0-ICE in Fig. 1). In initial evaluation, AAV1-CBA-GFP, AAV1-P0-GFP, and AAV1-P0-ICE vectors were used to infect human 293T cells and schwannoma HEI-193 cells. Using the constitutive promoter, CBA, both cell types showed a high degree of infectivity, that is, GFP-positive cells (Fig. 2A). When the P0 promoter (Brown and Lemke, 1997) was used, expression of GFP was seen only in HEI-193 cells, and not in 293T cells. Cell viability after infection with AAV1-P0-ICE and AAV1-P0-GFP vectors (or no vector) was assessed by CellTiter-Glo assay in 293T, human neuroblastoma SH-SY5Y, and HEI-193 cells. Infection with the AAV1-P0-ICE vector did not lead to any death of 293T or SH-SY5Y cells, whereas more than 90% of HEI-193 cells were dead by 72 hr postinfection (Fig. 2B). As an added control, we used TaqMan real-time RT-PCR to evaluate induction of the caspase-1 transgene after in vitro transduction of cultured HEI-193 cells with pAAV-P0-ICE. There was a 161-fold induction of caspase-1 mRNA (Supplementary Fig. S1; supplementary data are available online at www.liebertonline.com/hum).
FIG. 1.
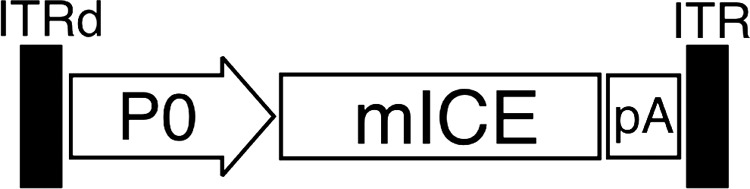
AAV vector encoding caspase-1 (interleukin-β-converting enzyme; ICE) under the control of the P0 promoter. Shown is a schematic diagram of the self-complementary AAV1-P0-ICE vector carrying the expression cassette for mouse ICE (caspase-1) under the control of the rat Schwann cell-specific P0 promoter (P0), and the bovine growth hormone polyadenylation signal (pA). One AAV2 inverted terminal repeat (ITRd) carries a deletion of the terminal resolution site that allows it to be packaged as a double-stranded DNA molecule.
FIG. 2.

The P0 promoter is selectively expressed in schwannoma cells. (A) 293T cells and HEI-193 schwannoma cells were infected with AAV1-CBA-GFP, AAV1-P0-GFP, or AAV1-P0-ICE vector in culture at a multiplicity of infection (MOI) of 10,000 GC/cell and, after 48 hr, were evaluated microscopically for GFP fluorescence and cell morphology at an original magnification of ×10. (B) Cell viability was assessed by Fluc bioluminescence in 293T, SH-SY5Y (neuroblastoma), and HEI-193 cells 72 hr after infection with AAV1-P0-ICE or AAV1-P0-GFP, or no infection, at the same MOI as in (A). Values for each cell line are normalized to viability of uninfected cells. *Significant difference compared with the control group (p=0.00006).
AAV1-P0-ICE causes regression of HEI-193FC schwannomas
The therapeutic efficacy of the AAV1-P0-ICE vector was tested in a mouse model in which schwannomas are generated via implantation of HEI-193FC cells in the distal region of the sciatic nerve of nude mice. Vector injections were carried out according to two paradigms—early in tumor growth and after establishment of schwannomas. The AAV1-P0-GFP vector was used for the control group. Two intratumoral injections of the AAV1-P0-ICE or AAV1-P0-GFP vector (1010 GC in 1 μl), spaced 1 week apart, at the site of early HEI-193FC tumor formation prevented further growth as assessed by in vivo bioluminescence imaging over a 14-week period after vector injections (Fig. 3A); one-way ANOVA revealing a significant effect of AAV1-P0-ICE treatment [F(1,11)=6.04, p=0.03]. In contrast, the majority of tumors (five of six) injected with the AAV1-P0-GFP vector continued to grow for the 9 weeks of postinjection evaluation. When intratumoral injections of AAV1-P0-ICE were carried out in larger, more established HEI-193FC tumors, AAV1-P0-ICE injections caused essentially complete regression of these tumors out to 12 weeks after vector injections [Fig. 3B; ANOVA: F(1,16)=8.27, p=0.01], whereas most of the AAV1-P0-GFP-injected tumors (five of six) continued to grow. In both panels of Fig. 3 the relatively large SEM in the AAV1-P0-GFP control group reflects the spontaneous tumor regression in one of six animals in each group. To avoid bias in our data, we used an intention-to-treat approach that included data from all vector-injected animals for statistical analysis.
Postmortem histological evaluation of formalin-fixed, paraffin-embedded sections stained with H&E was performed on animals from both experiments shown in Fig. 3. This revealed substantial tumor burden in nerves injected with control AAV vector (AAV1-P0-EGFP) (Fig. 4A and B), but only scant tumor cells in nerves injected with the AAV1-P0-ICE vector at 11 weeks after vector injection (Fig. 4C and D), as compared with normal nerve (Fig. 4E). Control animals implanted with HEI-193FC cells, but not injected with vector, also demonstrated significant tumor burden by H&E staining at this time point (data not shown).
FIG. 4.

Microscopic evaluation of AAV1-P0-ICE- and AAV1-P0-EGFP-injected HEI-193FC schwannomas. Hematoxylin and eosin (H&E) staining of a transverse section of (A) a representative sciatic nerve with abundant hematoxylin-positive tumor cells (darkly staining cells) 11 weeks after AAV1-P0-EGFP control vector injections (two injections spaced 1 week apart, 1×1010 GC in 1 μl) into an experimental schwannoma; and (C) a representative sciatic nerve of normal diameter with only a few hematoxylin-positive cells 11 weeks after AAV1-P0-ICE vector injections (two injections spaced 1 week apart, 1×1010 GC in 1 μl) into an experimental schwannoma. Higher power (original magnification, ×40) images of H&E-stained, longitudinal sections of sciatic nerves 11 weeks after vector injection of experimental schwannomas shows the presence of abundant, mitotically active tumor cells after AAV1-P0-EGFP injection (B), but scant numbers of tumor cells after AAV1-P0-ICE injection (D). Naive nerve (E) is shown for comparison. Color images available online at www.liebertpub.com/hum
AAV1-P0-ICE vector is not neurotoxic
Major concerns in using a vector encoding a proapoptotic protein under a Schwann cell-specific promoter are the potential toxicity to normal Schwann cells and consequent axonal injury of neurons. Neuropathological evaluation of sciatic nerve sections from nude mice collected 3 and 8 weeks after two injections of the AAV1-P0-ICE vector (1010 GC in 1 μl, 1 week apart) into non-tumor-bearing sciatic nerves revealed normal density of axons and no evidence of demyelination (Fig. 5B and C), as compared with normal uninjected nerve (Fig. 5A). Degenerating axons (Fig. 5B, inset) and macrophages (Fig. 5C, inset) were observed only rarely. H&E and toluidine staining of myelin failed to reveal evidence of acute or chronic inflammation (data not shown). Identical findings were obtained with immunocompetent C57BL/6 mice, that is, there was no significant neuropathology in AAV1-P0-ICE-injected sciatic nerves (Fig. 5D and E).
FIG. 5.

Neuropathological evaluation of sciatic nerve after AAV1-P0-ICE injection. Shown is toluidine blue staining of myelin in Epon-embedded sciatic nerve of nude mice (A–C) and immunocompetent C57BL/6 mice (D and E). Cross-sections of naive sciatic nerve (A), and 3 weeks (B and D), and 8 weeks (C and E) after the first of two weekly injection of AAV1-P0-ICE, show essentially normal nerves with rare axonal degeneration [(B), inset] and occasional macrophage infiltration [(C), inset] marked by arrows. Sciatic nerves of C57BL/6 mice (D and E) showed normal myelination and axonal integrity. Original magnification: (A)×20; (B–E): ×40; insets: ×100. Color images available online at www.liebertpub.com/hum
Sensory/pain and motor function tests were used to assess possible phenotypic alterations in C57BL/6 (immunocompetent) and nude (immunodeficient) mice resulting from intrasciatic nerve injections of AAV1-P0-ICE, AAV1-P0-GFP (both vectors at 1010 GC in 1 μl, two injections 1 week apart), or PBS (1 μl, two injections 1 week apart). Mechanical (“pain”) sensitivity (allodynia) was tested with von Frey filaments to establish withdrawal threshold of the hind paw ipsilateral to the injected sciatic nerve (Fig. 6A). Gross motor performance was assayed by the accelerating rotarod test (Fig. 6B). Although there was an expected temporary decrease in the von Frey withdrawal threshold (i.e., mechanical hyperalgesia interpreted as a painlike behavior) after the surgical manipulation required for injection, mechanical sensitivity did not differ between the AAV1-P0-ICE, AAV1-P0-GFP, and PBS groups over 5 weeks after injections, and the threshold for all groups returned to preinjection baseline within 2 weeks of vector injection. Thus, there was no sustained change in sensory function associated with vector injection. Further, there was no decrease in rotarod performance in any group at any time during the study. The data in Fig. 6 were obtained from C57BL/6 mice; statistically identical results were obtained with nude mice (data not shown).
FIG. 6.

Effects of AAV1-P0-ICE nerve injection on pain behavior and motor function. Normal sciatic nerves in immune-competent (C57BL/6) mice were injected with AAV1-P0-GFP vector or AAV1-P0-ICE vector (1×1010 GC in 1 μl) or PBS two times over a 1-week period (4 and 10 days, arrows). Pain (von Frey method) and motor control (rotarod) were evaluated at 4-day intervals for 41 days. Results are represented as the mean±SEM of n=12 mice per group. Arrows indicate times of vector injection. *Significantly different from baseline for all groups (p≤0.001). Color images available online at www.liebertpub.com/hum
Biodistribution studies were carried out after injection of AAV1-P0-ICE or AAV1-P0-GFP vector (1010 GC in 1 μl, 1 week apart) or PBS (1 μl, 1 week apart) into the sciatic nerve of nude mice (n=2 animals per group; AAV1-P0-ICE, AAV1-P0-GFP, or PBS). Sciatic nerve and DRG, both ipsilateral and contralateral to injection, as well as spinal cord were collected 3 weeks later and evaluated for the presence of vector by qPCR. Vector was found in the sciatic nerve and DRG in all samples ipsilateral, but not contralateral, to vector injections, as well as in all spinal cord samples (Table 1). Although vector was detectable in the DRG and spinal cord, immunohistological analysis of these tissues 8 weeks after a series of two intrasciatic AAV1-P0-GFP injections into adult mice (same method as described previously) failed to reveal any GFP fluorescence, suggesting that protein expression in these tissues is negligible (data not shown).
Table 1.
AAV Biodistribution as Determined by qPCR
| |
AAV1-P0-ICE |
AAV1-P0-GFP |
||
|---|---|---|---|---|
| Ipsilateral | Contralateral | Ipsilateral | Contralateral | |
| Sciatic nerve | + | – | + | – |
| DRG | + | – | + | – |
| Spinal cord | + | + | ||
DRG, dorsal root ganglia.
The observed absence of AAV-P0-ICE mediated neuronal and Schwann cell toxicity can be accounted for by P0 control of transgene expression. We hypothesized that the relatively low expression of P0 in the Schwann cells of adult mice would limit transgene expression after intrasciatic injection of our AAV vectors. P0 promoter expression in Schwann cells is greater in young than in adult mice, with expression peaking at 1 month of age and decreasing to about 50% of peak by 2 months (Shen et al., 2011). Further, neurotoxicity would not be expected to occur because the P0 promoter is not expressed in neurons (Prabhakar et al., 2010).
To test this hypothesis, we injected the sciatic nerves of young (postnatal day 21 [P21]) and adult (14-week-old) immune-competent C57BL/6 mice with AAV1-P0-GFP and collected sciatic nerves and DRG 10 days later for histological analysis of GFP expression (Fig. 7A, top four panels; and Fig. 7B, top two panels). In the sciatic nerve there was greater GFP expression after AAV1-P0-GFP injection in P21 mice compared with 14-week-old animals, consistent with lower P0 expression in adult mice. In DRG, there was no detectable GFP expression in either young or adult mice after AAV1-P0-GFP injection (Fig. 7B, top panels), consistent with constitutively low P0 expression in neurons. These results suggest that use of the P0 promoter in the AAV1-P0-ICE vector limits expression in Schwann cells and neurons after vector injection into the sciatic nerve of adult animals. This may explain the lack of observed vector-associated toxicity.
To further validate our results, we compared GFP expression after intrasciatic injection of AAV-P0-GFP with injection of AAV1-CBA-GFP. The CBA promoter is ubiquitously expressed (Gray et al., 2011). We found greater GFP expression when under CBA promoter control compared with P0 in both sciatic nerve (Fig. 7A) and DRG (Fig. 7B). Interestingly, we also found some indication that there was greater GFP expression in the sciatic nerve of young mice compared with adult mice after AAV1-CBA-GFP injection, suggesting that myelination, which is incomplete in young but complete in adult mice, may protect against adenoviral vector infection (Fig. 7A). However, given the high level of GFP transgene expression in both young and adult mice when GFP is under CBA control, we conclude that use of the P0 promoter is more important than degree of myelination in protection from AAV1-P0-ICE toxicity.
Proximal sciatic nerve implantation of schwannoma cells leads to painlike behavior that is relieved by AAV1-P0-ICE treatment
As persistent pain can be a major clinical issue in patients with schwannomatosis and NF2, and HEI-193FC tumors implanted in the distal sciatic nerve of mice did not produce painlike behavior (i.e., no decreased von Frey threshold), we endeavored to generate an experimental schwannoma model that would mimic clinical schwannoma-related pain. We hypothesized that more proximal implantation of HEI-193FC cells would lead to mechanical sensitization due to nerve compression, because in the proximal sciatic nerve expansion of tumors is limited by pelvic structures. As shown in Fig. 8, growth of proximal schwannomas was associated with a significant decrease in the von Frey threshold in the hind paw ipsilateral to tumor site starting at 3 weeks postimplantation. This mechanical sensitization persisted until approximately 3 weeks after initial AAV1-P0-ICE injection. As is evident in Fig. 8, AAV1-P0-ICE injection led to a statistically significant regression of tumors [one-way ANOVA: F(8,54)=2.80, p=0.01] and an associated return of von Frey threshold values to baseline (i.e., normalization of pain sensitivity) as indicated by one-way ANOVA [F(19,120)=7.8, p<0.0001].
FIG. 8.

Effect of AAV1-P0-ICE-mediated tumor regression on pain behaviors in proximally implanted sciatic nerve HEI-193FC schwannomas. Sixty thousand HEI-193FC cells were implanted into the sciatic nerve, at the level of the pelvis, in nude mice and tumor-associated bioluminescence was monitored at weekly intervals by in vivo bioluminescence imaging. Eight weeks after tumor implantation, at a time when painlike nocifensive behavior (i.e., decreased von Frey threshold in the plantar surface in foot ipsilateral to tumor) was well established, tumors were injected twice, 1 week apart, with AAV1-P0-ICE or AAV1-P0-GFP vector (1×1010 GC in 1 μl per injection). Arrows indicate times of vector injection. The solid line shows the von Frey threshold and the dotted line shows total flux, the former a measure of mechanical sensitivity and the latter a measure of tumor volume. As evident in the graph, AAV1-P0-ICE-mediated tumor regression is correlated with a return of the von Frey threshold to normal baseline. *Significantly different (p<0.05) from preimplantation baseline for von Frey threshold; #significantly different (p<0.05) from maximal total flux corresponding to tumor volume.
Discussion
We have validated a treatment strategy for schwannomas in which an AAV vector delivers an apoptotic protein, ICE, under the control of the Schwann-cell specific promoter P0, causing selective death of tumor cells. Using a xenograft model in which human NF2 schwannoma tumors form in the distal sciatic nerve of nude mice, we both prevented development of early schwannomas and caused regression of well-established tumors with minimal to no nerve damage through direct intratumoral injection of this AAV1-P0-ICE vector. In a related paradigm devised to model tumor-associated pain, the same human schwannoma cells were implanted more proximally in the sciatic nerve of mice with tumor development leading to mechanical hypersensitivity (i.e., pain behavior). Treatment of these tumors with AAV1-P0-ICE resulted in concurrent tumor regression and normalization of mechanical sensitivity. We have also shown that injection of the AAV1-P0-ICE vector into the sciatic nerve of both nude (nonimmune-competent) and immune-competent mice is not associated with any apparent neuropathology as indicated by histological evaluation and behavioral testing.
Several aspects of this study were surprising. The first was the selective killing of schwannoma cells by AAV1-P0-ICE while leaving resident Schwann cells (and neuronal axons) intact. ICE was the first mammalian caspase identified (Miura et al., 1993) and expression causes rapid lysis of cells associated with inflammatory activity (Miao et al., 2011). In some models ICE expression has been associated with neurodegeneration (Friedlander, 2000) and demyelination (Furlan et al., 1999). However, extensive evaluation of peripheral nerve integrity and function, including neuropathology, nerve conduction velocity, and behavior (pain sensitization and rotarod performance), revealed minimal to no damage after direct injection of AAV1-P0-mICE into the sciatic nerve (present study) (Prabhakar et al., 2010).
Protection of normal nerve may be attributed to the use of the P0 promoter, which normally regulates expression of a major myelin glycoprotein that is at its highest levels in Schwann cells during myelination, occurring at 1 month of age during normal development (Lee et al., 1997; Shen et al., 2011). Expression of P0 decreases by about 50% at 2 months of age (Shen et al., 2011)—the time of vector injection in our studies. Accordingly, our results indicate decreased GFP expression in the sciatic nerve when AAV-P0-GFP was injected into the nerve of 2-month-old mice compared with P21 mice. This decrease in transgene expression in older mice after AAV-CBA-GFP injection raises the possibility that degree of Schwann cell myelination reduces vector infectivity.
A second surprising finding was the extensive remission of tumors after injection of the AAV1-P0-ICE vector (two intratumoral injections of 1010 GC vector in 1 μl). On the basis of other tumor injection studies it is unlikely that all the tumor cells were infected with the vector, albeit infection efficiency varies with serotype and tumor type (Teschendorf et al., 2010). Although we estimate, based on the rate of growth as monitored by bioluminescence imaging (Saydam et al., 2011), that the MOI of tumor cells was about 3000 GC/cell, it is unlikely that the vector inoculum dispersed uniformly throughout the tumor. The high extent of tumor killing observed may be explained by the finding that ICE can be delivered from one cell to another via microvesicles, resulting in death of recipient cells (Sarkar et al., 2009), and the likelihood that schwannoma cells, like other tumor cells, are highly active in microvesicle release (Taylor and Gercel-Taylor, 2008; Balaj et al., 2011). The myelin sheath on Schwann cells may also block uptake of microvesicles. A third surprising finding was that we saw little to no sign of macrophage cell invasion into the nerve after AAV1-P0-ICE injection in nude and immune-competent mice, considering that ICE is a central component of the inflammasome complex (Lamkanfi and Dixit, 2009). It remains possible that some macrophages moved in and out quickly after AAV1-P0-ICE vector injection and were no longer present at the earliest time point we evaluated (3 weeks after the second of two vector injections).
Complete assessment of our AAV-P0-ICE therapeutic strategy for the treatment of schwannoma requires testing in immune-competent animals. We expect that in immune-competent mice we will observe a stronger inflammatory response compared with nude mice, and this may lead to greater neurotoxicity. However, AAV-P0-ICE tumor treatment in immune-competent mice also has the potential to establish both innate and adaptive antitumor immunity. If the latter host immune responses do occur, AAV-P0-ICE treatment of a single lesion may lead to systemic changes that cause regression of distal NF2 lesions and vaccinate animals from the development of tumors after implantation at a later time. This is a crucial issue in the treatment of patients with NF2 given the typical presence of multiple lesions in a given individual and the development of tumors over long time periods. We are currently testing this, using the NF2S-1 mouse schwannoma line (Prabhakar et al., 2007) implanted in the sciatic nerve of Swiss mice.
Current therapy for NF2 and schwannomatosis tumors involves surgical resection or radiosurgery of symptomatic lesions and symptomatic management of pain. There are several significant limitations to these available treatments. The location of tumors can make surgical resection infeasible because of the unacceptable risk of morbidity (e.g., lesioning of cervical nerve roots) or mortality (e.g., damage to the brainstem). Even when resection is possible, significant morbidity may be associated with the operation. Further, schwannomas—especially in schwannomatosis—can be associated with excruciating, persistent pain for which surgical resection may not be an option because of the number of tumors. Even if it is assumed that not all tumor cells would be destroyed by the vector, because schwannomas are slow growing and benign, a reduction in tumor mass would be expected to confer meaningful and prolonged symptomatic improvement.
These preclinical studies provide support for further progression toward phase 1 clinical trial evaluation of the AAV-P0-ICE vector for the treatment of schwannomas. AAV vectors have been shown to be safe for use in the human nervous system in a number of gene therapy trials (McPhee et al., 2006; Kaplitt et al., 2007; Eberling et al., 2008; Worgall et al., 2008; Bowers et al., 2011; McCown, 2011; Lentz et al., 2012). These vectors have also proven effective in treating malignant tumors in preclinical trials, although tumor cells that survive the initial infection continue to divide and thus lose AAV-delivered transgenes (Meijer et al., 2009; Maitituoheti et al., 2011; Tamai et al., 2012). Further, conditional expression of another caspase, caspase-9, has proven safe and effective in preventing graft-versus-host disease in patients with leukemia treated with genetically modified T cells in clinical trials (Di Stasi et al., 2011).
Our preclinical findings in an orthotopic mouse model demonstrate the efficacy and safety of treatment of schwannomas with an AAV1-P0-ICE vector. These studies will help form the basis of translational studies in support of transition to phase 1 human trials. The use of AAV1-P0-ICE vectors for the treatment of neurofibromatosis holds the promise of preventing and reversing the substantial disability and suffering associated with schwannomatosis and NF2 tumors. This approach, in which the mass of a benign tumor is reduced by injection of a vector that induces selective apoptosis via a cell-specific promoter, should have wide application for a variety of benign tumors, including adenomas, fibromas, hemangiomas, lipomas, meningiomas, myomas, moles (nevi), neuromas, osteochondromas, and papillomas—pending selective promoter identification. This would reduce the need for surgical resection, which is invasive and can have substantial associated morbidity. On the basis of the ease of vector injection as compared with surgery, we anticipate that vector treatment would be more frequently sought out by patients, thereby potentially improving prognosis (through early treatment), as many of these benign tumors undergo malignant transformation over time.
Supplementary Material
Acknowledgments
The authors thank Dr. Okay Saydam, Ozlem Senol, Arda Mirzak, and Mehmet Fatih Bolukbasi for help in generating the HEI-193FC line; Hae-Sook Shin for assistance with behavioral testing; and Ahmed Hassad for general technical assistance. The authors thank Kirsten E. Erger at the University of Florida Gene Therapy Center Toxicology Core for work on the DNA isolation and real-time PCR for vector biodistribution. The authors thank Leora Cramer at the Pathology Laboratory for work on the histology. The authors thank Ms. Suzanne McDavitt for skilled editorial assistance. This work was supported by NIH/NINDS grant NS24279 (X.O.B., A.S.) and DOD NF060106 (M.S. and G.J.B.).
Author Disclosure Statement
No competing financial interests exist for any of the authors.
References
- Agarwal N. Pacher P. Tegeder I., et al. Cannabinoids mediate analgesia largely via peripheral type 1 cannabinoid receptors in nociceptors. Nat. Neurosci. 2007;10:870–879. doi: 10.1038/nn1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antinheimo J. Sankila R. Carpén O., et al. Population-based analysis of sporadic and type 2 neurofibromatosis-associated meningiomas and schwannomas. Neurology. 2000;54:71–76. doi: 10.1212/wnl.54.1.71. [DOI] [PubMed] [Google Scholar]
- Balaj L. Lessard R. Dai L., et al. Tumour microvesicles contain retrotransposon elements and amplified oncogene sequences. Nat. Commun. 2011;2:180. doi: 10.1038/ncomms1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baser M.E. Friedman J.M. Evans D.G. Increasing the specificity of diagnostic criteria for schwannomatosis. Neurology. 2006;66:730–732. doi: 10.1212/01.wnl.0000201190.89751.41. [DOI] [PubMed] [Google Scholar]
- Bowers W.J. Breakefield X.O. Sena-Esteves M. Genetic therapy for the nervous system. Hum. Mol. Genet. 2011;20:R28–R41. doi: 10.1093/hmg/ddr110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broekman M.L. Comer L.A. Hyman B.T. Sena-Esteves M. Adeno-associated virus vectors serotyped with AAV8 capsid are more efficient than AAV-1 or −2 serotypes for widespread gene delivery to the neonatal mouse brain. Neuroscience. 2006;138:501–510. doi: 10.1016/j.neuroscience.2005.11.057. [DOI] [PubMed] [Google Scholar]
- Brown A.M. Lemke G. Multiple regulatory elements control transcription of the peripheral myelin protein zero gene. J. Biol. Chem. 1997;272:28939–28947. doi: 10.1074/jbc.272.46.28939. [DOI] [PubMed] [Google Scholar]
- Chen Q. Peto C.A. Shelton G.D., et al. Loss of modifier of cell adhesion reveals a pathway leading to axonal degeneration. J. Neurosci. 2009;29:118–130. doi: 10.1523/JNEUROSCI.3985-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Stasi A. Tey S.K. Dotti G., et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N. Engl. J. Med. 2011;365:1673–1683. doi: 10.1056/NEJMoa1106152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberling J.L. Jagust W.J. Christine C.W., et al. Results from a phase I safety trial of hAADC gene therapy for Parkinson disease. Neurology. 2008;70:1980–1983. doi: 10.1212/01.wnl.0000312381.29287.ff. [DOI] [PubMed] [Google Scholar]
- Eichler F.S. Hornemann T. McCampbell A., et al. Overexpression of the wild-type SPT1 subunit lowers desoxysphingolipid levels and rescues the phenotype of HSAN1. J. Neurosci. 2009;29:14646–14651. doi: 10.1523/JNEUROSCI.2536-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedlander R.M. Role of caspase 1 in neurologic disease. Arch. Neurol. 2000;57:1273–1276. doi: 10.1001/archneur.57.9.1273. [DOI] [PubMed] [Google Scholar]
- Furlan R. Martino G. Galbiati F., et al. Caspase-1 regulates the inflammatory process leading to autoimmune demyelination. J. Immunol. 1999;163:2403–2409. [PubMed] [Google Scholar]
- Gray S.J. Foti S.B. Schwartz J.W., et al. Optimizing promoters for recombinant adeno-associated virus-mediated gene expression in the peripheral and central nervous system using self-complementary vectors. Hum. Gene Ther. 2011;22:1143–1153. doi: 10.1089/hum.2010.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadfield K.D. Newman W.G. Bowers N.L., et al. Molecular characterisation of SMARCB1 and NF2 in familial and sporadic schwannomatosis. J. Med. Genet. 2008;45:332–339. doi: 10.1136/jmg.2007.056499. [DOI] [PubMed] [Google Scholar]
- Harris J.E. Sheean P.M. Gleason P.M., et al. Publishing nutrition research: A review of multivariate techniques. 2. Analysis of variance. J. Acad. Nutr. Diet. 2012;112:90–98. doi: 10.1016/j.jada.2011.09.037. [DOI] [PubMed] [Google Scholar]
- Huang J.H. Simon S.L. Nagpal S., et al. Management of patients with schwannomatosis: Report of six cases and review of the literature. Surg. Neurol. 2004;62:353–361. doi: 10.1016/j.surneu.2003.11.020. [DOI] [PubMed] [Google Scholar]
- Hulsebos T.J. Plomp A.S. Wolterman R.A., et al. Germline mutation of INI1/SMARCB1 in familial schwannomatosis. Am. J. Hum. Genet. 2007;80:805–810. doi: 10.1086/513207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung G. Li X. Faudoa R., et al. Establishment and characterization of a schwannoma cell line from a patient with neurofibromatosis 2. Int. J. Oncol. 2002;20:475–482. [PubMed] [Google Scholar]
- Jacoby L.B. Jones D. Davis K., et al. Molecular analysis of the NF2 tumor-suppressor gene in schwannomatosis. Am. J. Hum. Genet. 1997;61:1293–1302. doi: 10.1086/301633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessen K.R. Mirsky R. The origin and development of glial cells in peripheral nerves. Nat. Rev. Neurosci. 2005;6:671–682. doi: 10.1038/nrn1746. [DOI] [PubMed] [Google Scholar]
- Juan T.S. McNiece I.K. Jenkins N.A., et al. Molecular characterization of mouse and rat CPP32 β gene encoding a cysteine protease resembling interleukin-1β converting enzyme and CED-3. Oncogene. 1996;13:749–755. [PubMed] [Google Scholar]
- Kaplitt M.G. Feigin A. Tang C., et al. Safety and tolerability of gene therapy with an adeno-associated virus (AAV) borne GAD gene for Parkinson's disease: An open label, phase I trial. Lancet. 2007;369:2097–2105. doi: 10.1016/S0140-6736(07)60982-9. [DOI] [PubMed] [Google Scholar]
- Kirschbaum K.M. Hiemke C. Schmitt U. Rotarod impairment: Catalepsy-like screening test for antipsychotic side effects. Int. J. Neurosci. 2009;119:1509–1522. doi: 10.1080/00207450902984002. [DOI] [PubMed] [Google Scholar]
- Lamkanfi M. Dixit V.M. Inflammasomes: Guardians of cytosolic sanctity. Immunol. Rev. 2009;227:95–105. doi: 10.1111/j.1600-065X.2008.00730.x. [DOI] [PubMed] [Google Scholar]
- Lee M. Brennan A. Blanchard A., et al. P0 is constitutively expressed in the rat neural crest and embryonic nerves and is negatively and positively regulated by axons to generate non-myelin-forming and myelin-forming Schwann cells, respectively. Mol. Cell. Neurosci. 1997;8:336–350. doi: 10.1006/mcne.1996.0589. [DOI] [PubMed] [Google Scholar]
- Lentz T.B. Gray S.J. Samulski R.J. Viral vectors for gene delivery to the central nervous system. Neurobiol. Dis. 2012;48:179–188. doi: 10.1016/j.nbd.2011.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu-Emerson C. Plotkin S.R. The neurofibromatosis. 2. NF2 and schwannomatosis. Rev. Neurol. Dis. 2009;6:E81–E86. [PubMed] [Google Scholar]
- Maitituoheti M. Li Y. Wang W., et al. Adeno-associated virus-mediated local delivery of LIGHT suppresses tumorigenesis in a murine cervical cancer model. J. Immunother. 2011;34:581–587. doi: 10.1097/CJI.0b013e31822b9fe0. [DOI] [PubMed] [Google Scholar]
- McCarty D.M. Fu H. Monahan P.E., et al. Adeno-associated virus terminal repeat (TR) mutant generates self-complementary vectors to overcome the rate-limiting step to transduction in vivo. Gene Ther. 2003;10:2112–2118. doi: 10.1038/sj.gt.3302134. [DOI] [PubMed] [Google Scholar]
- McClatchey A.I. Giovannini M. Membrane organization and tumorigenesis—the NF2 tumor suppressor, Merlin. Genes Dev. 2005;19:2265–2277. doi: 10.1101/gad.1335605. [DOI] [PubMed] [Google Scholar]
- McCown T.J. Adeno-associated virus (AAV) vectors in the CNS. Curr. Gene Ther. 2011;11:181–188. doi: 10.2174/156652311795684759. [DOI] [PubMed] [Google Scholar]
- McPhee S.W. Janson C.G. Li C., et al. Immune responses to AAV in a phase I study for Canavan disease. J. Gene Med. 2006;8:577–588. doi: 10.1002/jgm.885. [DOI] [PubMed] [Google Scholar]
- Meijer D.H. Maguire C.A. LeRoy S.G. Sena-Esteves M. Controlling brain tumor growth by intraventricular administration of an AAV vector encoding IFN-β. Cancer Gene Ther. 2009;16:664–671. doi: 10.1038/cgt.2009.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messerli S.M. Tang Y. Giovannini M., et al. Detection of spontaneous schwannomas by MRI in a transgenic murine model of neurofibromatosis type 2. Neoplasia. 2002;4:501–509. doi: 10.1038/sj.neo.7900265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao E.A. Rajan J.V. Aderem A. Caspase-1-induced pyroptotic cell death. Immunol. Rev. 2011;243:206–214. doi: 10.1111/j.1600-065X.2011.01044.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura M. Zhu H. Rotello R., et al. Induction of apoptosis in fibroblasts by IL-1β-converting enzyme, a mammalian homolog of the C. elegans cell death gene ced-3. Cell. 1993;75:653–660. doi: 10.1016/0092-8674(93)90486-a. [DOI] [PubMed] [Google Scholar]
- Prabhakar S. Messerli S.M. Stemmer-Rachamimov A.O., et al. Treatment of implantable NF2 schwannoma tumor models with oncolytic herpes simplex virus G47Δ. Cancer Gene Ther. 2007;14:460–467. doi: 10.1038/sj.cgt.7701037. [DOI] [PubMed] [Google Scholar]
- Prabhakar S. Brennan G.J. Messerli S.M., et al. Imaging and therapy of experimental schwannomas using HSV amplicon vector-encoding apoptotic protein under Schwann cell promoter. Cancer Gene Ther. 2010;17:266–274. doi: 10.1038/cgt.2009.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouleau G.A. Merel P. Lutchman M., et al. Alteration in a new gene encoding a putative membrane-organizing protein causes neuro-fibromatosis type 2. Nature. 1993;363:515–521. doi: 10.1038/363515a0. [DOI] [PubMed] [Google Scholar]
- Sarkar A. Mitra S. Mehta S., et al. Monocyte derived microvesicles deliver a cell death message via encapsulated caspase-1. PLoS One. 2009;4:e7140. doi: 10.1371/journal.pone.0007140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saydam O. Ozdener G.B. Senol O., et al. A novel imaging-compatible sciatic nerve schwannoma model. J. Neurosci. Methods. 2011;195:75–77. doi: 10.1016/j.jneumeth.2010.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen D. Zhang Q. Gao X., et al. Age-related changes in myelin morphology, electrophysiological property and myelin-associated protein expression of mouse sciatic nerves. Neurosci. Lett. 2011;502:162–167. doi: 10.1016/j.neulet.2011.07.034. [DOI] [PubMed] [Google Scholar]
- Tamai H. Miyake K. Yamaguchi H., et al. AAV-8 vector expressing IL-24 efficiently suppresses tumor growth mediated by specific mechanisms in MLL/AF4-positive ALL model mice. Blood. 2012;119:64–71. doi: 10.1182/blood-2011-05-354050. [DOI] [PubMed] [Google Scholar]
- Taylor D.D. Gercel-Taylor C. MicroRNA signatures of tumor-derived exosomes as diagnostic biomarkers of ovarian cancer. Gynecol. Oncol. 2008;110:13–21. doi: 10.1016/j.ygyno.2008.04.033. [DOI] [PubMed] [Google Scholar]
- Teschendorf C. Emons B. Muzyczka N., et al. Efficacy of recombinant adeno-associated viral vectors serotypes 1, 2, and 5 for the transduction of pancreatic and colon carcinoma cells. Anticancer Res. 2010;30:1931–1935. [PubMed] [Google Scholar]
- Worgall S. Sondhi D. Hackett N.R., et al. Treatment of late infantile neuronal ceroid lipofuscinosis by CNS administration of a serotype 2 adeno-associated virus expressing CLN2 cDNA. Hum. Gene Ther. 2008;19:463–474. doi: 10.1089/hum.2008.022. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.