

Abstract
Chronic kidney disease (CKD) is recognized to cause pharmacokinetic changes in renally excreted drugs; however, pharmacokinetic changes are also reported for drugs that are non-renally eliminated. Few studies have investigated how uremic toxins may affect drug transporters and metabolizing enzymes and how these may result in pharmacokinetic/metabolic changes in CKD. Here, we investigated the effects of uremic toxins and human uremic serum on the transport of the prototypical transporter substrate [3H]-estrone sulfate and three BDDCS drugs, propranolol, losartan, and eprosartan. We observed a significant decrease in [3H]-estrone sulfate, losartan, and eprosartan uptake with some uremic toxins in both transfected cells and rat hepatocytes. The uptake of losartan was decreased in rat and human hepatocytes (28%, and 48% respectively) in the presence of hemodialysis (HD) serum. Time-course studies of losartan showed a 27%, 65% and 68% increase in AUC in the presence of HD serum, rifampin, and sulfaphenazole respectively. Intracellular losartan AUC decreased significantly in the treatment groups and the metabolite AUC decreased by 41% and 26% in rifampin and sulfaphenazole treated group. The intracellular AUC of eprosartan increased 190% in the presence of HD serum. These studies indicate that the uremic toxins contained in HD serum play an important role in drug disposition through drug transporters, and that there would be differential effects depending on the BDDCS classification of the drug.
Keywords: Disease effects, BDDCS, organic anion-transporter polypeptide transporters, hepatocytes, CYP enzymes, microsomes, phase I metabolism
Introduction
Drug metabolism has not been extensively studied in renal failure. Chronic kidney disease (CKD) causes pharmacokinetic changes of drugs that are renally excreted unchanged; thus, the dose for these drugs is usually adjusted for this patient population. However, dose adjustments for drugs that are eliminated primarily via hepatic metabolism or biliary excretion are rarely considered in CKD. In recent years, drug transporters have been recognized to have an important role in drug disposition. Several studies in our laboratory have shown that hepatic uptake transporters play a pivotal role in drug disposition by facilitating the uptake of drug into the hepatocyte and exposing the drug to the metabolizing enzyme 1–3. These studies demonstrated the importance of transporter-enzyme interplay in drug pharmacokinetics of drugs like atorvastatin, erythromycin and digoxin.
Uremic toxins are known to accumulate in patients with CKD, and they are categorized based on their size, protein binding, and water solubility 4. Indoxyl sulfate and 3-carboxy-4-methyl-5-propyl-2-furanpropanoic acid (CMPF) are two uremic toxins that have been previously studied for their involvement in hepatic drug metabolism and disposition 5–7. Human uremic serum from patients on hemodialysis has also been studied in vitro to determine how the toxins contained in uremic serum alter metabolizing enzymes. Studies have shown that uremic serum can inhibit CYPs in vitro in cultured rat hepatocytes 8,9. Organic anion transporters OATs have been investigated for their involvement in the transport of these uremic toxins in the kidney 10. The effect of these uremic toxins on hepatic drug transporters, however; has not been extensively investigated. Moreover, the effects of these uremic toxins on different drugs based on the Biopharmaceutics Drug Disposition Classification System (BDDCS) have never been investigated.
BDDCS is a drug classification system based on solubility, permeability (from the FDA’s Biopharmaceutics Classification System, BCS), and the extent of metabolism that predict the effects of drug transporters and metabolizing enzymes on drug disposition 11.
In this classification system, the route of elimination (metabolism or renal and biliary clearance), the involvement of transporters, and the permeability will place a drug in a certain class and will also determine the importance of transporters and enzyme-transporter interplay in the gut and the liver for that class of drug. For a Class 1 highly permeable and extensively metabolized drug, transporter effects in the liver will be minimal, (they would not require a transporter to enter or exit the hepatocyte). For a Class 2 drug, the low solubility of the drug may limit the amount of drug available, thus uptake transporters may be required to make more of the drug available to the cell and efflux transporters can affect efflux from the hepatocyte. For Class 3 and 4 drugs, transporters would be required because of the low permeability of the drug. Therefore, uptake and efflux transporters could become important in Class 2, 3, and 4 drugs in the liver. We hypothesized that if uremic toxins have effects on hepatic transporters, we may expect to see a difference in drug uptake and/or efflux of drug according to its BDDCS class. For a class 1 drug, since transporters are not clinically relevant, the presence of uremic toxins, even if they are affecting transporters, would not have an effect on uptake or efflux of the drug. On the other hand, uremic toxins could have an effect on drug uptake and/or efflux of Class 2, 3, and 4 drugs.
In our studies we investigated the effects of various uremic toxins, which accumulate in CKD and end-stage renal disease (ESRD), on the uptake of the model compound [3H]-estrone sulfate, which is a known substrate for uptake transporters, and three BDDCS (propranolol Class1, losartan Class 2, eprosartan Class 4) drugs in transfected cells and rat hepatocytes. We also studied the effects of human hemodialysis (HD) serum on drug uptake in transfected cells rat and human hepatocytes, and the effect of HD serum on the uptake and metabolism of propranolol, losartan, and eprosartan in rat hepatocytes in time course studies. It is worth noting that HEK293 cells, which are typically used in drug transport studies, have endogenous expression of these transporters, and they are used as over-expression systems. Also, hemodialysis patients take multiple medications, which may be present in the serum used, however, these patients were not dosed with the drugs we studies, and their blood chemistry labs indicated that their protein levels (albumin) were within normal range.
In our studies we analyzed the total concentration of parent compound and their major metabolite and also the intracellular concentration of the parent compound. In our studies we demonstrated that uremic toxins and uremic serum have an inhibitory effect on the uptake in cells expressing hepatic drug transporters, and that the changes in metabolism for these drugs is not due to uremic serum affecting liver enzymes. These studies show that HD serum affects uptake for losartan, a Class 2 drug, and may affect efflux for eprosartan, a Class 4 drug, but have no effect on transport of propranolol, a Class 1 drug. This is a proof of concept approach that can be applied to more drugs from Class 1–4 to establish how the transport of different classes of drugs is affected by uremic toxins/HD serum. This can be valuable information for pre-clinical studies in drug development.
Materials and Methods
Propranolol, rifampin, quinidine, sulfaphenazole, indoxyl sulfate, endothelin, quinolinic acid, indole-3-acetic acid, p-cresol, homocysteine, tumor necrosis factor- alpha (TNF-alpha), interleukin-6 (IL-6), and parathyroid hormone (PTH) as well as HPLC-grade dimethyl sulfoxide, tert-butyl-methyl-ether (MTBE), and acetonitrile (ACN) were purchased from Sigma-Aldrich (St. Louis, MO). Losartan potassium salt, EXP3174, 4-OH propranolol, and eprosartan were purchased from Toronto Research Chemicals (Ontario, Canada). GG918 (GF120918) was graciously donated by GlaxoSmithKline (Research Triangle Park, NC). [3H]-estrone sulfate was purchased from Perkin Elmer (Boston, MA). Plasmids containing transporter inserts or empty vector (pcMV6) were purchased from Origene Technologies Inc. (Rockville, MD). Lipofectamine was purchased from Invitrogen Corporation (Carlsbad, CA). CMPF was purchased from Cayman Chemicals (Ann Arbor, Michigan). Cell culture media was purchased from UCSF Cell Culture Facility (San Francisco, CA). Pooled male rat liver microsomes were acquired from BD Biosciences (Woburn, MA). Pooled human cryopreserved hepatocytes were obtained from Cellz Direct (Dallas, TX). Male Sprague-Dawley rats (200–350 g) from Charles River Laboratories (Wilmington, MA) were housed in the UCSF animal care facility with a 12-h light/dark cycle and allowed free access to water and food. The animal studies were approved by Committee on Animal Research, UCSF.
Hemodialysis Serum
A protocol to obtain blood from patients on hemodialysis was approved by the UCSF Committee on Human Research. Blood was obtained from eight patients from the outpatient hemodialysis unit at the UCSF Mount Zion clinic. All patients had been on hemodialysis for more than a year. The blood was obtained on dialysis day before initiating dialysis. The serum was separated from the blood and the serum from all patients was pooled into a single batch. The pooled serum was then used to carry out the in vitro experiments. All experiments using serum were carried out with 10% human serum (normal or hemodialysis).
Cell Transfection and Uptake Assay
HEK293 cells were cultured in Eagle’s minimal essential medium with Eagle’s balanced salt solution and L-glutamine (2mM) plus 10% heat-inactivated fetal bovine serum (FBS), penicillin/streptomycin (1μg/ml). A day before transfection, cells were seeded at a density of 0.5×106/cm2. The next day they were transiently transfected with OATP1B1-pcMV5, OATP1B3-pcMV6, OATP2B1-pcMV6 or pcMV6 empty vector control using Lipofectamine 2000 per the manufacturer’s protocol. After 24 hours the media was replaced, and 48 hours after transfection the cells were used for uptake studies. Before initiation of the uptake study, cells were washed once with Hanks buffer pre-warmed to 37°C. The uptake study was initiated by adding 1ml of Hanks buffer containing 10% of FBS plus 17.45μM [3H]-estrone sulfate (1:4000 dilution) alone as a control or with 10μM rifampin, 100μM MK-571, or a uremic toxin as inhibitors and incubating at 37°C for 2 min. Preliminary experiments had shown that the uptake rate was linear over this time period (data not shown). For the inhibition studies, inhibitors and substrates were added simultaneously. Uremic toxin concentration was chosen based on a preliminary dose-response curve and literature review of concentrations previously used. Uremic toxin concentrations vary depending on the toxin. The concentrations used here were concentrations reported to be physiologically present in serum of renally impaired patients. Using these clinically relevant concentrations is appropriate in studying the inhibitory effects on transporters in a physiologically relevant scenario. After two minutes, the buffer was removed to terminate the reaction and the cells were washed three times with ice-cold PBS. Cells were scraped from the wells and homogenized. An aliquot of 200 μL was transferred to a scintillation vial and quenched with liquid scintillation counting solution. The intracellular radioactivity was measured using a scintillation counter (LS6000TA; Beckman Coulter, Fullerton, CA). The results are reported as pM/cell count.
Microsome Incubations
The incubation conditions were as described previously12. In brief, each reaction mixture contained 0.5 mg/ml microsomes, 5 mM NADPH, phosphate buffer, 10μM losartan or 10μM propranolol, plus 10μM of rifampin, or 1μM of sulfaphenazole as a CYP2C9 inhibitor (negative control for losartan metabolism), or 30μM of quinidine as CYP2D6 inhibitor (negative control for propranolol metabolism). The total DMSO concentration used to solubilize rifampin was less than 1% for all in vitro studies, all other compounds were solubilized in methanol. The total reaction volume was 250μl. Microsomes from the same batch were used; thus, the protein content was the same for all samples in each experiment for each drug tested. The reaction period was 30 minutes at 37°C. For each sample, the reaction was stopped via protein precipitation by addition of an equal volume of ACN containing the internal standard (IS), warfarin (1μM). The supernatants were stored at −80°C for LC/MS-MS analysis.
Hepatocyte Isolation
Anesthesia was induced in rats by intraperitoneal injection with a 1 ml/kg dose of ketamine:xylazine (80 mg/ml:12 mg/ml) before surgery1. The portal vein was cannulated with an i.v. catheter (catalog number 2007-04; Becton Dickinson, Sandy, UT) and perfused with oxygenated liver perfusion buffer (Invitrogen, Carlsbad, CA) for 5 min at 30 ml/min, followed by perfusion with an oxygenated hepatocyte washing buffer (Invitrogen) modified with 2 mM L-glutamine, 10 mM HEPES, and 1.2 U/ml collagenase (Sigma-Aldrich) for 5 min at 20 ml/min. The digested liver was excised and homogenized in a beaker. Hepatocytes were then washed twice with an ice-cold hepatocyte wash buffer containing 2 mM L-glutamine and 10 mM HEPES and were centrifuged at 50g for 2 to 3 min. Cell viability was determined using the trypan blue exclusion method. Cells with viability of greater than 80% were used for further studies.
Hepatocyte Incubations
Hepatocyte incubations were carried out immediately after cell isolation. Cells were resuspended and diluted to 2 ×106 per ml in Krebs-Henseleit buffer (pH 7.4) containing 0.21 g/l sodium bicarbonate and supplemented with 1% BSA and 10 mM glucose. Cell suspensions for all hepatocyte incubations were pre-warmed for 5 min in a 37°C shaking water bath before initiation of incubations. For the uptake studies, propranolol (10 μM), losartan (10 μM), or eprosartan (10μM) with and without 10 μM rifampin, or 100 μM MK571, was added to the cell suspensions in the 37°C water bath. At 2 min, the reactions were terminated by transferring 1×106 hepatocytes into a centrifuge tube containing 700 μl of a mixture of silicone oil and mineral oil 13 and centrifuged at 13,000g for 10 seconds. After removing the buffer layer and the oil layer, each cell pellet was resuspended in 100 μl of water and sonicated for 15 min to ensure a thorough cell lysis. This was followed by adding 200 μl of ACN containing IS and spinning at 13,000g for 15 min to precipitate protein. The supernatant was then transferred into a HPLC vial (Hewlett Packard, Palo Alto, CA) for LC-MS/MS analysis. The same experimental conditions were used with radiolabeled compound, and uremic toxins. Intracellular radioactivity was measured with a liquid scintillation counter (LS6000TA; Beckman Coulter, Fullerton, CA).
For the time course studies, 15 ml of hepatocytes (2 ×106 cells/ml) were warmed in a 50 ml flask and shaken in a 37°C water bath for 5 min. Each study was initiated by concomitantly adding propranolol (1μM), losartan (1μM), or eprosartan (10μM) with DMSO (control), 10 μM rifampin, 1 μM sulfaphenazole, or 30μM of quinidine. Sulfaphenazole was used as the CYP2C9 inhibitor for losartan and quinidine was used as the CYP2D6 inhibitor for propranolol. Rifampin was used as the OATP inhibitor. At 5, 10, 15, 20, 30, 45, and 60 min, 0.5 ml of cells were transferred into a centrifuge tube containing 700 μl of a mixture of silicone oil and mineral oil and centrifuged at 13,000g for 10 s to stop the reaction. Ten seconds later, 1 ml of cells was transferred into a glass tube containing MTBE and IS followed by vortexing to stop the reaction. Sample preparation for the intracellular measurement of propranolol and losartan was the same as for the uptake studies. Half a million hepatocytes were transferred to a centrifuge tube to stop the reaction. The samples were centrifuged immediately, supernatant removed, and washed three times with PBS. The pellet was then dissolved in 200 μL of ACN, vortexed and centrifuged for 10 s at 13,000 g, 100 uL of the supernatant were transferred to HPLC vials for LC/MS-MS measurements. Sample preparation for the measurement of propranolol and losartan metabolism was the same as that for the inhibition studies.
For evaluating the inhibition of metabolism, propranolol or losartan was co incubated with quinidine (30 μM) or sulfaphenazole (1 μM) and a 0.5ml sample was taken at 5, 10, 15, 20, 30, 45, and 60 minutes. The reaction was stopped by transferring 1 ml of cells to a fresh glass tube containing MTBE and IS followed by vortexing. All samples were spun down at 2000g for 10 min. After quick-freezing the aqueous layer in a methanol/dry ice bath, the organic layer was poured into a new tube and evaporated under nitrogen gas. Each sample was reconstituted with 300 μl of ACN/water (50/50, v/v) for LC/MS-MS analysis of the parent compound and its primary metabolite. Results are expressed as μM/cell count
LC/MS-MS Measurement of Propranolol, Losartan, and Eprosartan and their Metabolites
A PESCIEX triple quadrupole instrument (PESCIEX API4000) was used with electro spray-positive ionization mode. The multiple reaction monitor was set at transitions 260.1-116.0 m/z for propranolol, 276.0-116.0 m/z for 4-OH-propranolol, 423.0-207.0 m/z for losartan, 473.0-235.0 m/z for EXP3179, 425.1-207.3 m/z for eprosartan, and 309.1-251.1 m/z for warfarin. The ionspray voltage was set at 5500 and the temperature at 450°C. The collision energy was set at 27eV for propranolol, 25eV for 4-OH-propranolol and EXP3179, 50eV for losartan, and 35eV for eprosartan. An analytical Waters Symmetry Column C18 (2.1×50mm, 5μm particle size; Symmetry Columns, Milford, Massachusetts) was used in a Shimadzu liquid chromatography system. The mobile phase consisted of 70% methanol:30% water containing 0.1% formic acid for propranolol and 4-OH-propranolol. For losartan and EXP3179, the mobile phase consisted of 55%acetonitrile:45% water containing 0.2% of formic acid. Finally, for eprosartan, the mobile phase consisted of 28%acetonitrile:72%water containing 0.5% of formic acid. Twenty microliter aliquots were injected, and the flow rate was set at 0.3ml/min into the mass system.
Data/Statistical Analysis
ANOVA statistical analysis was used to compare the differences between groups, subsequently a t-test was carried out if ANOVA showed statistical significance, a difference was considered statistically significant different if p< 0.05. Area under the concentration-time curve (AUC) for parent and metabolite was determined from time 0 to tlast by the trapezoidal rule.
Results
Effect of uremic toxins on uptake of [3H]-estrone sulfate into transfected cells
To determine the effect of uremic toxins on drug transport we carried out uptake studies on transfected cells systems, using [3H]-estrone sulfate (4.3nM) as a prototypical substrate for uptake transporters. Rifampin (10μM), a potent inhibitor of OATPs was used as a positive control. MK-571 (100μM), an inhibitor of OATP and MRPs, was also used as a positive control. The uptake of [3H]-estrone sulfate in OATP1B1 transfected cells was decreased (p<0.05) on average by 50% in the pcMV6 empty vector transfected cells suggesting some passive absorption. In Fig. 2a we depict absolute uptake concentrations and statistically significant differences compared to controls. However, relative to pcMV6 empty vector, inhibition of active uptake was significantly decreased by 100%, 80%, 100% and 90% in the presence of rifampin (10 μM), indole-3-acetic acid (8 μM), p-cresol (300μM) and homocystein (200μM, respectively.
Figure 2.
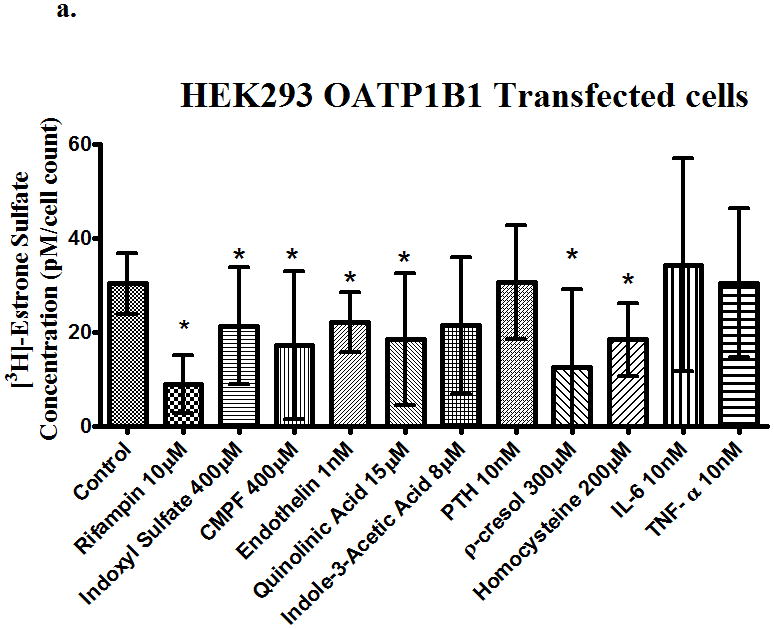
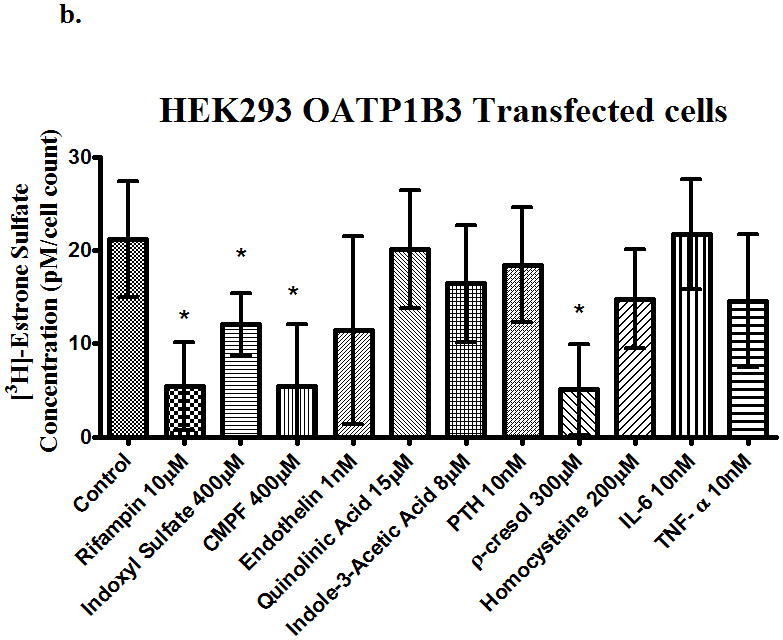
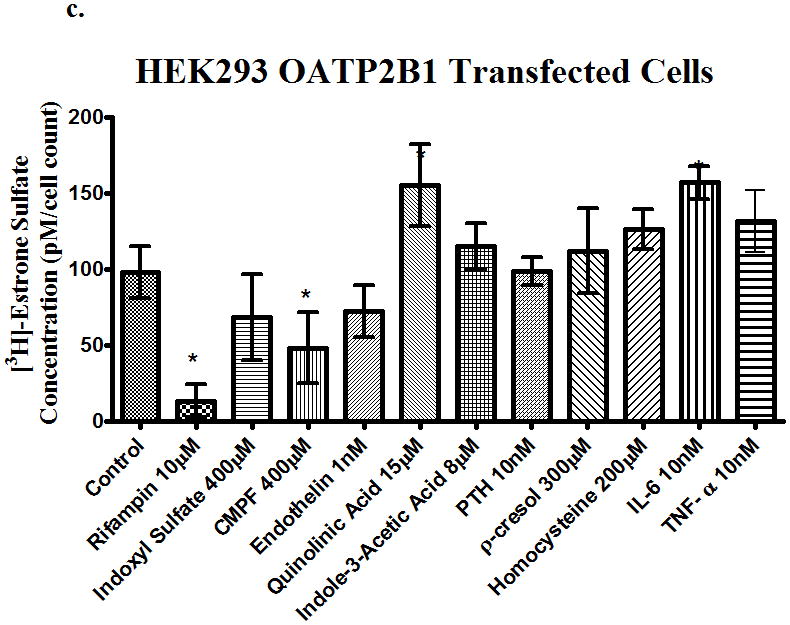
Uptake of [3H]-estrone sulfate in (a) OATP1B1, (b) OATP2B1, (c) OATP1B3 and transfected cells in the presence of uremic toxins. Control is transporter-expressing transfected cell without uremic toxin or inhibitor. Values corrected by subtracting pc-MV6-empty vector uptake values. Average ± SD, * p<0.05 n=6.
The uptake of [3H]-estrone sulfate in OATP1B3 transfected cells was also decreased (p<0.05) on average by 50% in the pcMV6 empty vector control. From Fig. 2b, inhibition of active relative to pcMV6 empty vector was significantly decreased by 100%, 79%, 100%, 85%, and 100% in the presence of rifampin (10μM), indoxyl sulfate(400μM), CMPF (400μM), endothelin (1nM), and p-cresol (300μM), respectively.
The uptake of [3H]-estrone sulfate in OATP2B1 transfected cells was decreased (p<0.05) on average by 80% in pcMV6 empty vector control. From Fig. 2c, inhibition of active uptake relative to pcMV6 empty vector was significantly decreased by 100%, 67%, 82%, and 65%, in the presence of rifampin (10μM), indoxyl sulfate(400μM), CMPF (400μM), and endothelin (1nM), respectively.
Uptake of BDDCS drugs by rat hepatocytes in the presence of uremic toxins
Using a prototypical substrate such as [3H]-estrone sulfate allowed us to investigate the effects of uremic toxins on drug transporters. Next, we investigated the effects of uremic toxins on different BDDCS drugs. We used propranolol (10 μM) as our Class 1 drug (highly soluble, highly permeable, and extensively metabolized), losartan (10 μM) as our Class 2 drug (low solubility, high permeability, and extensively metabolized), and eprosartan (10 μM) as our Class 4 drug (high solubility, low permeability, and poorly metabolized). The uptake of propranolol in rat hepatocytes (Figure 3a) was not significantly changed with any of the uremic toxins or with the known uptake inhibitors rifampin and MK-571. The uptake of losartan 10μM (Figure 3b) was inhibited (p<0.05) by 52%, 48%, 22%, 23%, 22%, 26%, and 28% in the presence of rifampin, MK-571, endothelin, para thyroid hormone (PTH), p-cresol, homocysteine, and IL-6, respectively. This indicates that losartan is a substrate for uptake transporters since rifampin and MK-571 inhibited the uptake of losartan and the uremic toxins also had inhibitory effects. Lastly, the uptake of eprosartan (Figure 3c) was also shown to be inhibited by (p<0.05) 22%, 30%, 19%, 21%, and 17% in the presence of rifampin, MK-571, quinolinic acid, indole-3-acetic acid, and homocysteine, respectively. This suggests that the uptake of eprosartan is also mediated by uptake drug transporters expressed in hepatocytes. Uptake of [3H]-estrone sulfate in the presence and absence of rifampin in hepatocytes was also assessed to confirm the active transport of the compound into the hepatocytes (data not shown).
Figure 3.
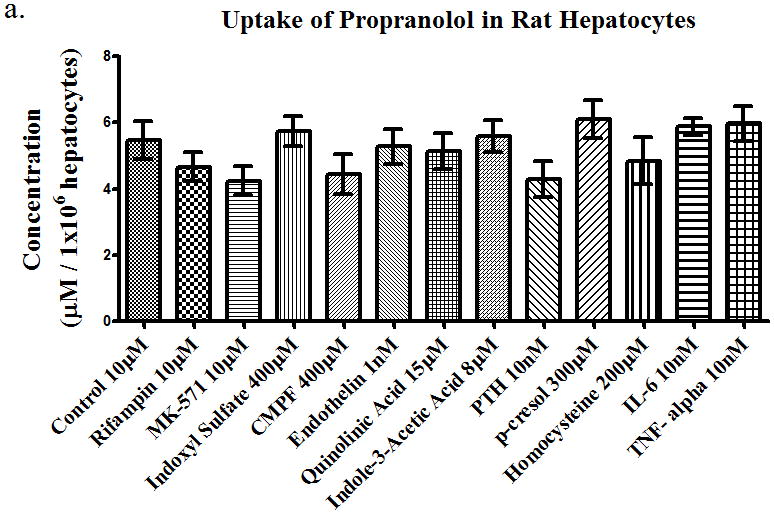
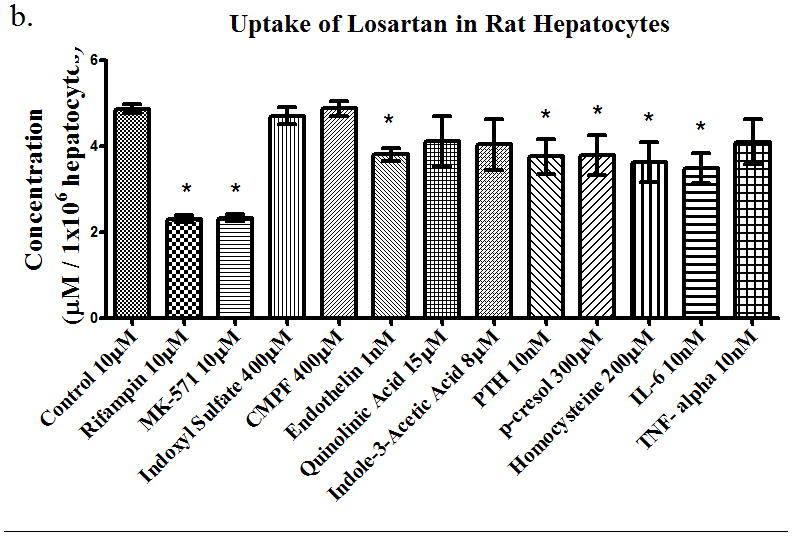
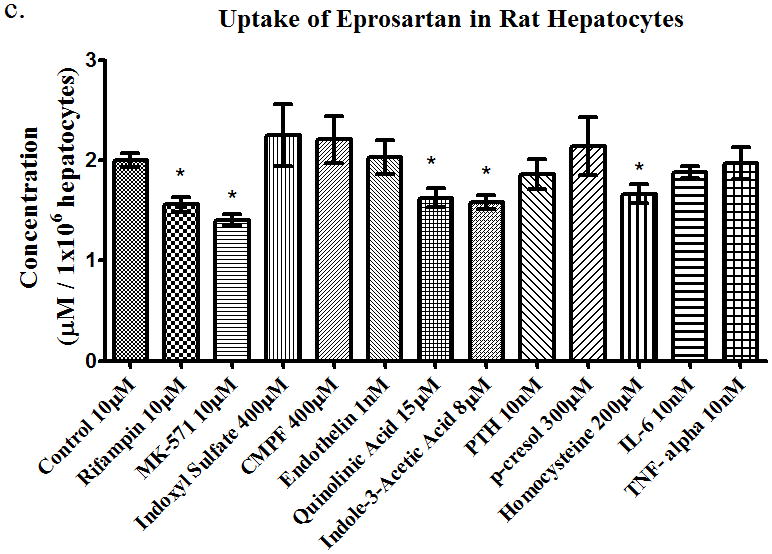
Uptake of (a) propranolol 10μM, (b) losartan 10μM, and (c) eprosartan 10μM in rat hepatocytes in the presence of uremic toxins or uptake transporter inhibitors. Average ± SD *p<0.05, n=4.
Effects of hemodialysis serum on drug uptake
Serum from patients on hemodialysis was obtained from the outpatient hemodialysis unit at UCSF to carry out in vitro uptake studies, to determine the collective effects of uremic toxins on the uptake of the three BDDCS drugs. Propranolol (10μM), losartan (10μM), and eprosartan (10μM) were assayed for their uptake into rat heptocytes (Figure 4a) in the presence of normal serum, hemodialysis serum or normal serum plus rifampin (10μM) and the CYP inhibitors quinidine (30μM) for propranolol and sulfaphenazole (1μM) for losartan. There were no significant differences in uptake of propranolol in the presence of hemodialysis serum or rifampin, which agrees with our in vitro studies of the uptake of propranolol in the presence or uremic toxins. The uptake of losartan was decreased by 28% and 66% in the presence of HD serum and rifampin respectively, compared to normal serum. There were also no significant differences in the uptake of eprosartan in the presence of HD serum or rifampin. The same experiment was repeated using cryopreserved human hepatocytes (Figure 4b). We observed the same results, with no significant changes in uptake of propranolol or eprosartan in the presence of HD serum or rifampin, but the uptake of losartan was decreased (p<0.05) by 48% and 55% in the presence of HD serum and rifampin, respectively. However, the uptake of eprosartan in human hepatocytes was much lower than that observed in rat hepatocytes, while the opposite result was seen with propranolol. These studies indicated that the uremic toxins contained in the HD serum do have an effect on uptake of a Class 2 drug, therefore we investigated the effects of HD serum in longer time-course studies to determine the effects it may have in metabolism and uptake over an hour.
Figure 4.
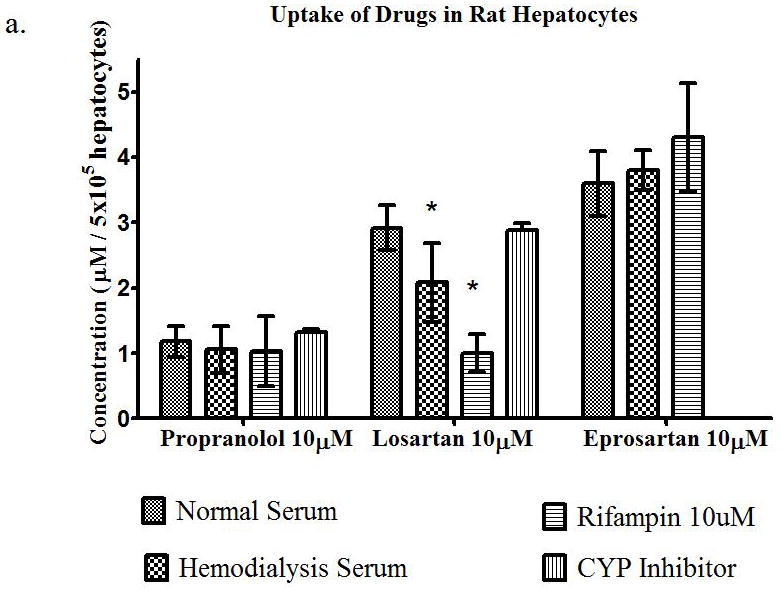
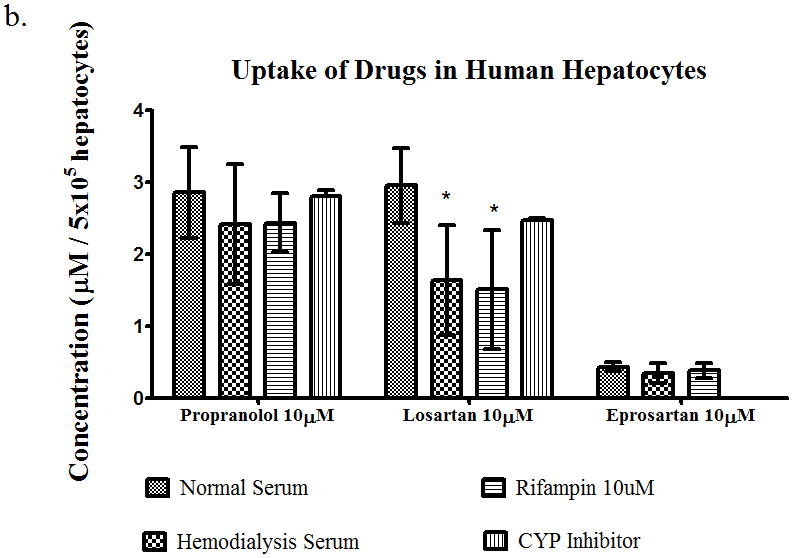
Uptake of propranolol 10μM, losartan 10μM, or eprosartan 10μM in (a) rat hepatocytes and (b) human hepatocytes in the presence of normal serum, hemodyalisis serum, rifampin 10μM, or CYP inhibitor. Average ± SD, *p<0.05, n=4.
Time-course studies in rat hepatocytes and microsome studies
Time-course studies were carried out using freshly isolated rat hepatocytes over an hour for propranolol (10μM), losartan (1μM), or eprosartan (10μM). These concentrations were chosen since they are representative of the plasma concentrations in humans. We analyzed the total drug concentration over time, the intracellular drug concentration, and the total metabolite concentration over time. The intracellular metabolite concentrations were below the limit of quantitation. Quinidine (30μM) was used as a CYP2D6 inhibitor for propranolol and sulfaphenazole (1μM) was used as a CYP2C9 inhibitor for losartan. For the total concentration of propranolol over time, we saw no significant change in parent drug in the presence of rifampin or HD serum. We observed a 37% increase in AUC (area under the curve) in the quinidine treated time course samples vs. those treated with normal serum (Table 1). The intracellular AUC of propranolol in hepatocytes (Table 1) increased over time by 243% in the quinidine treated group. The metabolite AUC (Table 1) did not change significantly in any of the groups. The microsomes studies showed that rifampin and quinidine both inhibit metabolism in rat microsomes (Figure 5a), whereas only quinidine inhibited propranolol metabolism in human microsomes (Figure 5b).
Table 1.
Propranolol and 4-OH-propranolol AUC 0→60min for time-course studies and/fold increase for significant changes vs. normal serum in rat hepatocytes. * p<0.05, n=4.
| Propranolol 1μM | Normal Serum | Hemodialysis Serum | Rifampin 10μM | Quinidine 30μM |
|---|---|---|---|---|
| AUC of Cumulative Drug (μM* min) | 410 ± 38 | 403 ± 87 | 421 ± 39 | *561 ± 18/1.37 |
| AUC of Intracellular Drug (μM* min) | 2.67 ± 1.14 | 3.48 ± 2.49 | 3.20 ± 0.46 | *6.51±0.44/2.44 |
| AUC of Cumulative Metabolite (μM* min) | 31.9 ± 17 | 40.3 ± 26.0 | 36.1 ± 24.0 | 31.0 ± 10.0 |
Results shown as average ± SD, /ratio compared to normal serum,
p<0.05 by ANOVA
Figure 5.
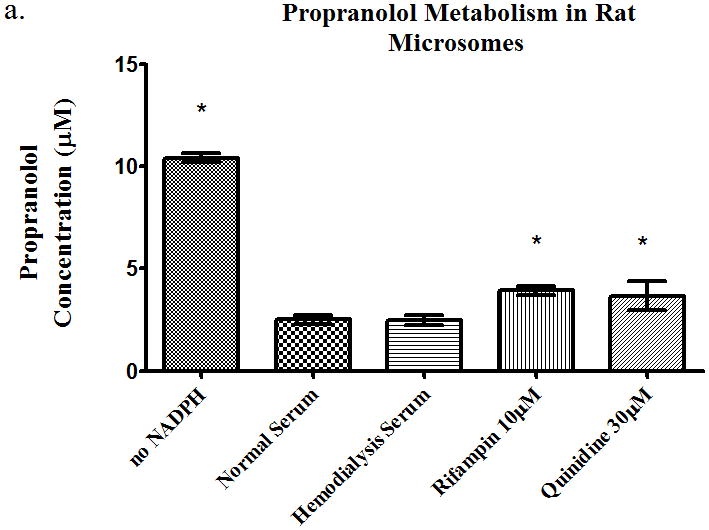
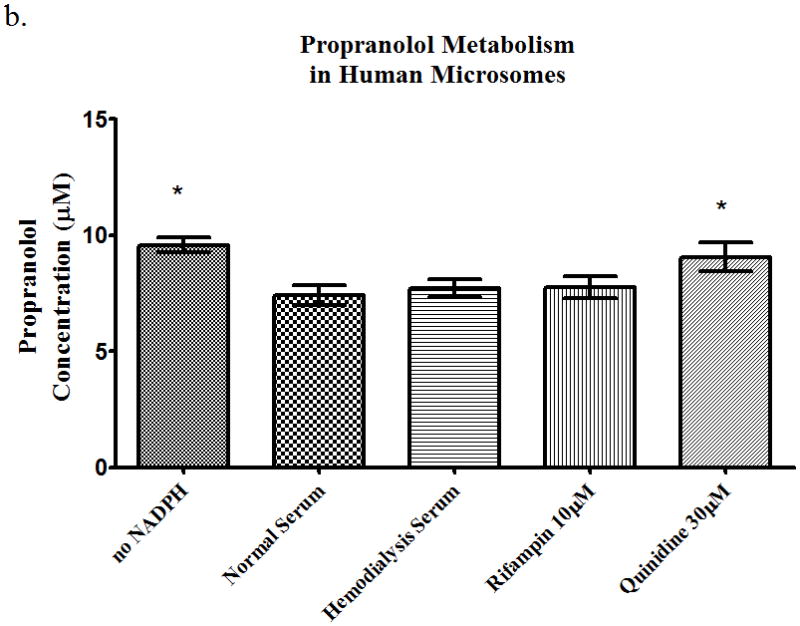
Metabolism of propranolol 10μM in pooled (a) rat microsomes and (b) human microsomes in the presence of nomal serum, hemodialysis serum, uptake transporter inhibitor, or CYP inhibitor. Average ± SD, * p<0.05 (relative to normal serum group), n=4.
The losartan time-course study showed a 27%, 65%, and 68% increase (p<0.05) in AUC in the presence of hemodialysis serum, rifampin, and sulfaphenazole, respectively (Table 2). The intracellular AUC of losartan (Table 2) decreased (p<0.05) by 26%, 47%, and 33% in the presence of HD serum, rifampin and sulfaphenazole, respectively. The metabolite AUC (Table 2) decreased (p<0.05) 41% and 26% in the presence of rifampin, and sulfaphenazole. The rat microsome studies showed a decrease in metabolism in the presence of sulfaphenazole (Figure 6a), i.e. significant increase in parent drug. Sulfaphenazole is reported to be a strong inhibitor of CYP2C9. In contrast, human microsome studies showed no inhibition of metabolism of losartan in the presence of sulfaphenazole at 1μM and 100μM (Figure 6b).
Table 2.
Losartan and EXP3174 AUC 0→60min for time-course studies and/fold increase or decrease for significant changes vs. normal serum in rat hepatocytes. n=4.
| Losartan 1μM | Normal Serum | Hemodialysis Serum | Rifampin 10μM | Sulfaphenazole 1μM |
|---|---|---|---|---|
| AUC of Cumulative Drug (μM* min) | 32.1 ± 9.4 | *40.8 ± 6.2/1.27 | *53.0 ± 18.7/1.65 | *54.0 ± 19.8/1.68 |
| AUC of Intracellular Drug (μM* min) | 44.6 ± 17.1 | *33.1 ± 20.4/0.74 | *23.7 ± 14.1/0.53 | *30.1 ± 20.9/0.67 |
| AUC of Cumulative Metabolite (μM* min) | 1.47 ± 0.97 | 1.66±1.05 | *0.87 ± 0.47/0.59 | *1.09 ± 0.66/0.74 |
Results shown as average ± SD, /ratio compared to normal serum,
p<0.05 by ANOVA, n=3
Figure 6.
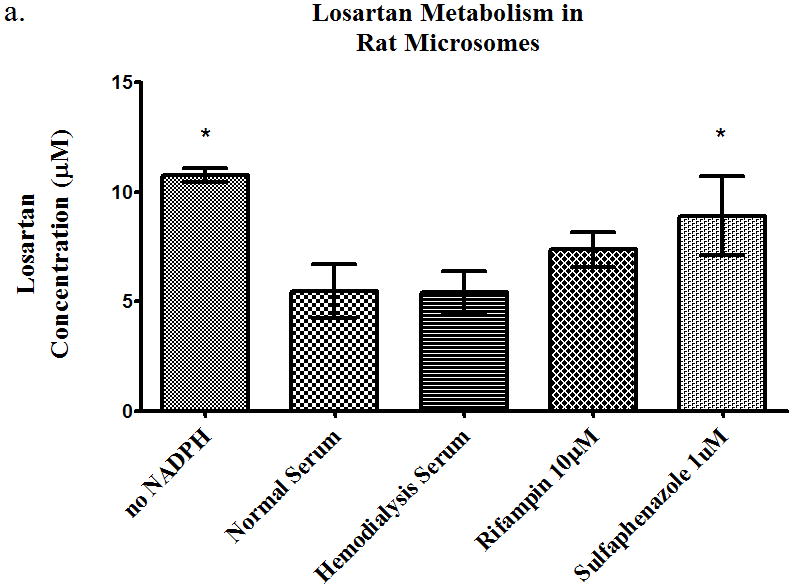
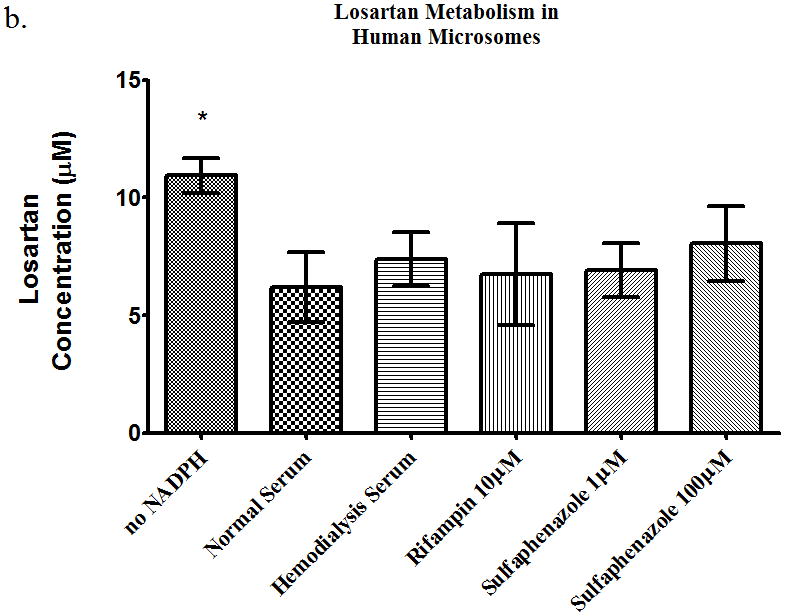
Metabolism of losartan 10 μM in pooled (a) rat microsomes and (b) human microsomes in the presence of nomal serum, hemodialysis serum, uptake transporter inhibitor, or CYP inhibitor. Average ± SD, * p<0.05 (relative to normal serum group), n=4.
The eprosartan time-course study showed no change in cumulative AUC of drug in the presence of hemodialysis serum and rifampin (Table 3), which corresponds to the drug not undergoing metabolism. The intracellular AUC increased by 91% in the presence of hemodialysis serum (Table 3). This indicates that eprosartan was taken up into the cell either actively and/or passively, but once inside the cell, it did not readily exit the cell in the presence of HD serum.
Table 3.
Eprosartan AUC 0→60min for time-course studies and/fold increase for significant changes vs. normal serum in rat hepatocytes.
| Eprosartan 10μM | Normal Serum | Hemodialysis Serum | Rifampin 10μM |
|---|---|---|---|
| AUC of Cumulative Drug (μM* min) | 575 ± 32 | 615 ± 60 | 572 ± 37 |
| AUC of Intracellular Drug (μM* min) | 59.7 ± 18.2 | *114 ± 23/1.91 | 74.7 ± 27.2 |
Results shown as average ± SD, /ratio compared to normal serum,
p<0.05 by ANOVA, n=4
Discussion
Hepatic drug transporters have been widely recognized to play a significant role in the disposition of drugs that undergo hepatic metabolism and elimination 14,15. These transporters are known to transport endogenous substances such as hormones and other compounds that are classified as cations and anions. In the last 15 years transporters have been studied for their role in transporting xenobiotics. In the liver, uptake transporters facilitate the drug’s transport from the basolateral side of the cell membrane (the blood side) into the hepatocyte, and efflux transporters facilitate the exit of the drug from inside the cell back into the blood through a basolateral efflux transporter or into the bile through an apical (canalicular) efflux transporter. Studies have shown that these transporters play a role in drug-drug interactions and also affect the metabolism of drugs by altering the access of the drug to the enzyme 14,16–19. Polymorphisms in these transporters have been shown to be of clinical relevance, by which alterations in the transporter expression or function due to mutations can lead to a clinically significant change in drug pharmacokinetics or drug response 20–22 These transporters are of clinical relevance due to their effects on drug pharmacokinetics, drug pharmacodynamics, and drug-drug interactions.
The role of drug transporters in disease, such as chronic kidney disease or renal failure, has not been investigated extensively. Studies show that in patients with chronic kidney disease there are pharmacokinetic changes of non-renally eliminated drugs23–28. Studies have investigated the effect of uremic toxins on drug metabolism, and it has been reported that some uremic toxins inhibit some CYP450 metabolizing enzyme but not others 8,29–33. However, since drug transporters have a significant effect on drug disposition, we investigated whether the uremic toxins and HD serum from patients with chronic kidney disease have an effect on drug transporters. Furthermore, we investigated whether different drugs would be differentially affected by uremic toxins depending on the drug’s properties that would likely require them to be transported by a hepatic drug transporter. In the BDDCS classification system, the drug’s solubility, permeability and extent of metabolism, predicts whether a transporter could have a significant effect on drug disposition. In these studies, we showed that some uremic toxins have an inhibitory effect on the prototypical OATP substrate [3H]-estrone sulfate, in cells transfected with OATP1B1, OATP2B1 and OATP1B3. The uptake by the OATP transfected cells was approximately 50% greater compared to the pcMV6-empty vector control, most likely due to estrone sulfate’s affinity for multiple transporters. The differences in substrate uptake in different OATPs may also be due to differences in transfection efficiency from one transporter to another.
Losartan and eprosartan have been previously shown to be substrates for uptake and/or efflux transporters 34,35. Uptake studies were carried out using the drugs propranolol, losartan, and eprosartan, in the presence of uremic toxins in rat hepatocytes. In these studies we found no significant inhibition or increase of uptake for propranolol in the presence of uremic toxins or rifampin. Since propranolol is a Class 1 drug, highly soluble and permeable, BDDCS predicts that transporters do not play a significant role in the drug’s intestinal and hepatic disposition. Hence, if uremic toxins are having an effect on hepatic transporters, this would not affect the uptake of propranolol, which is consistent with our results. On the other hand, the uptake of losartan and eprosartan was inhibited by some uremic toxins as well as rifampin (OATP inhibitor). This suggests that both are substrates for uptake transporters and that uremic toxins have an inhibitory effect on these transporters.
In these studies, uremic toxins were tested separately, however; since some uremic toxins had an effect on one transporter but not other, we obtained human HD serum from patients on hemodialysis to account for in vivo protein binding of the uremic toxins and for any synergistic effects due to uremic toxins. Preliminary studies we did showed that there were no changes in [3H]-estrone sulfate in the presence of 1% or 10% BSA or HD serum. Thus, we decided to use 10% serum for hepatocyte studies to represent a closer physiological condition since the liver is bathed by blood, which contains approximately 50% serum. When we tested propranolol, losartan, and eprosartan in the presence of HD serum in rat hepatocytes, we observed a significant decrease in losartan uptake in the presence of HD serum and rifampin. However we did not see this inhibitory effect on the uptake of propranolol or eprosartan. We repeated the experiment using human cryopreserved hepatocytes and observed the same results. We predicted that there would be no inhibitory effect in the uptake of the Class 1 drug propranolol in the presence of HD serum; however, we had expected to see an inhibitory effect in the uptake of eprosartan. We cannot explain why eprosartan uptake was not affected in rat or human hepatocytes in the presence of HD serum, and further studies are necessary to understand this result. The experiments from Figure 3 used uremic toxins individually, whereas these experiments used HD serum, in which many more uremic toxins may be present, which may be influencing rifampin’s activity to block uptake transporters.
The hepatocyte time-course studies were carried out in order to study the influence of metabolism and uptake in liver cells rather than only transfected cell systems. In addition, using hepatocytes would show how HD serum affects the enzyme-transporter interplay in drug disposition, whereas microsomes would only show the effects of HD serum on metabolism but not transport since no drug membrane transporters are present in microsomes. In the hepatocyte time-course studies of Class 1 drug, propranolol, the cumulative propranolol AUC increased in the quinidine (CYP inhibitor) treated group only compared to the normal serum group. As expected, the AUC of intracellular propranolol increased in the quinidine treated group only, but there were no changes in the cumulative AUC of 4-OH-propranolol. In the quinidine treated groups, the increase in intracellular propranolol is due to the decrease in metabolism as indicated by the rat and human microsome studies. The rat and human hepatocyte 2 min uptake studies indicate that HD serum, rifampin, or quinidine, have no effect in the uptake of propranolol (Figure 4). In the quinidine treated group, we suspect that we did not see a change in metabolite formation, due to the increase in intracellular parent compound (secondary to the inhibition of metabolism by quinidine). The increase in the amount of intracellular propranolol, apparently was sufficient to produce enough metabolite and not cause a significant difference in metabolite AUC compared to the normal serum treated group. These findings are evidence that in the presence of hemodialysis serum or rifampin there is no change in uptake or metabolism of propranolol.
In the time-course studies of the class 2 drug, losartan; the AUC of cumulative losartan was higher in the presence of hemodialysis serum, rifampin, and sulfaphenazole compared to the normal serum group. This suggests that the metabolism in those groups was decreased either due to a decrease in CYP450 activity or decrease in uptake of the drug, which would lead to less drug being exposed to the enzyme for metabolism. The rat microsome studies showed that there was no change in metabolism in the presence of HD serum or rifampin, but there was inhibition of metabolism in the presence of sulfaphenazole. Also, the intracellular AUC of losartan decreased in the presence of HD serum, rifampin, and sulfaphenazole. These observations suggest that HD serum is decreasing the uptake of losartan into the hepatocytes but not affecting the metabolism of losartan. Also, sulfaphenazole is affecting only the metabolism and not the uptake of losartan (as indicated by the rat and human hepatocyte 2min uptake results in the presence of sulfaphenazole, Figure 4). The AUC of the cumulative metabolite showed that there was a decrease in metabolite in the presence of rifampin and sulfaphenazole, however there was no change in metabolite formation in the presence of hemodialysis serum. The rat microsome studies indicate a slight inhibition (but not statistically significant) of metabolism of losartan in the presence of rifampin. The decrease in metabolite in the rifampin treated group is due to the inhibition of uptake, which in turn will expose less of the drug to the metabolizing enzymes and decrease the metabolite formation. In the sulfaphenazole treated group, the decrease in metabolite formation is a direct effect of metabolism inhibition but not uptake. The HD treated group showed no significant change in the metabolite formation, although there was a significant decrease in uptake.
For the time course studies of the Class 4 drug, eprosartan, the cumulative AUC of the drug in all groups remained the same since this drug is not metabolized. The intracellular AUC of eprosartan increased in the presence of hemodialysis serum. This indicates that eprosartan was transported into the cell or passively diffused into the cell, but once inside the cell it was unable to be effluxed, leading us to believe that efflux transporters are being inhibited by hemodialysis serum and not allowing the drug to readily exit the cell.
Conclusions
We have demonstrated that uremic toxins and hemodialysis serum can have inhibitory effects in drug transport, but not necessarily on phase I metabolism. Uremic toxins and hemodialysis serum did not have an effect on propranolol uptake in hepatocytes or transfected cells. On the other hand, uremic toxins and hemodialysis serum did show inhibitory effects on the transport of losartan and eprosartan. These findings indicate that in chronic kidney disease patients, the pharmacokinetics of certain drugs may be greatly affected by their disease state and that dose adjustment for non-renally excreted drugs may be necessary for prevention of drug toxicity or to reach the desired therapeutic effect. Also, these findings indicate that studying the effect of uremic toxins on drug transport is important in drug development in order to dose properly the patients in a special population such as kidney disease. Further studies will be carried out in our laboratory to determine the hepatic and biliary clearance of these drugs in rats in the presence of hemodialysis serum and normal serum.
New recommendations from the Food and Drug Administration suggest a full pharmacokinetic study for new drugs in chronic kidney disease patients, even when renal excretion is minimal 36. Being able to predict based on BDDCS class if a drug will be affected by uremic toxins can be useful in deciding what studies may be necessary for a new drug with respect to various patient populations. It is important to consider different disease states and how they can affect drug transporter activity and/or expression and how these changes can have consequences in drug pharmacokinetics and pharmacodynamics.
Figure 1.
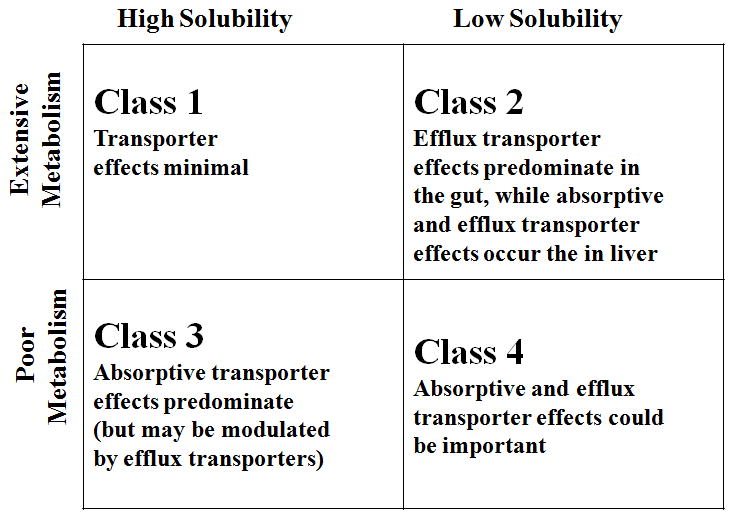
Biopharmaceutics Drug Disposition Classification System (BDDCS). Transporter effects followed by oral dosing (Shugarts and Benet, 2010).
Acknowledgments
We thank GlaxoSmithKline for the gracious donation of compound GG918 (GF120918). This work was supported in part by NIH grants from GM075900 and GM061390 during portions of this work Maribel Reyes received support as an AFPE fellow and as UCSF research mentorship fellow.
Nonstandard Abbreviations
- BDDCS
Biopharmaceutics Drug Disposition Classification System
- CKD
chronic kidney disease
- HD
hemodialysis
References
- 1.Lau YY, Okochi H, Huang Y, Benet LZ. Multiple transporters affect the disposition of atorvastatin and its two active hydroxy metabolites: application of in vitro and ex situ systems. The Journal of pharmacology and experimental therapeutics. 2006;316(2):762–771. doi: 10.1124/jpet.105.093088. [DOI] [PubMed] [Google Scholar]
- 2.Lau YY, Wu CY, Okochi H, Benet LZ. Ex situ inhibition of hepatic uptake and efflux significantly changes metabolism: hepatic enzyme-transporter interplay. The Journal of pharmacology and experimental therapeutics. 2004;308(3):1040–1045. doi: 10.1124/jpet.103.061770. [DOI] [PubMed] [Google Scholar]
- 3.Lam JL, Okochi H, Huang Y, Benet LZ. In vitro and in vivo correlation of hepatic transporter effects on erythromycin metabolism: characterizing the importance of transporter-enzyme interplay. Drug metabolism and disposition: the biological fate of chemicals. 2006;34(8):1336–1344. doi: 10.1124/dmd.106.009258. [DOI] [PubMed] [Google Scholar]
- 4.Vanholder R, De Smet R, Glorieux G, Argiles A, Baurmeister U, Brunet P, Clark W, Cohen G, De Deyn PP, Deppisch R, Descamps-Latscha B, Henle T, Jorres A, Lemke HD, Massy ZA, Passlick-Deetjen J, Rodriguez M, Stegmayr B, Stenvinkel P, Tetta C, Wanner C, Zidek W. Review on uremic toxins: classification, concentration, and interindividual variability. Kidney international. 2003;63(5):1934–1943. doi: 10.1046/j.1523-1755.2003.00924.x. [DOI] [PubMed] [Google Scholar]
- 5.Tsujimoto M, Kinoshita Y, Hirata S, Otagiri M, Ohtani H, Sawada Y. Effects of Uremic Serum and Uremic Toxins on Hepatic Uptake of Digoxin. Therapeutic drug monitoring. 2008 doi: 10.1097/FTD.0b013e3181838077. [DOI] [PubMed] [Google Scholar]
- 6.Sun H, Huang Y, Frassetto L, Benet LZ. Effects of uremic toxins on hepatic uptake and metabolism of erythromycin. Drug metabolism and disposition: the biological fate of chemicals. 2004;32(11):1239–1246. doi: 10.1124/dmd.104.000521. [DOI] [PubMed] [Google Scholar]
- 7.Sun H, Frassetto LA, Huang Y, Benet LZ. Hepatic clearance, but not gut availability, of erythromycin is altered in patients with end-stage renal disease. Clinical pharmacology and therapeutics. 2010;87(4):465–472. doi: 10.1038/clpt.2009.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guevin C, Michaud J, Naud J, Leblond FA, Pichette V. Down-regulation of hepatic cytochrome p450 in chronic renal failure: role of uremic mediators. British journal of pharmacology. 2002;137(7):1039–1046. doi: 10.1038/sj.bjp.0704951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Michaud J, Dube P, Naud J, Leblond FA, Desbiens K, Bonnardeaux A, Pichette V. Effects of serum from patients with chronic renal failure on rat hepatic cytochrome P450. British journal of pharmacology. 2005;144(8):1067–1077. doi: 10.1038/sj.bjp.0706138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deguchi T, Ohtsuki S, Otagiri M, Takanaga H, Asaba H, Mori S, Terasaki T. Major role of organic anion transporter 3 in the transport of indoxyl sulfate in the kidney. Kidney international. 2002;61(5):1760–1768. doi: 10.1046/j.1523-1755.2002.00318.x. [DOI] [PubMed] [Google Scholar]
- 11.Wu CY, Benet LZ. Predicting drug disposition via application of BCS: transport/absorption/elimination interplay and development of a biopharmaceutics drug disposition classification system. Pharmaceutical research. 2005;22(1):11–23. doi: 10.1007/s11095-004-9004-4. [DOI] [PubMed] [Google Scholar]
- 12.Salphati L, Benet LZ. Metabolism of digoxin and digoxigenin digitoxosides in rat liver microsomes: involvement of cytochrome P4503A. Xenobiotica. 1999;29(2):171–185. doi: 10.1080/004982599238722. [DOI] [PubMed] [Google Scholar]
- 13.Shitara Y, Li AP, Kato Y, Lu C, Ito K, Itoh T, Sugiyama Y. Function of uptake transporters for taurocholate and estradiol 17beta-D-glucuronide in cryopreserved human hepatocytes. Drug metabolism and pharmacokinetics. 2003;18(1):33–41. doi: 10.2133/dmpk.18.33. [DOI] [PubMed] [Google Scholar]
- 14.Shugarts S, Benet LZ. The role of transporters in the pharmacokinetics of orally administered drugs. Pharmaceutical research. 2009;26(9):2039–2054. doi: 10.1007/s11095-009-9924-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamazaki M, Suzuki H, Sugiyama Y. Recent advances in carrier-mediated hepatic uptake and biliary excretion of xenobiotics. Pharmaceutical research. 1996;13(4):497–513. doi: 10.1023/a:1016077517241. [DOI] [PubMed] [Google Scholar]
- 16.Lu C, Gallegos R, Li P, Xia CQ, Pusalkar S, Uttamsingh V, Nix D, Miwa GT, Gan LS. Investigation of drug-drug interaction potential of bortezomib in vivo in female Sprague-Dawley rats and in vitro in human liver microsomes. Drug metabolism and disposition: the biological fate of chemicals. 2006;34(4):702–708. doi: 10.1124/dmd.105.008060. [DOI] [PubMed] [Google Scholar]
- 17.Zamek-Gliszczynski MJ, Kalvass JC, Pollack GM, Brouwer KL. Relationship between drug/metabolite exposure and impairment of excretory transport function. Drug metabolism and disposition: the biological fate of chemicals. 2009;37(2):386–390. doi: 10.1124/dmd.108.023648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith NF, Figg WD, Sparreboom A. Role of the liver-specific transporters OATP1B1 and OATP1B3 in governing drug elimination. Expert opinion on drug metabolism & toxicology. 2005;1(3):429–445. doi: 10.1517/17425255.1.3.429. [DOI] [PubMed] [Google Scholar]
- 19.Zheng HX, Huang Y, Frassetto LA, Benet LZ. Elucidating rifampin’s inducing and inhibiting effects on glyburide pharmacokinetics and blood glucose in healthy volunteers: unmasking the differential effects of enzyme induction and transporter inhibition for a drug and its primary metabolite. Clinical pharmacology and therapeutics. 2009;85(1):78–85. doi: 10.1038/clpt.2008.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Degorter MK, Kim RB. Hepatic drug transporters, old and new: pharmacogenomics, drug response, and clinical relevance. Hepatology (Baltimore, Md. 2009;50(4):1014–1016. doi: 10.1002/hep.23233. [DOI] [PubMed] [Google Scholar]
- 21.Oswald S, Konig J, Lutjohann D, Giessmann T, Kroemer HK, Rimmbach C, Rosskopf D, Fromm MF, Siegmund W. Disposition of ezetimibe is influenced by polymorphisms of the hepatic uptake carrier OATP1B1. Pharmacogenetics and genomics. 2008;18(7):559–568. doi: 10.1097/FPC.0b013e3282fe9a2c. [DOI] [PubMed] [Google Scholar]
- 22.Kivisto KT, Niemi M. Influence of drug transporter polymorphisms on pravastatin pharmacokinetics in humans. Pharmaceutical research. 2007;24(2):239–247. doi: 10.1007/s11095-006-9159-2. [DOI] [PubMed] [Google Scholar]
- 23.Gonzalez-Martin G, Thambo S, Paulos C, Vasquez I, Paredes J. The pharmacokinetics of nifurtimox in chronic renal failure. European journal of clinical pharmacology. 1992;42(6):671–673. doi: 10.1007/BF00265935. [DOI] [PubMed] [Google Scholar]
- 24.Hilger RA, Richly H, Grubert M, Kredtke S, Thyssen D, Eberhardt W, Hense J, Schuler M, Scheulen ME. Pharmacokinetics of sorafenib in patients with renal impairment undergoing hemodialysis. International journal of clinical pharmacology and therapeutics. 2009;47(1):61–64. doi: 10.5414/cpp47061. [DOI] [PubMed] [Google Scholar]
- 25.Small DS, Wrishko RE, Ernest CS, 2nd, Ni L, Winters KJ, Farid NA, Li YG, Brandt JT, Salazar DE, Borel AG, Kles KA, Payne CD. Prasugrel pharmacokinetics and pharmacodynamics in subjects with moderate renal impairment and end-stage renal disease. Journal of clinical pharmacy and therapeutics. 2009;34(5):585–594. doi: 10.1111/j.1365-2710.2009.01068.x. [DOI] [PubMed] [Google Scholar]
- 26.Nolin TD, Frye RF, Le P, Sadr H, Naud J, Leblond FA, Pichette V, Himmelfarb J. ESRD impairs nonrenal clearance of fexofenadine but not midazolam. J Am Soc Nephrol. 2009;20(10):2269–2276. doi: 10.1681/ASN.2009010082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martin DE, Chapelsky MC, Ilson B, Tenero D, Boike SC, Zariffa N, Jorkasky DK. Pharmacokinetics and protein binding of eprosartan in healthy volunteers and in patients with varying degrees of renal impairment. Journal of clinical pharmacology. 1998;38(2):129–137. doi: 10.1002/j.1552-4604.1998.tb04401.x. [DOI] [PubMed] [Google Scholar]
- 28.Nolin TD. Altered nonrenal drug clearance in ESRD. Current opinion in nephrology and hypertension. 2008;17(6):555–559. doi: 10.1097/MNH.0b013e3283136732. [DOI] [PubMed] [Google Scholar]
- 29.Nolin TD, Naud J, Leblond FA, Pichette V. Emerging evidence of the impact of kidney disease on drug metabolism and transport. Clinical pharmacology and therapeutics. 2008;83(6):898–903. doi: 10.1038/clpt.2008.59. [DOI] [PubMed] [Google Scholar]
- 30.Pichette V, Leblond FA. Drug metabolism in chronic renal failure. Current drug metabolism. 2003;4(2):91–103. doi: 10.2174/1389200033489532. [DOI] [PubMed] [Google Scholar]
- 31.Leblond F, Guevin C, Demers C, Pellerin I, Gascon-Barre M, Pichette V. Downregulation of hepatic cytochrome P450 in chronic renal failure. J Am Soc Nephrol. 2001;12(2):326–332. doi: 10.1681/ASN.V122326. [DOI] [PubMed] [Google Scholar]
- 32.Dreisbach AW, Lertora JJ. The effect of chronic renal failure on hepatic drug metabolism and drug disposition. Seminars in dialysis. 2003;16(1):45–50. doi: 10.1046/j.1525-139x.2003.03011.x. [DOI] [PubMed] [Google Scholar]
- 33.Michaud J, Nolin TD, Naud J, Dani M, Lafrance JP, Leblond FA, Himmelfarb J, Pichette V. Effect of hemodialysis on hepatic cytochrome P450 functional expression. Journal of pharmacological sciences. 2008;108(2):157–163. doi: 10.1254/jphs.08042fp. [DOI] [PubMed] [Google Scholar]
- 34.Flynn C, BAH, Reed G. Losartan is a Substrate of Organic Anion Transporting Polypeptide 2B1. FASEB Meeting abstract, #758.2; Anaheim, California. April 24–28.2010. [Google Scholar]
- 35.Sun H, Huang Y, Okochi H, Frassetto L, Benet LZ. Uremic Toxins Inhibit Hepatic Uptake of Eprosartan. Clinical pharmacology and therapeutics. 2005 Abstract. [Google Scholar]
- 36.Zhang Y, Zhang L, Abraham S, Apparaju S, Wu TC, Strong JM, Xiao S, Atkinson AJ, Jr, Thummel KE, Leeder JS, Lee C, Burckart GJ, Lesko LJ, Huang SM. Assessment of the impact of renal impairment on systemic exposure of new molecular entities: evaluation of recent new drug applications. Clinical pharmacology and therapeutics. 2009;85(3):305–311. doi: 10.1038/clpt.2008.208. [DOI] [PubMed] [Google Scholar]