

Abstract
Early, vigorous intrahepatic induction of interferon (IFN)-stimulated gene (ISG) induction is a feature of hepatitis C virus (HCV) infection, even though HCV inhibits the induction of type I IFNs in vitro. To identify the cytokines and cells that drive ISG induction and mediate antiviral activity during acute HCV infection, type I and III IFN responses were studied in (1) serial liver biopsies and plasma samples obtained from 6 chimpanzees throughout acute HCV infection and (2) primary human hepatocyte (PHH) cultures upon HCV infection. Type I IFNs were minimally induced at the messenger RNA (mRNA) level in the liver and were undetectable at the protein level in plasma during acute HCV infection of chimpanzees. In contrast, type III IFNs, in particular, interleukin (IL)-29 mRNA and protein, were strongly induced and these levels correlated with ISG expression and viremia. However, there was no association between intrahepatic or peripheral type III IFN levels and the outcome of acute HCV infection. Infection of PHH with HCV recapitulated strong type III and weak type I IFN responses. Supernatants from HCV-infected PHH cultures mediated antiviral activity upon transfer to HCV-replicon–containing cells. This effect was significantly reduced by neutralization of type III IFNs and less by neutralization of type I IFNs. Furthermore, IL-29 production by HCV-infected PHH occurred independently from type I IFN signaling and was not enhanced by the presence of plasmacytoid dendritic cells.
Conclusion
Hepatocyte-derived type III IFNs contribute to ISG induction and antiviral activity, but are not the principal determinant of the outcome of HCV infection.
HCV poses a significant health problem, with more than 170 million chronically infected people worldwide.1 Treatment is based on interferon (IFN)-α in combination with ribavirin and direct antiviral agents,2 but the role of IFN-α and other IFNs in the spontaneous outcome of infection is unclear.
Type I IFNs (13 IFN-α proteins plus IFN-β, IFN-ε, IFN-κ, and IFN-ϖ) form the frontline of innate host defenses by inducing an antiviral state in infected and neighboring cells, and by modulating adaptive immune responses directly and by the induction of IFN-stimulated genes ISGs.3 Whereas ISGs are strongly induced during HCV infection,4,5 neither the ISG-inducing cyto-kines nor their cellular sources have been defined. HCV has been shown to interfere with Toll-like receptor (TLR)3- and retinoic-acid–inducible gene (RIG) I–mediated induction of IFN-β and with Janus kinase/signal transducer and activator of transcription (JAK-STAT) signaling downstream of the IFN-α/β receptor,6 thus reducing IFN-α/β production to levels that are undetectable in HCV-infected patients. In vitro studies suggest that plasmacytoid dendritic cells (pDCs) may be the source of the ISG-inducing type I IFNs,7 but the role of pDCs has not been studied in the HCV-infected liver.
In this context, type III IFNs have become of interest. This family is composed of interleuking (IL)-29, IL-28A, and IL-28B, and induced in response to several viral pathogens.8 Although signaling by the JAK-STAT pathway is shared with type I IFNs and similar sets of ISGs are induced,9 receptors for type III IFNs are distinct from those for type I IFNs10 and are expressed in a cell-type–specific manner.11 In the liver, type III IFN receptors are expressed at significant levels as a functional full-length form,10,11 suggesting intact type III IFN signaling as part of the intrahepatic innate immune response. Furthermore, single nucleotide polymorphisms (SNPs) near and within IL28B are strong predictive markers for spontaneous, treatment-induced HCV clearance,12–15 suggesting that variations in type III IFN expression or function affect the outcome of HCV infection. In this context, Langhans et al. reported that IL-29 serum levels do not differ between patients with acute HCV infection and healthy controls, but that they are lower in chronically infected patients.16 Whereas recombinant type III IFNs are known to suppress HCV replication in vitro,17–19 their expression level in the liver has never been studied prospectively during acute HCV infection. Thus, the relative antiviral effect of endogenously produced type I and type III IFNs is not known.
Chimpanzees constitute the sole animal model for HCV infection and recapitulate many features of the human immune responses to HCV. To examine the role of IFNs in acute HCV infection, we studied the expression of type I (IFN-β and IFN-α2) and III (IL28-A/B and IL-29) IFNs in relation to ISGs and viremia using serial liver biopsies and plasma samples throughout the first 6 months of HCV infection. To further evaluate the HCV-induced IFN profile, we infected primary human hepatocytes (PHHs) with HCV in the presence and absence of pDCs, identified the induced IFNs and the requirements for their induction, and studied their antiviral potency relative to their expression level.
Materials and Methods
Chimpanzees and HCV Challenge
Chimpanzees were intravenously (IV) inoculated with 100 chimpanzee infectious dose 50 HCV (H77 strain) and studied under standard conditions for humane care, in compliance with National Institutes of Health guidelines, at an Association for Assessment and Accreditation of Animal Care accredited facility under a protocol approved by the New Iberia Research Center Animal Care and Use Committee. The clinical and virologic course of HCV infection were described previously.20,21 Liver biopsies and plasma were cryopre-served for the present study.
HCV Infection and Polyinosinic/Polycytidylic Acid Transfection of PHH
The purity of PHH (Invitrogen, Carlsbad, CA and Celsis, Chicago, IL) was confirmed by intracellular staining with anti-albumin (clone 2F11; Sigma-Aldrich, St. Louis, MO), followed by staining with anti-mouse immunoglobulin G-phycoerythrin (Invitrogen), treatment with 10% mouse serum (Innovative Research, Southfield, MI), staining with ethidium monoazide (EMA; Sigma-Aldrich), anti-CD14-Pacific Blue (clone M5E2), anti-CD45RA-fluorescein isothio-cyanate (FITC) (clone HI100), anti-CD45RO-FITC (clone UCHL1), anti-CD11c-allophycocyanin (clone B-ly6), and anti-CD16-PE-cyanine 5 (clone 3G8; all from BD Biosciences, Bedford, MA) and analysis on an LSRII flow cytometer (BD Biosciences). Hepatocytes constituted >95% and EMA-CD16-CD45+ hematopoietic cells <0.4% of the cells. Of the latter, 85% were CD14+ and 18.6% were CD11c+ .
PHHs shipped in plates were incubated with InVitroGRO HI medium (Celsis) for 4 hours at 37° C (5% CO2), followed by polyinosinic/polycytidylic acid (polyI:C) transfection or HCV infection, as described below.
PHHs shipped in suspension were centrifuged at 50×g for 5 minutes, incubated in InVitroGRO CP medium (Celsis) in 12- (7 × 105/well) or 24-well (3.5 × 105/well) plates overnight, and transfected with 5 μg of polyI:C (InvivoGen, San Diego, CA) and 6.4 μL of Lipofectamine 2000 in Opti-MEM medium (Invitrogen), or infected with HCV (Japanese fulminant hepatitis type I [JFH-1] strain)22 at a multiplicity of infection (MOI) of 0.4–2.7. After 3–6 hours, culture medium was replaced with InVitroGRO HI including Torpedo antibiotic mix (Celsis).
In some experiments, PHHs were incubated 30 minutes before HCV infection with neutralizing antibodies (Abs) against type I IFNs (10 μg/mL each of anti-IFN-α [clone MMHA-2; PBL Interferon Source, Piscataway, NJ] and polyclonal anti-IFN-β [R&D Systems, Minneapolis, MN]) or type III IFNs (10 μg/ mL each of polyclonal anti-IL-29, polyclonal anti-IL-28A, and anti-IL-29/IL-28B [clone 247801;, R&D Systems]).
Extraction of RNA, Complementary DNA synthesis, and Quantitative Polymerase Chain Reaction
Total RNA was isolated from snap-frozen, mechanically homogenized liver biopsies or from PHH using the RNeasy Mini Kit (Qiagen, Valencia, CA) with on-column DNase digestion.
A complementary DNA (cDNA) equivalent to 20–80 ng of total RNA, generated with the Monster-Script 1st-Strand cDNA Synthesis Kit (EPICENTRE Biotechnologies, Madison, WI), was used to determine IFN-induced protein with tetratricopeptide repeats 1 (IFIT1), myxovirus resistance 1 (MX1), chemokine (C-X-C motif) ligand (CXCL)10, CXCL11, IFN-α2, IFN-β, IL-29, glyceraldehyde 3-phosphate dehydrogenase (GAPDH), and proteasome subunit, beta type, 4 (PSMB4) messenger RNA (mRNA) levels with pre-designed human TaqMan Gene Expression Assays (Applied Biosystems, Foster City, CA). Because IL28A and IL28B share 98% of their nucleotide sequence, their mRNA levels were quantitated with shared forward primer 5′-TGAGGCAGCTGCAGGTGA-3′, reverse primer 5′-CTCCAGAACCTTCAGCGTCAG-3′, and probe 5′-FAM-TGGCTTTGGAGGCTGA-MGB-3′ designed with Primer Express (Applied Biosystems). The specific mRNA amount was quantitated using comparative cycle threshold values and 1–107 copies/well standard curves, and normalized to mean GAPDH and PSMB4 mRNA levels. Relative mRNA levels represent fold-increase over pre-infection or pre-transfection samples.
For HCV RNA quantitation, we used TaqMan EZ RTPCR Core Reagents (Applied Biosystems) and forward primer 5′-CGGGAGAGCCATAGTGG-3′, reverse primer 5′AGTACCACAAGGCCTTTCG-3′, and probe 5′-FAM-CTGCGGAACCGGTGAGTA CAC -TAMRA-3′ (Sigma-Aldrich). RT-PCR conditions are 2 minutes at 50° C, 30 minutes at 60° C, 5 minutes at 95° C, followed by 50 cycles at 95° C for 20 seconds and at 60° C for 1 minute. HCV RNA levels were normalized to microgram of total input RNA.
Enzyme-Linked Immunosorbent Assay
IFN-α and IFN-β (PBL Interferon Source, Piscataway, NJ), IL-28A, CXCL10, and CXCL11 (R&D Systems), and IL-29 (eBioscience, San Diego, CA) enzyme-linked immunosorbent assays (ELISAs) were used to quantitate cytokines in chimpanzee plasma or PHH culture supernatants. IL-28A exhibited 30% cross-reactivity with IL-28B.
Antiviral Activity Assay
Huh7-Lunet cells harboring a subgenomic, luciferase-expressing HCV JFH-1 replicon (Luc-ubi-neo/JFH-1 cells,23 here designated Huh7-HCV-Luc cells) were treated for 30 hours with the indicated amounts of recombinant IL-29, IL-28A, IL-28B (R&D Systems), IFN-αA2, and IFN-β (PBL Interferon Source) or with 72-hour supernatants from HCV-infected PHH. Luciferase activity was measured with the Luciferase Assay System (Promega, Madison, WI) using a POLARstar Omega instrument (BMG Labtech, Cary, NC). For some experiments, the neutralizing Abs to type I or III IFNs described above were added to either (1) supernatants from HCV-infected PHH that had been treated with the same neutralizing Abs or (2) recombinant cytokines, 30 minutes before adding the mixture to Huh7-HCV-Luc cells.
Isolation of pDCs and Coculture With PHH
Peripheral blood mononuclear cells were isolated from buffy coats, using density gradient centrifugation, and pDCs were selected with BDCA-4 magnetic beads (Miltenyi Biotec, Auburn, CA). Purity of CD123+ BDCA2+ pDCs was 86%–95% by flow cytometry. PHHs (3 × 105/well) were infected with HCV JFH-1 at an MOI of 2 in a 24-well plate for 30 hours, followed by removal of culture supernatants and the addition of 3 × 104 pDCs/200 μL directly or into a transwell. The total coculture volume was 500 μL/ well. Released IFN-α, IL-29, CXCL10, and CXCL11 were quantitated after 24 hours by ELISA.
Genotyping
DNA samples of 90 chimpanzees, including the 6 of this study, were genotyped by Sanger sequencing with primers, designed by Primer Express software to flank human SNPs associated with HCV-related traits12–15 (Table 1). Cleaned polymerase chain reaction (PCR) products were sequenced on a 3730 Automatic Sequencer (Applied Biosystems). Genetic variants were analyzed in relation to human sequences using Sequencher 4.1 software.
Table 1.
Analysis of Human SNPs Associated With HCV-Related Traits
| rs12979860 | rs12980275 | rs8099917 | rs8103142/rs11881222 | |
|---|---|---|---|---|
| Primers | ||||
| Forward | 5′-AGCTCAGCGCCTCTTCCT-3′ | 5′-GAATTCTGCCCATCCAGAGA-3′ | 5′-TCACCATCCTCCTCTCATCC-3′ | 5′-GGGTCAGGAGCTGTCTGG-3′ |
| Reverse | 5′-AGGGACCGCTACGTAAGTCA-3’ | 5′-CGACCTCCTAGGCTCAAGTG -3’ | 5′-TGCTGGGCCCTAACTGATAC-3’ | 5′-CATTCCCTCAGCTCCCT-3′ |
| Risk allele | T | G | G | C/G |
| Genotype | ||||
| All chimpanzees (n = 90) | TT | GG | TT | CC/GG |
| PHH donors | ||||
| Donor 1 | CT | AG | TG | CT/AG |
| Donor 2 | CT | AG | TG | CT/AG |
| Donor 3 | CC | AA | TT | TT/AA |
| Donor 4 | CT | AG | TT | CT/AG |
| Donor 5 | CT | AG | TT | CT/AG |
| Donor 6 | CC | AA | TT | TT/AA |
| Donor 7 | CT | AG | TG | CT/AG |
| Donor 8 | CT | AG | TT | CT/AG |
| Donor 9 | CT | AG | TG | CT/AG |
| Donor 10 | CT | AG | TT | CT/AG |
| Donor 11 | CC | AA | TT | TT/AA |
| Donor 12 | CC | AA | TT | TT/AA |
| Donor 13 | CC | AA | TT | TT/AA |
| Donor 14 | TT | AG | TT | CT/AG |
| Donor 15 | CC | AA | TT | TT/AA |
Results
Rapid Induction of ISGs in the Liver During Acute HCV Infection
Six chimpanzees were intravenously infected with HCV and developed acute hepatitis, which was self-limited for chimpanzees 97A009, 97A014, and 98A005 and chronically evolving for chimpanzees 97A015, 98A002, and A3A025 (Fig. 1A).20,21 Qualitative nested reverse-transcription (RT)-PCR (for chimpanzee A3A025) and transcription-mediated amplification (TMA) assays (for the other chimpanzees) confirmed that HCV was cleared from the blood of chimpanzees with self-limited, but not chronic infection.
Fig. 1.

ISG induction in the liver during acute HCV infection parallels viremia. (A) Serum HCV RNA titers (filled circles) and ALT levels (gray-colored area) were previously reported20,21 and are shown for reference purposes. HCV+ /− above each graph indicates qualitative nested RT-PCR (for chimpanzee A3A025) and TMA results (for all other chimpanzees). Presence or absence of viremia was assessed for 77 weeks (not shown) to confirm outcome of infection. (B–D) Intrahepatic mRNA levels of the ISGs IFIT1 (B), MX1 (C), and CXCL11 (D), correlate with serum HCV RNA titers.
HCV infection rapidly induced the ISGs IFIT1, MX1, and CXCL11 in the livers of all chimpanzees (Fig. 1B–D), and ISG expression kinetics correlated with viremia. The induction of CXCL11, a T-cell-recruiting chemokine, was slightly delayed, compared with IFIT1 and MX1, and immediately followed by the peak in alanine aminotransferase (ALT) activity, which is attributed to T-cell-induced liver injury in this model.21 Consistent with previous microarrays,4,5 intrahepatic ISGs were induced regardless of infection outcome.
Minimal Type I IFN, but Robust Type III IFN Induction in Liver and Blood During Acute HCV Infection
To identify factors that trigger intrahepatic ISG expression during acute HCV infection, we analyzed serial liver and plasma samples for type I and III IFNs. Both IFN-α2 and IFN-β were minimally induced at the RNA level in liver (1.6 to 3.3-fold induction of IFN-α2 and 1.2- to 3.6-fold induction of IFN-β; Fig. 2B) and remained undetectable in plasma (not shown).
Fig. 2.

Type I IFNs are minimally, but type III IFNs are robustly induced during acute HCV infection. (A) Serum HCV RNA titers (filled circles) and ALT levels (gray-colored area) were previously reported20,21 and are shown for reference purposes. (B–D) Intrahepatic type I (B) and III IFN (C) mRNA levels were determined by qPCR, normalized to endogenous references, and presented as fold-increase over preinfection levels. Type III (D), but not type I, IFNs (not shown) were detectable by ELISA in plasma.
Intrahepatic IL-28 mRNA levels peaked as early as week 4 postinfection (chimpanzee 97A015), but relative induction levels varied widely (1.5- to 24-fold induction, compared with preinfection levels; Fig. 2C). This was mirrored by peak IL-28 protein levels of 600 pg/mL in plasma of chimpanzee 97A009 and no induction in chimpanzee A3A025. In contrast, intrahepatic IL-29 mRNA levels were more consistently and more strongly induced in all chimpanzees, reaching 33- to 53-fold induction, compared with preinfection levels (Fig. 2D). Likewise, IL-29 was detectable in plasma of all chimpanzees. Peak IL-29 protein levels in blood followed peak mRNA levels in liver in all chimpanzees, except A3A025 (which had a higher baseline level), suggesting the liver as the main source of IL-29 during HCV infection.
To examine whether variations in IL-28 expression were related to SNPs in the IL28B gene region that predict spontaneous and treatment-induced HCV clearance in humans,12–15 genomic DNA was sequenced. All 6 chimpanzees displayed invariable sequences that combined the human risk allele for chronic outcome of HCV infection in rs12979860, rs12980275, rs8103142, and rs11881222 with the nonrisk allele in rs8099917. These results, which were confirmed for a larger cohort of 90 chimpanzees (Table 1), indicate that these five IL28B SNPs are specific to humans.
Collectively, the results show that intrahepatic ISG induction correlated with a strong type III, but not type I IFN response. Type III IFN levels were independent of the known human IL28B SNPs and did not predict the outcome of HCV infection.
ISG Response of PHH to polyI:C and HCV is Associated With Weak Type I IFN, But Strong Type III IFN Induction
To determine the cellular source of ISGs and IFNs, we asked whether the ISG as well as type I and III IFN expression profiles that we observed in vivo could be recapitulated in vitro in PHH cultures. PHHs were first transfected with polyI:C to mimic intracellular viral RNA. MX1 and IFIT1 mRNA levels significantly increased 6 hours, and peaked 24 hours, after transfection. As observed in vivo in the HCV-infected liver, CXCL10 and CXCL11 mRNA and protein levels peaked later (i.e., 48 hours after polyI:C transfection; Fig. 3A,B). Furthermore, consistent with in vivo results, type III IFNs were induced to significantly higher mRNA (Fig. 3C) and protein levels (Fig. 3D) than type I IFNs.
Fig. 3.

ISG response of PHH to dsRNA is associated with weak type I, but strong type III IFN induction. PHHs were trans-fected with polyI:C to mimic intracellular viral RNA. ISG (A) and IFN mRNA (C) and protein levels (B and D) were determined by qPCR in transfected cells and by ELISA in culture supernatants. mRNA levels were normalized to endogenous references and presented as fold-increase over pretrans-fection levels. Mean and standard error of the mean are shown (A and C).
Next, we infected PHH with HCV and investigated ISG induction in relation to type I and III IFN expression. IFIT1, MX1, CXCL10, and CXCL11 mRNA levels peaked 48 hours, and CXCL10 and CXCL11 protein levels peaked 72 hours after HCV infection (Fig. 4A,B). Type I IFN mRNA was induced less than 100-fold (Fig. 4C), IFN-β protein levels remained below 4 pg/mL, and IFN-α protein was undetectable (Fig. 4D). In contrast, IL-28 and IL-29 peaked to significantly higher mRNA levels within 24–48 hours and protein levels reached up to 250 pg/mL 72 hours after HCV infection (Fig. 4C–D). Variations in ISG and IFN responses among donors did not correlate with HCV RNA levels in PHH cultures (Fig. 4E,F and Supporting Fig. 1) nor with IL28B SNPs (Supporting Fig. 2). Overall, our results demonstrate that transfection of PHH with double-stranded RNA (dsRNA) and infection with HCV reproduced the ISG and IFN expression profile that we observed in the liver during acute HCV infection.
Fig. 4.

ISG response of PHH to HCV infection is associated with weak type I, but strong type III IFN induction. PHHs were infected with HCV at an MOI of 0.4–2.7. ISG mRNA (A) and protein (B) levels as well as IFN mRNA (C) and protein (D) levels are presented as in Fig. 3. (E and F) HCV RNA levels in PHHs were compared to ISG (E) and IFN levels (F). Mean and standard error of the mean are shown (A, C, and E).
Type III IFNs Produced by HCV-Infected PHH Inhibit HCV replication
To study the functional relevance of the strong type III IFN response during HCV infection, we examined its effect on HCV replication using Huh7-Lunet cells with a subgenomic HCV luciferase replicon as a readout.23 Recombinant type I (500 pg/mL) and type III IFNs (100 ng/mL) inhibited HCV replication by more than 90%, and this inhibition was almost completely abrogated by cytokine-specific neutralizing Abs (Fig. 5A). IL-29, IL-28A, and IL-28B showed 50% inhibition of HCV replication at 200–500 pg/mL (Fig. 5B), which is the maximum amount of type III IFNs that PHH produced 72 hours postinfection (Figs. 4D and 6). Because 50% antiviral activity can also be mediated by very low concentrations of type I IFNs below the ELISA detection limit, we used an indirect method to further evaluate the relative effect of type I and III IFNs on HCV replication.
Fig. 5.
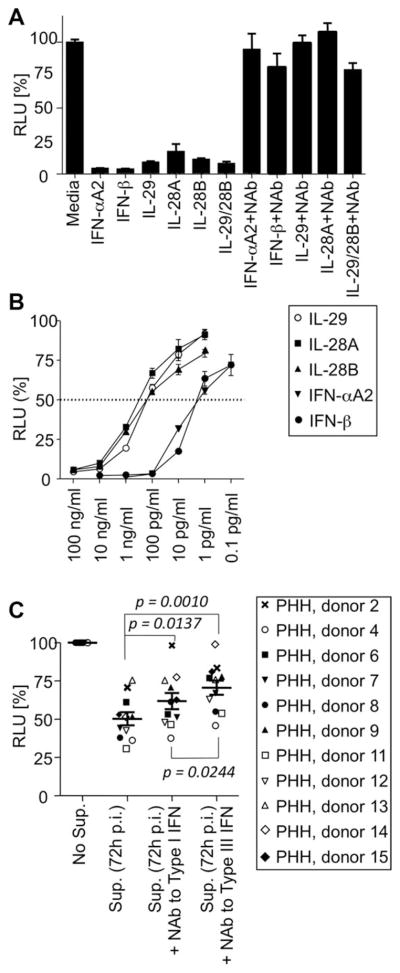
Type III IFNs produced by HCV-infected PHH inhibit HCV replication. (A) Recombinant type I (IFN-αA2 and IFN-β; 500 pg/mL) and type III (IL-28A, IL-28B, and IL-29; 100 ng/mL) IFNs decreased luciferase activity of Huh7-HCV-Luc cells, and this effect was blocked by Ab-mediated neutralization of the respective cytokines. An isotype control Ab did not change RLU values (not shown). (B) Recombinant type I and III IFNs mediated a dose-dependent antiviral effect when added for 30 hours to Huh7-HCV-Luc cells. (C) Supernatant of HCV-infected PHH, harvested 72 hours postinfection and containing an average of 526 pg/ mL of type III IFN proteins, reduced HCV replication of Huh7-HCV-Luc cells by 50%. NAbs against IL28A, IL-28B, and IL-29 (type III IFN) reversed antiviral activity to a significantly greater extent than NAbs against IFN-α and IFN-β (type I IFN; P = 0.0244). Statistical analysis: mean plus standard error of the mean, paired Wilcoxon’s signed-rank test. RLU, relative light units; Sup, supernatant; Nabs, neutralizing Abs.
Fig. 6.

IL-29, but not IL-28, is produced in a type I IFN-independent manner in HCV-infected hepatocytes. Treatment of PHH before and during HCV infection with NAb against IFN-α and IFN-β reduced the level of secreted IL-28, but not IL-29, in most PHH cultures. Statistical analysis: paired Wilcoxon’s signed-rank test. Nab, neutralizing Ab.
Supernatants from PHH cultures harvested 72 hours post-HCV infection reduced HCV replication by 50% (Fig. 5C). Whereas neutralization of the supernatant with Abs against either type I (IFN-α and IFN-β) or III IFNs (IL-28A, IL-28B, and IL-29) significantly increased HCV replication, neutralization of type III IFNs had a greater effect than neutralization of type I IFNs (P = 0.0244). This result suggests that type III IFNs contribute significantly to antiviral activity during HCV infection.
IL-29 Can Be Produced in a Type I IFN-Independent Manner in HCV-Infected Hepatocytes
Experiments with hepatoma cell lines and TLR-stimulated macrophages suggest that type III IFNs are ISGs themselves and can be induced in a type I IFN-mediated manner.8,24 To evaluate whether this holds true for HCV-infected PHH, we infected PHH with HCV in the presence or absence of neutralizing Abs against IFN-α and IFN-β. Whereas IL-28 secretion decreased in the presence of neutralizing Abs against type I IFNs (P= 0.0078), IL-29 production was not affected in the majority of cultures (P = not significant; Fig. 6). Thus, IL-29 can be induced independently from type I IFN signaling in HCV-infected hepatocytes.
Production of IL-29 by PHH is Independent of Direct Contact With pDCs
pDCs were recently shown to contribute to the induction of IFN-α and ISGs when cocultured with HCV-replicating hepatoma cells.7 We therefore asked whether this result extended to pDCs cocultured with HCV-infected PHH and to the production of type III IFNs. Less than 0.075% of cells in PHH cultures were CD11c+ (not shown), and IFN-α was not detected upon HCV infection in this setting (Figs. 4D and 7). The addition of pDCs induced IFN-α and depended on PHH/ pDC contact because IFN-α production was substantially reduced when PHHs and pDCs were separated by transwells (Fig. 7B). The chemokines CXCL10 and CXCL11, which are confirmed ISGs, were produced in HCV-infected PHH cultures in the absence of pDCs, but their production further increased in the presence of pDCs in 2 of 3 donors, likely the result of TLR7-dependent7 induction of IFN-α21 by pDCs. In contrast, IL-29 production was barely changed in PHH/pDC coculture and was not affected by trans-wells. This suggests that IL-29 was mainly produced by HCV-infected hepatocytes, rather than by pDCs, in HCV infection.
Fig. 7.

Production of IL-29 by PHH is independent of direct contact with pDCs. (A) Experimental strategy: PHHs were infected with HCV for 30 hours, followed by medium change and a 24-hour coculture with or without pDCs or transwells. (B) IFN-α secretion depended on direct contact between pDCs and HCV-infected PHHs. CXCL10 and CXCL11, but not IL-29, secretion was enhanced by PHH/pDC contact. PHH/ pDC coculture in the absence of HCV infection did not induce cytokines (not shown). Mean and standard error of the mean are shown for CXCL10 and CXCL11.
Discussion
This is the first study to analyze type III IFN levels in serial liver and blood samples during acute HCV infection. Type III IFNs were robustly induced, both in liver and blood, and their expression kinetics paralleled viremia and ISG levels. In contrast, type I IFNs were barely detectable at the RNA level in liver and were undetectable at the protein level in blood. Robust type III and minimal type I IFN expression was recapitulated in vitro in HCV-infected PHH. The strong induction of IL-29, the main type III IFN in the acutely HCV-infected liver, may be attributed to a type I IFN-independent production, because neutralization of type I IFNs did not affect IL-29 production in most of the tested PHH cultures, and because IL-29 production was not further enhanced in the presence of pDCs that secrete IFN-α when they are in direct contact with HCV-infected PHH.
Type I IFN-independent IL-29 induction likely depends on the promoter structure of the IL29 gene. Promoter analysis demonstrated that IL-29 and IFN-β expression require both IFN regulatory factor (IRF)3 and IRF7.25 In contrast, IL-28 and IFN-α depend solely on IRF725 (i.e., on JAK-STAT signaling downstream of the IFN-β/α receptor). In addition, the IL-29 promoter displays distinct differences to the IFN-β promoter. Although both promoters contain spatially separate enhancer regions, namely, an IRF3/7-binding site, and proximal and distal nuclear factor kappa light-chain enhancer of activated B cells (NF-κB) binding sites,26 each element in the IL-29 promoter acts independently, whereas those in the IFN-β promoter work in a highly cooperative manner.27 To further clarify the molecular mechanism of this type I IFN-independent IL-29 induction, it would therefore be important to examine the availability of these transcription factors, especially those of the NF-κB family, in the HCV-infected liver and to determine which enhancer elements preferentially contribute to the observed induction of IL-29.
Given that recombinant type III IFNs produce an almost identical set of ISGs as type I IFNs,18,19 the robustly produced type III IFNs may contribute to ISG induction during acute HCV infection, particularly if type I IFN production is impaired or effective only in a paracrine manner in close vicinity of pDCs that have direct contact with HCV-infected hepatocytes. In addition, type III IFNs may contribute to overall antiviral activity. Whereas recombinant type I and III IFNs are known to have antiviral activity,17–19,28 their relative effects on replication, based on the respective production levels in HCV infection, are not known. Culture supernatants of HCV-infected PHH showed a decrease of HCV replication by approximately 50%, which was the same level of antiviral activity that can be achieved with 200–500 pg/ mL of recombinant type III IFN proteins, the maximum amount detected in PHH culture supernatants. This type III IFN concentration was also consistent with the peak type III IFN level (approximately 550 pg/mL of plasma) in acutely HCV-infected chimpanzees. However, the antiviral activity in the supernatant of HCV-infected PHH cultures was only partially, reversed by neutralization of type III IFNs, and neutralization of type I IFNs had a lesser effect. There are at least two possible explanations. First, the complexity of HCV-induced ISGs is large and includes not only antiviral, but also proviral genes, such as ISG15 and ubiquitin-specific peptidase 18, that may be induced in this setting.29,30 Second, IFN-independent pathways may contribute to antiviral activity.31 The latter hypothesis is consistent with our in vivo results, where the increase in type III IFNs mirrored viremia, but was not associated with HCV clearance.
An important final aspect of this study is that all chimpanzees were monomorphic for the known human IL28B SNPs associated with spontaneous and treatment-induced outcome of HCV infection.12–15 This implies that human IL28B SNPs developed after the split of the human and chimpanzee lineages. Although chimpanzees may have other SNPs, our study shows that type III IFN levels in the liver do not correlate with the spontaneous outcome of acute HCV infection. In addition, type III IFN levels from HCV-infected PHHs were not associated with IL28B SNPs. This corroborates a cross-sectional study of chronically HCV-infected humans in which intrahepatic IL28B mRNA levels were not affected by IL28B SNPs and not related to treatment-induced HCV clearance.32 Collectively, these data suggest that the identified IL28B SNPs affect the outcome of HCV infection by other, still to be determined, mechanisms.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgments
The authors thank Lauren Holz for her analysis of PHH purity and Su Hyung Park, Thomas O’Brien, and Sukanya Raghuraman for their discussion.
This study was supported by the intramural research programs of the National Institute of Diabetes and Digestive and Kidney Diseases and National Cancer Institute of the National Institutes of Health. The authors thank Volker Lohmann for the Huh7-HCV-Luc cells.
Abbreviations
- Abs
antibodies
- ALT
alanine aminotransferase
- APC
allophycocyanin
- cDNA
complementary DNA
- CXCL
chemokine (C-X-C motif) ligand
- dsRNA
double-stranded RNA
- ELISA
enzyme-linked immunosorbent assays
- EMA
ethidium monoazide
- FITC
fluorescein isothiocyanate
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- HCV
hepatitis C virus
- IFIT1
IFN-induced protein with tetratricopeptide repeats 1
- IFN
interferon
- IRF
IFN regulatory factor
- ISG
interferon-stimulated genes
- IV
intravenously
- JAK
Janus kinase
- JFH-1
Japanese fulminant hepatitis type I
- MOI
multiplicity of infection
- mRNA
messenger RNA
- MX1
myxovirus resistance 1
- NF-κB
nuclear factor kappa light-chain enhancer of activated B cells
- pDCs
plasmacytoid dendritic cells
- PHH
primary human hepatocyte
- polyI:C
polyinosinic/polycytidylic acid
- PSMB4
proteasome subunit, beta type, 4
- qPCR
quantitative polymerase chain reaction
- RT-PCR
reverse-transcription PCR
- SNPs
single-nucleotide polymorphisms
- STAT
signal transducer and activator of transcription
- TCR
Toll-like receptor
- TMA
transcription-mediated amplification assay
Footnotes
Additional Supporting Information may be found in the online version of this article.
Potential conflict of interest: Nothing to report.
References
- 1.Rehermann B. Hepatitis C virus versus innate and adaptive immune responses: a tale of coevolution and coexistence. J Clin Invest. 2009;119:1745–1754. doi: 10.1172/JCI39133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ciesek S, Manns MP. Hepatitis in 2010: the dawn of a new era in HCV therapy. Nat Rev Gastroenterol Hepatol. 2011;8:69–71. doi: 10.1038/nrgastro.2010.219. [DOI] [PubMed] [Google Scholar]
- 3.Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, Rice CM. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature. 2011;472:481–485. doi: 10.1038/nature09907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bigger CB, Brasky KM, Lanford RE. DNA microarray analysis of chimpanzee liver during acute resolving hepatitis C virus infection. J Virol. 2001;75:7059–7066. doi: 10.1128/JVI.75.15.7059-7066.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Su AI, Pezacki JP, Wodicka L, Brideau AD, Supekova L, Thimme R, et al. Genomic analysis of the host response to hepatitis C virus infection. Proc Natl Acad Sci U S A. 2002;99:15669–15674. doi: 10.1073/pnas.202608199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Horner SM, Gale M., Jr Intracellular innate immune cascades and interferon defenses that control hepatitis C virus. J Interferon Cytokine Res. 2009;29:489–498. doi: 10.1089/jir.2009.0063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Takahashi K, Asabe S, Wieland S, Garaigorta U, Gastaminza P, Isogawa M, Chisari FV. Plasmacytoid dendritic cells sense hepatitis C virus-infected cells, produce interferon, and inhibit infection. Proc Natl Acad Sci U S A. 2010;107:7431–7436. doi: 10.1073/pnas.1002301107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ank N, West H, Bartholdy C, Eriksson K, Thomsen AR, Paludan SR. Lambda interferon (IFN-lambda), a type III IFN, is induced by viruses and IFNs and displays potent antiviral activity against select virus infections in vivo. J Virol. 2006;80:4501–4509. doi: 10.1128/JVI.80.9.4501-4509.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou Z, Hamming OJ, Ank N, Paludan SR, Nielsen AL, Hartmann R. Type III interferon (IFN) induces a type I IFN-like response in a restricted subset of cells through signaling pathways involving both the Jak-STAT pathway and the mitogen-activated protein kinases. J Virol. 2007;81:7749–7758. doi: 10.1128/JVI.02438-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kotenko SV, Gallagher G, Baurin VV, Lewis-Antes A, Shen M, Shah NK, et al. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat Immunol. 2003;4:69–77. doi: 10.1038/ni875. [DOI] [PubMed] [Google Scholar]
- 11.Sommereyns C, Paul S, Staeheli P, Michiels T. IFN-lambda (IFN-lambda) is expressed in a tissue-dependent fashion and primarily acts on epithelial cells in vivo. PLoS Pathog. 2008;4:e1000017. doi: 10.1371/journal.ppat.1000017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ge D, Fellay J, Thompson AJ, Simon JS, Shianna KV, Urban TJ, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature. 2009;461:399–401. doi: 10.1038/nature08309. [DOI] [PubMed] [Google Scholar]
- 13.Suppiah V, Moldovan M, Ahlenstiel G, Berg T, Weltman M, Abate ML, et al. IL28B is associated with response to chronic hepatitis C interferon-alpha and ribavirin therapy. Nat Genet. 2009;41:1100–1104. doi: 10.1038/ng.447. [DOI] [PubMed] [Google Scholar]
- 14.Tanaka Y, Nishida N, Sugiyama M, Kurosaki M, Matsuura K, Sakamoto N, et al. Genome-wide association of IL28B with response to pegylated interferon-alpha and ribavirin therapy for chronic hepatitis C. Nat Genet. 2009;41:1105–1109. doi: 10.1038/ng.449. [DOI] [PubMed] [Google Scholar]
- 15.Thomas DL, Thio CL, Martin MP, Qi Y, Ge D, O’Huigin C, et al. Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature. 2009;461:798–801. doi: 10.1038/nature08463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Langhans B, Kupfer B, Braunschweiger I, Arndt S, Schulte W, Nischalke HD, et al. Interferon-lambda serum levels in hepatitis C. J Hepatol. 2011;54:859–865. doi: 10.1016/j.jhep.2010.08.020. [DOI] [PubMed] [Google Scholar]
- 17.Robek MD, Boyd BS, Chisari FV. Lambda interferon inhibits hepatitis B and C virus replication. J Virol. 2005;79:3851–3854. doi: 10.1128/JVI.79.6.3851-3854.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang L, Jilg N, Shao RX, Lin W, Fusco DN, Zhao H, et al. IL28B inhibits hepatitis C virus replication through the JAK-STAT pathway. J Hepatol. 2011;55:289–298. doi: 10.1016/j.jhep.2010.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marcello T, Grakoui A, Barba-Spaeth G, Machlin ES, Kotenko SV, MacDonald MR, Rice CM. Interferons alpha and lambda inhibit hepatitis C virus replication with distinct signal transduction and gene regulation kinetics. Gastroenterology. 2006;131:1887–1898. doi: 10.1053/j.gastro.2006.09.052. [DOI] [PubMed] [Google Scholar]
- 20.Folgori A, Capone S, Ruggeri L, Meola A, Sporeno E, Ercole BB, et al. A T-cell HCV vaccine eliciting effective immunity against heterologous virus challenge in chimpanzees. Nat Med. 2006;12:190–197. doi: 10.1038/nm1353. [DOI] [PubMed] [Google Scholar]
- 21.Shin EC, Park SH, Demino M, Nascimbeni M, Mihalik K, Major M, et al. Delayed induction, not impaired recruitment, of specific CD8 T cells causes the late onset of acute hepatitis C. Gastroenterology. 2011;141:686–695. 695, e681. doi: 10.1053/j.gastro.2011.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med. 2005;11:791–796. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jo J, Aichele U, Kersting N, Klein R, Aichele P, Bisse E, et al. Analysis of CD8+ T-cell-mediated inhibition of hepatitis C virus replication using a novel immunological model. Gastroenterology. 2009;136:1391–1401. doi: 10.1053/j.gastro.2008.12.034. [DOI] [PubMed] [Google Scholar]
- 24.Siren J, Pirhonen J, Julkunen I, Matikainen S. IFN-alpha regulates TLR-dependent gene expression of IFN-alpha, IFN-beta, IL-28, and IL-29. J Immunol. 2005;174:1932–1937. doi: 10.4049/jimmunol.174.4.1932. [DOI] [PubMed] [Google Scholar]
- 25.Osterlund PI, Pietila TE, Veckman V, Kotenko SV, Julkunen I. IFN regulatory factor family members differentially regulate the expression of type III IFN (IFN-lambda) genes. J Immunol. 2007;179:3434–3442. doi: 10.4049/jimmunol.179.6.3434. [DOI] [PubMed] [Google Scholar]
- 26.Thomson SJ, Goh FG, Banks H, Krausgruber T, Kotenko SV, Foxwell BM, Udalova IA. The role of transposable elements in the regulation of IFN-lambda1 gene expression. Proc Natl Acad Sci U S A. 2009;106:11564–11569. doi: 10.1073/pnas.0904477106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Iversen MB, Paludan SR. Mechanisms of type III interferon expression. J Interferon Cytokine Res. 2010;30:573–578. doi: 10.1089/jir.2010.0063. [DOI] [PubMed] [Google Scholar]
- 28.Marukian S, Andrus L, Sheahan TP, Jones CT, Charles ED, Ploss A, et al. Hepatitis C virus induces interferon-lambda and interferon-stimulated genes in primary liver cultures. HEPATOLOGY. 2011 Jul 28; doi: 10.1002/hep.24580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Randall G, Chen L, Panis M, Fischer AK, Lindenbach BD, Sun J, et al. Silencing of USP18 potentiates the antiviral activity of interferon against hepatitis C virus infection. Gastroenterology. 2006;131:1584–1591. doi: 10.1053/j.gastro.2006.08.043. [DOI] [PubMed] [Google Scholar]
- 30.Chen L, Sun J, Meng L, Heathcote J, Edwards AM, McGilvray ID. ISG15, a ubiquitin-like interferon-stimulated gene, promotes hepatitis C virus production in vitro: implications for chronic infection and response to treatment. J Gen Virol. 2010;91:382–388. doi: 10.1099/vir.0.015388-0. [DOI] [PubMed] [Google Scholar]
- 31.DeWitte-Orr SJ, Mehta DR, Collins SE, Suthar MS, Gale M, Jr, Mossman KL. Long double-stranded RNA induces an antiviral response independent of IFN regulatory factor 3, IFN-beta promoter stimulator 1, and IFN. J Immunol. 2009;183:6545–6553. doi: 10.4049/jimmunol.0900867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Honda M, Sakai A, Yamashita T, Nakamoto Y, Mizukoshi E, Sakai Y, et al. Hepatic ISG expression is associated with genetic variation in interleukin 28B and the outcome of IFN therapy for chronic hepatitis C. Gastroenterology. 2010;139:499–509. doi: 10.1053/j.gastro.2010.04.049. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.