

Abstract
Endoplasmic reticulum stress resulting from abnormal folding of newly synthesized proteins impairs metabolism, transcriptional regulation, and gene expression, and it is a key mechanism of cell injury. Endoplasmic reticulum stress plays an important role in cardiovascular and neurodegenerative diseases, cancer, and diabetes. We evaluated the role for this phenomenon in diabetic peripheral neuropathy. Endoplasmic reticulum stress manifest in upregulation of multiple components of unfolded protein response was identified in neural tissues (sciatic nerve, spinal cord) of streptozotocin diabetic rats and mice. A chemical chaperone, trimethylamine oxide, administered for 12 weeks after induction of diabetes (110 mg⋅kg−1⋅d−1, a prevention paradigm) attenuated endoplasmic reticulum stress, peripheral nerve dysfunction, intraepidermal nerve fiber loss, and sciatic nerve and spinal cord oxidative-nitrative stress in streptozotocin diabetic rats. Similar effects on diabetes-induced endoplasmic reticulum stress and peripheral nerve dysfunction were observed with a structurally unrelated chemical chaperone, 4-phenylbutyric acid (100 mg⋅kg−1⋅d−1, intraperitoneal). CCAAT/enhancer-binding protein homologous protein (CHOP)−/− mice made diabetic with streptozotocin displayed less severe sciatic nerve oxidative-nitrative stress and peripheral neuropathy than the wild-type (C57Bl6/J) mice. Neither chemical chaperones nor CHOP gene deficiency reduced diabetic hyperglycemia. Our findings reveal an important role of endoplasmic reticulum stress in the development of diabetic peripheral neuropathy and identify a potential new therapeutic target.
Diabetic peripheral neuropathy (DPN) affects at least 50% of patients with type 1 and type 2 diabetes and is a leading cause of foot amputation (1,2). Clinical indications of DPN include increased vibration and thermal perception thresholds that progress to sensory loss, occurring in conjunction with degeneration of all fiber types in the peripheral nerve. A proportion of patients with DPN also describe abnormal sensations such as paresthesias, allodynia, hyperalgesia, and spontaneous pain (3). The pathogenetic mechanisms of DPN include, but are not limited to, increased aldose reductase activity (4), advanced glycation/glycoxidation (5), oxidative-nitrative stress (6–12), activation of protein kinase C (13), poly(ADP-ribose) polymerase (14,15), cyclooxygenase-2 (16), 12/15-lipoxygenase (17), and impaired neurotrophic support (18). Unfortunately, all drug candidates for DPN studied so far, including aldose reductase and protein kinase C inhibitors, acetyl carnitine, nerve growth factor, and the antioxidant α-lipoic acid, showed modest efficacy in clinical trials or adverse side effects. Identification of principally novel therapeutic targets for DPN therefore is highly warranted.
The endoplasmic reticulum (ER) plays a pivotal role in the folding and processing of newly synthesized proteins. Damage to ER and resultant ER stress lead to aberrant transcriptional regulation and gene expression, ion channel function, metabolism, and signaling (19–21). To counteract ER stress, the ER mounts the unfolded protein response (UPR). Three canonical arms of UPR, such as: PKR-like eukaryotic initiation factor 2A kinase (PERK), which phosphorylates eukaryotic initiation factor-2α (eIF2α) to suppress general protein translation; inositol-requiring enzyme-1 (IRE1) involved in recruitment of several signaling molecules, splicing, and production of an active transcription factor called X-box binding protein 1, ER chaperones such as glucose-regulated protein BiP/GRP78 (BiP) and glucose-regulated protein 94 (GRP94), as well as CCAAT/enhancer-binding protein homologous protein (CHOP) and other components of the ER-associated degradation process; and activating transcription factor-6 (ATP-6), which translocates to the Golgi apparatus and produces an active transcription factor ATP-6N stimulating expression of chaperones and X-box binding protein 1, act together to reduce general protein synthesis, to facilitate protein degradation, and to increase folding capacity to resolve ER stress (19–21). However, the excessive and long-term upregulation of UPR and, in particular, X-box binding protein 1, CHOP, and ATF-4 leads to cell injury and death (19–21).
ER stress plays a key role in cardiovascular (21–23) and neurodegenerative (24) diseases, cancer (25), obesity, and diabetes (19,20,26–28). Recent experimental studies implicate ER stress in the development of diabetes complications, such as nephropathy (29), early retinopathy (30), and cognitive decline (31). We evaluated the contribution of ER stress to functional and morphological changes associated with experimental DPN.
RESEARCH DESIGN AND METHODS
Reagents.
Unless otherwise stated, all chemicals were of reagent-grade quality and were purchased from Sigma-Aldrich Chemical (St. Louis, MO). For immunohistochemistry, rabbit polyclonal antiprotein gene product 9.5 (PGP 9.5) antiserum for assessment of intraepidermal nerve fiber density (INFD) (UltraClone, Isle of Wight, U.K.), Alexa Fluor 488 goat anti-rabbit highly cross-adsorbed IgG (H+L; Invitrogen, Eugene, OR), SuperBlock blocking buffer (Thermo Scientific, Rockford, IL), and the optimum cutting temperature compound (Sakura Finetek USA, Torrance, CA) were used. VECTASHIELD Mounting Medium was obtained from Vector Laboratories (Burlingame, CA). Other reagents for immunohistochemistry have been purchased from Dako Laboratories (Santa Barbara, CA). For Western blot analysis, rabbit polyclonal anti-GRP78/BiP and anti-GRP94 antibodies and mouse monoclonal horseradish peroxidase–conjugated antiβ-actin antibody were obtained from Abcam (Cambridge, MA), rabbit polyclonal anti-human IRE1p and phosphorylated (Ser724) anti-human IRE1p antibodies were obtained from USBiological (Swampscott, MA), rabbit polyclonal antiphospho-PERK and anti-PERK antibodies and mouse monoclonal anti- ER oxidase 1 (ERO1)-Lα antibody were obtained from Santa Cruz Biotechnology (Santa Cruz, CA), rabbit polyclonal antiphospho-eIF2α (ser51) and antieIF2α antibodies, mouse monoclonal anti-CHOP antibody, and anti-mouse and anti-rabbit IgG horseradish peroxidase-linked antibodies were obtained from Cell Signaling 9(Danvers, MA), and mouse monoclonal (clone 1A6) antinitrotyrosine (NT) antibody was obtained from Millipore (Billerica, MA).
Animals.
We used two models of DPN, i.e., male Wistar rats and male C57Bl6/J mice that have development of robust peripheral nerve dysfunction and intraepidermal nerve fiber loss when made diabetic with streptozotocin (STZ) (4–9,12–15,17,18). In the rat model, we conducted pharmacological studies with trimethyloxide (TMAO) and 4-phenylbutyric acid (PBA), two structurally unrelated chemical chaperones. Restoring ER function by chemical chaperones, i.e., compounds that counteract ER stress completely by promoting normal protein folding, is an emerging therapeutic approach for metabolic diseases recently used in a first clinical trial (27). Identification of the important role for ER stress completely in peripheral nerve dysfunction and degeneration in the chemical chaperone experiments consequently led us to the studies of individual components of ER stress and UPR using the specific inhibitors and transgenic mouse models. Herein, we report our findings in CHOP-deficient mice. The experiments were performed in accordance with regulations specified by the Guide for the Care and Handling of Laboratory Animals (National Institutes of Health publication 85–23) and Pennington Biomedical Research Center Protocol for Animal Studies. Male Wistar rats, body weight 250–300 g, were purchased from Charles River (Wilmington, MA). They were fed a standard rat chow (PMI Nutrition International, Brentwood, MO) and had access to water ad libitum throughout the experiment. Immediately after assessment of motor nerve conduction velocity (MNCV) and sensory nerve conduction velocity (SNCV) and small sensory nerve fiber function in 10-week-old rats, diabetes was induced by a single injection of STZ (50 mg⋅kg−1 intraperitoneal). Blood samples for glucose measurements were taken from the tail vein ∼48 h after the STZ injection and the day before the study termination. All rats with blood glucose levels ≥3.8 mmol/L were considered diabetic. Diabetic rats were maintained on suboptimal doses of insulin (∼1–2 units every second day) to prevent ketoacidosis and weight loss. The experimental groups comprised control and diabetic rats maintained with or without TMAO (110 mg⋅kg−1⋅d−1) or PBA (100 mg⋅kg−1⋅d−1, intraperitoneal) treatments for 12 weeks, after which nerve functional measurements were performed again. In the second experiment, male C57Bl6/J mice and several breeding pairs of B6.129S-Ddit3tm1Dron/J (CHOP−/−) mice were obtained from The Jackson Laboratories (Bar Harbor, ME). The colony of CHOP−/− mice was established at Pennington Biomedical Research Center. All the mice had ad libitum access to water. Male wild-type (body weights 25–27 g) and age-matched CHOP−/− mice were fed standard mouse chow (PMI Nutrition International, Brentwood, MO) and were randomly assigned to nondiabetic or diabetic groups. Diabetes was induced in 8-week-old mice by STZ (one injection of 100 mg⋅kg−1⋅d−1 intraperitoneal, followed by, if necessary, one or several consecutive injections of 40 mg⋅kg−1⋅d−1 intraperitoneal). Diabetes was defined as a nonfasting blood glucose ≥13.8 mmol/L in tail vein blood 48 h after STZ injection and on the day before sacrifice. Evaluations of MNCV, SNCV, thermal and mechanical algesia, and tactile response thresholds were performed at a baseline (before STZ administration) and at 12 weeks.
Anesthesia, euthanasia, and tissue sampling.
The animals were sedated by CO2 and immediately killed by cervical dislocation. Sciatic nerves and spinal cords were rapidly isolated, immediately frozen in liquid nitrogen, and stored at −80°C before assessment of variables of UPR and nitrated proteins by Western blot analysis and NT and 4-hydroxynonenal (4-HNE) adducts by ELISA. The footpads were fixed in ice-cold Zamboni fixative for 3 h, washed in 100 mmol/L PBS overnight, and then washed in PBS containing increasing concentrations of sucrose, i.e., 10%, 15%, and 20%, for 3 h in each solution. After washing, the samples were snap-frozen in optimum cutting temperature compound and stored at −80°C for further assessment of intraepidermal nerve fiber density.
Nerve functional studies.
Nerve functional studies included measurements of sciatic MNCV and hindlimb digital SNCV, thermal response latency, and mechanical and tactile response thresholds.
For assessment of sciatic MNCV, the left sciatic nerve was stimulated proximally at the sciatic notch and distally at the ankle via bipolar electrodes with supramaximal stimuli (8 V) at 20 Hz. The latencies of the compound muscle action potentials were recorded via bipolar electrodes from the first interosseous muscle of the hindpaw and measured from the stimulus artifact to the onset of the negative M-wave deflection. MNCV was calculated by subtracting the distal latency from the proximal latency, and the result was divided into the distance between the stimulating and recording electrode.
Hindlimb SNCV was recorded in the digital nerve to the second toe by stimulating with a square-wave pulse of 0.05 ms duration using the smallest intensity current that resulted in a maximal amplitude response. The sensory nerve action potential was recorded behind the medial malleolus. Sixteen responses were averaged to obtain the position of the negative peak. The maximal SNCV was calculated by measuring the latency to the onset/peak of the initial negative deflection and the distance between stimulating and recording electrodes.
In all nerve conduction measurements, TCAT-2 Temperature Controller with RET-3 Temperature probe and HL-1 Heat Lamp (Physitemp Instruments, Clifton, NJ) was used to maintain body and hindlimb temperature at 37°C.
To determine the sensitivity to noxious heat, rats or mice were placed within a plexiglass chamber on a transparent glass surface and allowed to acclimate for at least 20 min. A thermal stimulation meter (IITC model 336 TG Combination Tail Flick & Paw algesia meter; IITC Life Science, Woodland Hills, CA) was used. The device was activated after placing the stimulator directly beneath the plantar surface of the hindpaw. The paw withdrawal latency in response to the radiant heat (a heating rate of ∼1.3°C per second, cut-off time of 35 s for rats and 30 s for mice) was recorded. Floor temperature was set at ∼32–33°C (manufacturer’s set-up). Individual measurements were repeated four to five times and the mean value was calculated.
Tail pressure thresholds were registered with a paw/tail pressure analgesia meter for the Randall–Selitto test (37215, Analgesy-Meter; UGO-Basile, Comerio VA, Italy). Pressure increasing at a linear rate of 10 g with the cut-off of 250 g to avoid tissue injury was applied to the dorsal surface of the paw (rats) or base of the tail (mice). The applied tail pressure that evoked biting or licking behavior was registered by an analgesia meter and expressed in grams. Three tests separated by at least 15 min were performed for each animal, and the mean value of these tests was calculated.
Tactile responses were evaluated by quantifying the withdrawal threshold of the hindpaw in response to stimulation with flexible von Frey filaments as described in detail previously (14,17).
Intraepidermal nerve fiber density.
Three longitudinal 50-μm-thick footpad sections from each rat were cut on Leica CM1950 cryostat (Leica Microsystems, Nussloch, Germany). Nonspecific binding was blocked by 3% goat serum containing 0.5% porcine gelatin and 0.5% Triton X-100 in SuperBlock blocking buffer at room temperature, for 2 h The sections were incubated overnight with PGP 9.5 antiserum in 1:400 dilution at 4°C, after which secondary Alexa Fluor 488 IgG (H+L) in 1:1,000 was applied at room temperature for 1 h. Sections were then coverslipped with VECTASHIELD mounting medium. Intraepidermal nerve fiber profiles were counted blindly by three independent investigators under Axioplan 2 microscope (Zeiss) at 40× magnification, and the average values were used. The length of epidermis was assessed on the microphotographs of stained sections taken at 5× magnification with a 3I Everest imaging system (Intelligent Imaging Innovations, Denver, CO) equipped with Axioplan 2 microscope (Zeiss) using the National Institutes of Health ImageJ software (version 1.42q). An average of 2.8 ± 0.3 mm of the sample length was investigated to calculate a number of nerve fiber profiles per millimeter of epidermis. Representative images of intraepidermal nerve fibers were obtained by confocal laser scanning microscopy at 400× magnification using Leica TCS SP5 confocal system (Leica Microsystems, Mannheim, Germany).
Western blot analysis of UPR components in rat sciatic nerve and spinal cord and of nitrated proteins in mouse sciatic nerve.
Total and phosphorylated PERK, total and phosphorylated eIF2α, total and phosphorylated IRE1α, and ERO1α, CHOP, BiP/GRP78, and GRP94 in rat sciatic nerve and spinal cord, and nitrated protein levels in mouse sciatic nerve were evaluated by Western blot analysis. We used 7.5% (phosphorylated and total PERK, and phosphorylated and total IRE1α) and 10% (phosphorylated and total eIF2α, ERO1α, BiP/GRP78, GRP94, and nitrated proteins) sodium dodecyl sulfate (SDS) polyacrylamide gels, and the electrophoresis was conducted for 2 h. After blocking free binding sites as described, primary antibodies against phosphorylated PERK, eIF2α, and IRE1α, as well as ERO1α, CHOP, GRP78/BiP, GRP94, or nitrated proteins, were applied overnight at 4°C. Then, the corresponding (anti-rabbit or anti-mouse) secondary antibodies were applied at room temperature for 1 h. Protein bands detected by the antibodies were visualized with Amersham ECL Western Blotting Detection Reagent (Little Chalfont, Buckinghamshire, UK). For assessment of total PERK, eIF2α, and IRE1α, the membranes used for detection of phosphorylated PERK, eIF2α, and IRE1α, were stripped and reprobed with primary antibodies against total PERK, eIF2α, and IRE1α, respectively. After incubation with secondary antibody and visualization of total PERK, eIF2α, and IRE1α protein bands as described, the membranes were stripped again and reprobed with β-actin antibody to confirm equal protein loading. Free binding sites were blocked in 5% (wt/vol) BSA (BiP/GRP78, phospho-PERK, phospho-eIF2α, ERO1α), 3% (wt/vol) BSA (CHOP, GRP94, nitrated proteins), or 5% (wt/vol) nonfat dry milk (total PERK and eIF2α, phosphorylated and total IRE1α), all diluted in 20 mmol/L Tris-HCl buffer, pH 7.5, containing 150 mmol/L NaCl and 0.05% Tween 20, for 1 h. Stripping was conducted in 25 mmol/L glycine-HCl, pH 2.5, buffer containing 2% SDS.
ELISA measurements of 4-HNE adducts and nitrotyrosine in rat sciatic nerve and spinal cord.
For 4-HNE adduct measurements, the samples were homogenized in 20 mmol/L PBS, pH 7.4 (1:10, wt/vol), on ice. Homogenates were centrifuged at 14,000g (4°C, 20 min). Supernatants were used for measurements of 4-HNE adducts with the OxiSelect HNE-His Adduct ELISA kit (Cell BioLabs, San Diego, CA). For NT measurements, the samples were homogenized on ice in RIPA buffer (1:10 wt/vol) containing 50 mmol/L Tris-HCl, pH 7.2; 150 mmol/L NaCl; 0.1% sodium dodecyl sulfate; 1% NP-40; 5 mmol/L EDTA; 1 mmol/L EGTA; 1% sodium deoxycholate; and containing the protease/phosphatase inhibitors leupeptin (10 μg/mL); aprotinin (20 μg/mL); benzamidine (10 mmol/L); phenylmethylsulfonyl fluoride (1 mmol/L); and sodium orthovanadate (1 mmol/L). Homogenates were sonicated (3 × 5 s) and centrifuged at 14,000g (4°C, 20 min). Supernatants were used for measurements of NT concentrations with the OxiSelect Nitrotyrosine ELISA kit (Cell Biolabs). 4-HNE adducts and NT concentrations were normalized per milligram of protein. Protein was measured with the bicinchoninic acid protein assay (Pierce Biotechnology, Rockford, IL).
Statistical analysis.
The results are expressed as mean ± standard errors. The data were subjected to equality of variance F test, and then to log transformation, if necessary, before one-way ANOVA. When overall significance (P < 0.05) was attained, individual between-group comparisons were made using the Student-Newman-Keuls multiple range test. Significance was defined at P ≤ 0.05. When between-group variance differences could not be normalized by log transformation (datasets for final body weights and plasma glucose), the data were analyzed by the nonparametric Kruskal-Wallis one-way ANOVA, followed by the Bonferroni-Dunn or Fisher protected least significant difference tests for multiple comparisons.
RESULTS
Experiment 1.
Experiment 1 involved ER stress and activation of UPR in neural tissues of STZ diabetic rats and their prevention by TMAO and PBA. Rats with 12-week duration of STZ diabetes displayed upregulation of UPR indicative of ER stress in sciatic nerve (Fig. 1) and spinal cord (Fig. 2). Sciatic nerve phospho-PERK level and phospho-PERK:total PERK ratio were increased by 32% and 37% (Fig. 1A and B). Phospho-eIF2α was increased by 27% (Fig. 1C and D). The diabetic condition was associated with a 31% increase in phospho-IRE1α:total IRE1α ratio (Fig. 1E and F). Increased phospho-IRE1α:total IRE-1α ratio is consistent with increased levels of CHOP (Fig. 1G and H), the molecular chaperones BiP (Fig. 1I and J), and GRP94 (Fig. 1K and L), as well as CHOP-inducible ERO1α (Fig. 1M and N) by 18%, 41%, 19%, and 22%, respectively. TMAO, a chemical chaperone, blunted diabetes-induced activation of URP, indicative of prevention of ER stress in peripheral nerve. In the spinal cord, the diabetic condition was associated with 42 and 20% increase in CHOP and ERO1α levels (Fig. 2A–D), whereas BiP (Fig. 2E and F), and GRP94 (Fig. 2G and H) levels were indistinguishable between control and diabetic rats. TMAO-treated diabetic rats preserved normal CHOP and ERO1α levels consistent with prevention of ER stress.
FIG. 1.

Representative Western blot analyses of total and phosphorylated PERK (A), total and phosphorylated eIF2α (C), total and phosphorylated IRE1α (E), CHOP (G), GRP78/BiP (I), GRP94 (K), and ERO1α (M), and their contents (densitometry; B, D, F, H, J, L, and N) and, when applicable, phosphorylation states, in sciatic nerve of nondiabetic and diabetic rats maintained with or without trimethylamine oxide treatment for 12 weeks after induction of STZ diabetes (a prevention study). C, control; D, diabetic; TMAO, trimethylamine oxide. Mean ± SEM, n = 6–12 per group. *P < 0.05 and **P< 0.01 vs. nondiabetic controls; #P < 0.05 and ##P< 0.01 vs. diabetic rats maintained without trimethylamine oxide treatment.
FIG. 2.

Representative Western blot analyses of CHOP (A), ERO1α (C), GRP78/BiP (E), and GRP94 (G), and their content (densitometry; B, D, F, and H) in spinal cord of nondiabetic and diabetic rats maintained with or without trimethylamine oxide treatment for 12 weeks after induction of STZ diabetes (a prevention study). C, control; D, diabetic. Mean ± SEM, n = 8–12 per group. *P < 0.05 and **P < 0.01 vs. nondiabetic controls; #P < 0.05 and ##P < 0.01 vs. diabetic rats maintained without trimethylamine oxide treatment.
The effects of PBA on sciatic nerve phospho-eIF2α (Supplementary Fig. 1A), ERO1α (Supplementary Fig. 1B), CHOP (Supplementary Fig. 1C), and BiP/GRP78 (Fig. 1D) closely mimicked those of TMAO, consistent with the prevention of ER stress and activation of UPR by both chemical chaperones.
Chemical chaperones did not reduce diabetic hyperglycemia but attenuated DPN. Blood glucose concentrations were increased by 319%, 276%, and 319% in untreated and TMAO-treated and PBA-treated diabetic rats, respectively, compared with nondiabetic controls (Table 1). Rats with 12-week duration of STZ diabetes had development of MNCV (26%) and SNCV (13%) deficits (Table 2). They also displayed small sensory fiber neuropathy manifested in thermal and mechanical hypoalgesia as well as tactile allodynia, i.e., a condition when the light touch is perceived as painful. TMAO-treated and PBA-treated diabetic rats preserved normal SNCV and displayed less severe MNCV deficit and small sensory nerve fiber dysfunction than the untreated diabetic group. Diabetic rats exhibited 34% intraepidermal nerve fiber loss (Fig. 3A–D), which was attenuated, but not completely prevented, by TMAO.
TABLE 1.
Body weights and blood glucose concentrations in nondiabetic and STZ diabetic rats maintained with or without trimethylamine oxide or 4-phenylbutyric acid treatments
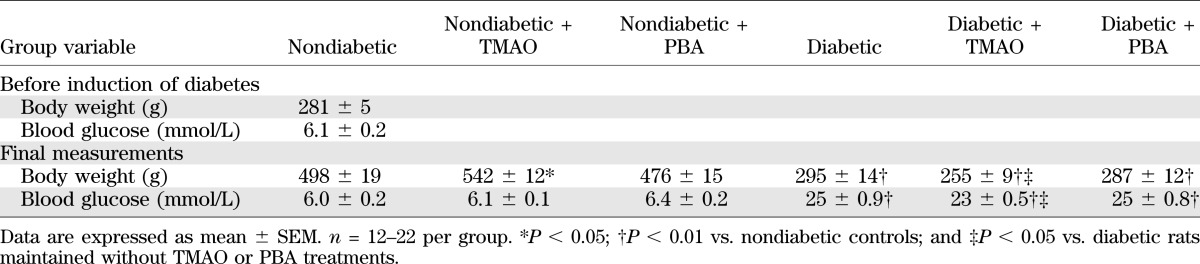
TABLE 2.
Variables of peripheral nerve function in nondiabetic and STZ diabetic rats maintained with or without trimethylamine oxide or PBA treatments
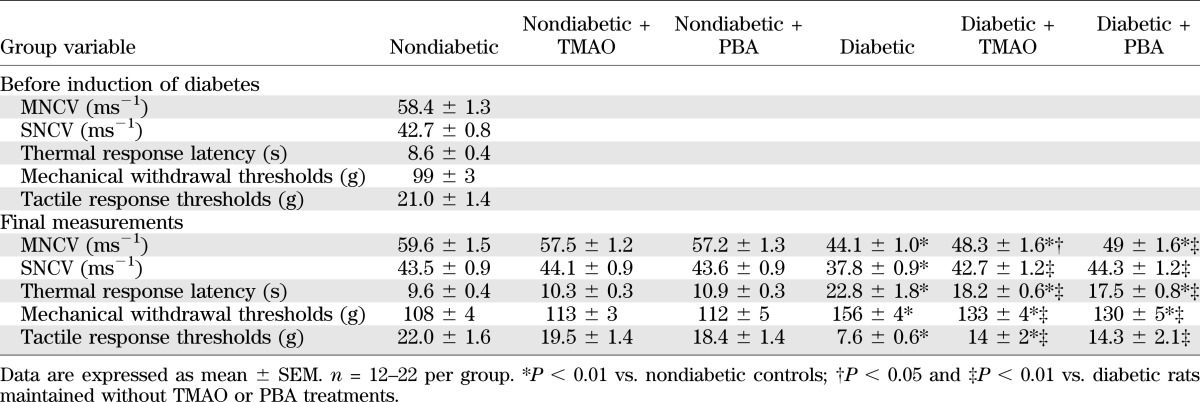
FIG. 3.

Representative images of intraepidermal nerve fiber profiles (A) magnification ×400, intraepidermal nerve fiber densities (B), and indices of oxidative-nitrative stress in sciatic nerve and spinal cord (C–F) in nondiabetic and diabetic rats maintained with or without trimethylamine oxide treatment for 12 weeks after induction of STZ diabetes (a prevention study). C, control; D, diabetic; INFD, intraepidermal nerve fiber density. Mean ± SEM, n = 8–12 per group. *P < 0.05 and **P < 0.01 vs. nondiabetic controls; #P < 0.05 and ##P < 0.01 vs. diabetic rats maintained without trimethylamine oxide treatment.
Inhibition of ER stress attenuated oxidative-nitrative stress in the sciatic nerve and spinal cord of TMAO-treated rats. Rats with 12-week duration of STZ diabetes displayed oxidative-nitrative stress manifest by accumulation of 4-HNE adducts (66% and 42%) and nitrotyrosine (97% and 44%) in sciatic nerve (Fig. 3E and F) and spinal cord (Fig. 3G and H). TMAO treatment attenuated oxidative-nitrative stress in both tissue targets for DPN.
Experiment 2.
CHOP gene deficiency reduced the severities of diabetes-associated oxidative-nitrative stress in the sciatic nerve, as well as MNCV and SNCV deficits, thermal hypoalgesia, and intraepidermal nerve fiber loss. Nonfasting blood glucose concentrations were increased by 235% and by 190% in diabetic wild-type and diabetic CHOP−/− mice, respectively (Table 3). Nondiabetic CHOP−/− mice displayed normal MNCV, SNCV, thermal algesia, and tactile sensitivity, and had slightly increased mechanical withdrawal thresholds and INFD (Table 4, Fig. 4A and B). The wild-type mice with 12-week duration of STZ diabetes exhibited 22% increase in the sciatic nerve CHOP level (Fig. 4C and D), MNCV and SNCV deficits, increased thermal response latencies and mechanical withdrawal thresholds, and reduced tactile response thresholds and INFD. CHOP−/− mice did not manifest any expression of CHOP in the sciatic nerve. CHOP gene deficiency reduced the severities of diabetes-associated oxidative-nitrative stress in the sciatic nerve, MNCV and SNCV deficits, thermal hypoalgesia, and intraepidermal nerve fiber loss, but did not have any beneficial effect on mechanical or tactile sensation.
TABLE 3.
Body weights and blood glucose concentrations in nondiabetic and diabetic CHOP-deficient mice
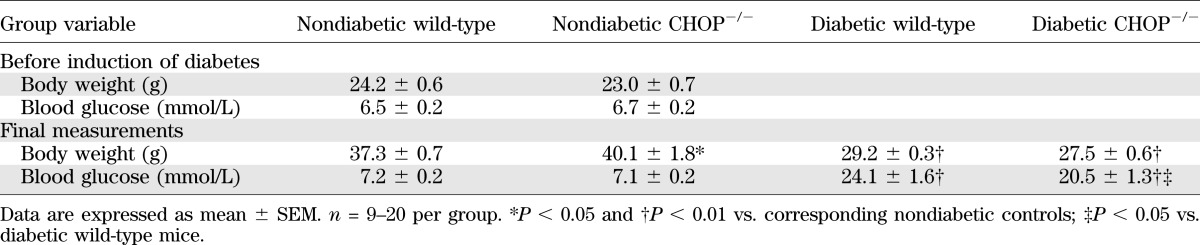
TABLE 4.
Variables of peripheral nerve function in nondiabetic and diabetic CHOP-deficient mice
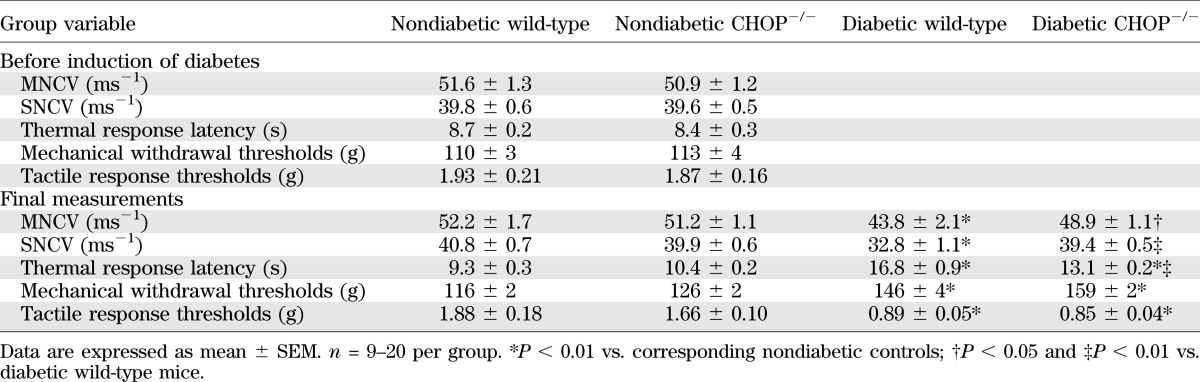
FIG. 4.

Representative images of intraepidermal nerve fiber profiles (A), magnification ×400, intraepidermal nerve fiber densities (B), representative Western blot analyses of sciatic nerve CHOP and nitrated proteins (C, E), and CHOP and nitrated protein contents (D, F) in nondiabetic and diabetic (12-week duration) wild-type (C57Bl6/J) and CHOP−/− mice. C, control; D, diabetic; INFD, intraepidermal nerve fiber density. Mean ± SEM, n = 7–12 per group. *P < 0.05 and **P < 0.01 vs. nondiabetic wild-type mice; ##P < 0.01 vs. diabetic wild-type mice.
DISCUSSION
The findings reported herein implicate ER stress, manifested in upregulation of UPR, in the development of DPN. In particular, ER stress leads to MNCV and SNCV deficits, small sensory nerve fiber dysfunction and degeneration, and oxidative-nitrative stress in peripheral nerve.
In the large-scale Diabetes Control and Complications Trial/Epidemiology of Diabetes Intervention and Complications study (32), improved blood glucose control reduced the risk of DPN associated with type 1 diabetes, thereby implicating hyperglycemia as a causative factor. Because severity of DPN strongly depends on blood glucose control, STZ diabetic rats exhibiting β-cell necrosis and irreversible hyperglycemia 2–3 days after induction of STZ comprise a suitable model for exploration of new pathogenetic mechanisms of DPN and drug discovery. Previous studies in STZ diabetic rats identified multiple pathogenetic mechanisms of DPN (6–9,13–15). The present findings demonstrating prevention of ER stress and attenuation of MNCV and SNCV deficits and small sensory nerve fiber dysfunction and degeneration, in the absence of alleviation of hyperglycemia, in chemical chaperone--treated STZ diabetic rats add ER stress to the list of pathogenetic mechanisms triggered either downstream of hyperglycemia or by other factors in the diabetic milieu such as nonesterified fatty acids (33) or altered tissue lipid composition (34). This conclusion is supported by our data in STZ diabetic CHOP−/− mice that displayed reduced severity of DPN, but not of the systemic hyperglycemia.
Many clinical neuropathy trials failed because of a lack of robust end points sensitive to pharmacological interventions (35). Reduction in INFD is emerging as a sensitive marker of diabetes-induced small sensory nerve fiber degeneration from both clinical (36–38) and experimental (14,16–18,39) studies, but the biochemical mechanisms underlying this phenomenon are not well understood. The present findings of attenuation of diabetes-induced intraepidermal nerve fiber loss by TMAO treatment or CHOP gene deficiency identify ER stress as an important contributor to small sensory nerve fiber loss in DPN.
Dissection of the contribution of long-term upregulation of the individual components of UPR to pathological conditions including diabetes and its complications is quite challenging because of the lack of specific inhibitors suitable for in vivo administration. Female CHOP−/− mice on C57Bl6/J background preserved normal glucose tolerance and insulin sensitivity despite marked obesity (28). CHOP deletion in insulin-resistant mice profoundly increased pancreatic β-cell mass and their ability to forestall the progression of diabetes (40,41). Aged STZ diabetic CHOP−/− mice did not have development of renal mesangial expansion and albuminuria (29). Our findings of diabetes-associated increase in peripheral nerve CHOP level in wild-type mice and of reduced severity of MNCV and SNCV deficits, thermal hypoalgesia, and intraepidermal nerve fiber degeneration in STZ diabetic CHOP−/− mice implicate CHOP in DPN.
It is well-established that intracellular oxidative-nitrative stress induces ER stress (42–44). In contrast, the role of ER stress in the development of oxidative-nitrative stress and, in particular, in diabetes is not understood properly. Under normal conditions, generation of ∼25% of reactive oxygen species in mammalian cells is a consequence of formation of disulfide bonds in the ER during oxidative protein folding (41,44). In this process, ER oxidoreductases, including protein disulphide isomerase (PDI), catalyze formation, isomerization, and reduction of disulfide bonds. During disulfide bond formation, cysteine residues within the PDI active site accept two electrons from thiol residues in the polypeptide chain substrate. Reduced PDI transfers its electrons through ERO1α to molecular oxygen as the final electron acceptor (41,44). Hydrogen peroxide is formed as a byproduct from the sequential action of PDI and ERO1α. Upregulation of PDI and ERO1α under conditions of ER stress therefore should result in excessive hydrogen peroxide formation. Hydrogen peroxide neutralization may be impaired because of glutathione depletion that occurs in the process of glutathione oxidation during reduction of disulfide bonds. ER causes induction of NAD(P)H oxidase-2 and NAD(P)H oxidase-dependent oxidative stress via CHOP and ERO1α-mediated release of ER Ca++ (45). ERO1α-mediated release of ER Ca++ at the mitochondria-associated membrane, with concomitant mitochondrial fission and Ca++ overload, also may lead to increased mitochondrial superoxide generation (46–48). Whereas CHOP overexpression has been reported to perturb the cellular redox state (49) and to increase superoxide generation (50), CHOP gene deficiency induced expression of the genes encoding antioxidant response and reduced protein carbonyl and hydroxyoctadecadienoic acid production (40). Attenuation of 4-HNE adducts and NT accumulation in peripheral nerve and spinal cord of TMAO-treated diabetic rats in the current study suggest that ER stress provides an important contribution to oxidative-nitrative stress in the peripheral nervous system in diabetes. This observation consistent with less pronounced nitrated protein accumulation in diabetic CHOP−/− mice is highly significant because oxidative-nitrative stress is one of the key mechanisms underlying MNCV and SNCV deficits, neurovascular dysfunction, and small sensor fiber neuropathy in diabetes (6–9,12,15,39).
In conclusion, our findings demonstrate an important contribution of ER stress completely and of the specific component of UPR, CHOP, to both large and small fiber neuropathies, and identify new therapeutic direction for this devastating complication of diabetes. With consideration of a key role of reduced nerve blood flow and endoneurial hypoxia in diabetes-induced MNCV and SNCV deficits (6–8,13), and of involvement of ER stress in other diabetic microvascular complications, such as nephropathy (29) and retinopathy (30), future studies should dissect the role for ER stress/UPR in neurovascular dysfunction associated with diabetes. Interactions among ER stress and other biochemical mechanisms implicated in the pathogenesis of DPN also need to be explored. In addition, our work provides rationale for evaluation of variables of ER stress in easily accessible biological materials (peripheral blood monocytes, skin) as potential biomarkers of DPN with diagnostic and prognostic value.
Supplementary Material
ACKNOWLEDGMENTS
S.L., P.W., R.S., H.S., and I.G.O. were partially supported by the National Institutes of Health Grants RO1-DK-074517, RO1-DK-077141, and RO1-DK-081147 and American Diabetes Association Research Grant 7-08-RA-102 (all to I.G.O.). The Cell Biology and Bioimaging Core used in this work is supported in part by COBRE (NIH P20 RR0-21945) and CNRU (NIH 1P30-DK-072476) center grants from the National Institutes of Health.
No potential conflicts of interest relevant to this article were reported.
S.L. performed most of Western blot analyses and participated in MNCV and SNCV measurements. P.W. was responsible for animal treatment, CHOP−/− mouse colony development and maintenance, and behavioral tests. R.S. performed the remaining part of Western blot analyses as well as ELISA assays. H.S. performed assessment of intraepidermal nerve fiber density. All authors participated in the data discussion. I.G.O. wrote the manuscript. H.S. is the guarantor of this work and, as such, had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
The authors thank Dr. Douglas E. Wright from the University of Kansas Medical Center, Kansas City, Kansas, for valuable recommendations regarding intraepidermal nerve fiber density measurements.
NOTE ADDED IN PROOF
The authors are deeply saddened by the loss of dear colleague and mentor, Dr. Irina Obrosova. Dr. Obrosova died on December 4, 2012. She continued to work until the very last moment despite her difficult fight with a terminal disease. Her selfless devotion and invaluable contribution to the field of diabetic complications will be remembered by everyone who knew her.
Footnotes
†Deceased.
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db12-0716/-/DC1.
See accompanying commentary, p. 696.
REFERENCES
- 1.Sinnreich M, Taylor BV, Dyck PJ. Diabetic neuropathies. Classification, clinical features, and pathophysiological basis. Neurologist 2005;11:63–79 [DOI] [PubMed] [Google Scholar]
- 2.Boulton AJ, Vinik AI, Arezzo JC, et al. American Diabetes Association Diabetic neuropathies: a statement by the American Diabetes Association. Diabetes Care 2005;28:956–962 [DOI] [PubMed] [Google Scholar]
- 3.Veves A, Backonja M, Malik RA. Painful diabetic neuropathy: epidemiology, natural history, early diagnosis, and treatment options. Pain Med 2008;9:660–674 [DOI] [PubMed] [Google Scholar]
- 4.Yagihashi S, Yamagishi SI, Wada Ri R, et al. Neuropathy in diabetic mice overexpressing human aldose reductase and effects of aldose reductase inhibitor. Brain 2001;124:2448–2458 [DOI] [PubMed] [Google Scholar]
- 5.Bierhaus A, Fleming T, Stoyanov S, et al. Methylglyoxal modification of Na(v)1.8 facilitates nociceptive neuron firing and causes hyperalgesia in diabetic neuropathy. Nat Med 2012;18:1445DOI: 10.1038/nm.2750 [DOI] [PubMed] [Google Scholar]
- 6.Nagamatsu M, Nickander KK, Schmelzer JD, et al. Lipoic acid improves nerve blood flow, reduces oxidative stress, and improves distal nerve conduction in experimental diabetic neuropathy. Diabetes Care 1995;18:1160–1167 [DOI] [PubMed] [Google Scholar]
- 7.Coppey LJ, Gellett JS, Davidson EP, Dunlap JA, Lund DD, Yorek MA. Effect of antioxidant treatment of streptozotocin-induced diabetic rats on endoneurial blood flow, motor nerve conduction velocity, and vascular reactivity of epineurial arterioles of the sciatic nerve. Diabetes 2001;50:1927–1937 [DOI] [PubMed] [Google Scholar]
- 8.Cameron NE, Tuck Z, McCabe L, Cotter MA. Effect of the hydroxyl radical scavenger, dimethylthiourea, on peripheral nerve tissue perfusion, conduction velocity and nociception in experimental diabetes. Diabetologia 2001;44:1161–1169 [DOI] [PubMed] [Google Scholar]
- 9.Schmeichel AM, Schmelzer JD, Low PA. Oxidative injury and apoptosis of dorsal root ganglion neurons in chronic experimental diabetic neuropathy. Diabetes 2003;52:165–171 [DOI] [PubMed] [Google Scholar]
- 10.Ziegler D, Sohr CG, Nourooz-Zadeh J. Oxidative stress and antioxidant defense in relation to the severity of diabetic polyneuropathy and cardiovascular autonomic neuropathy. Diabetes Care 2004;27:2178–2183 [DOI] [PubMed] [Google Scholar]
- 11.Ziegler D, Low PA, Litchy WJ, et al. Efficacy and safety of antioxidant treatment with α-lipoic acid over 4 years in diabetic polyneuropathy: the NATHAN 1 trial. Diabetes Care 2011;34:2054–2060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Obrosova IG, Mabley JG, Zsengellér Z, et al. Role for nitrosative stress in diabetic neuropathy: evidence from studies with a peroxynitrite decomposition catalyst. FASEB J 2005;19:401–403 [DOI] [PubMed] [Google Scholar]
- 13.Cameron NE, Cotter MA, Jack AM, Basso MD, Hohman TC. Protein kinase C effects on nerve function, perfusion, Na(+), K(+)-ATPase activity and glutathione content in diabetic rats. Diabetologia 1999;42:1120–1130 [DOI] [PubMed] [Google Scholar]
- 14.Obrosova IG, Xu W, Lyzogubov VV, et al. PARP inhibition or gene deficiency counteracts intraepidermal nerve fiber loss and neuropathic pain in advanced diabetic neuropathy. Free Radic Biol Med 2008;44:972–981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Drel VR, Lupachyk S, Shevalye H, et al. New therapeutic and biomarker discovery for peripheral diabetic neuropathy: PARP inhibitor, nitrotyrosine, and tumor necrosis factor-alpha. Endocrinology 2010;151:2547–2555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kellogg AP, Wiggin TD, Larkin DD, Hayes JM, Stevens MJ, Pop-Busui R. Protective effects of cyclooxygenase-2 gene inactivation against peripheral nerve dysfunction and intraepidermal nerve fiber loss in experimental diabetes. Diabetes 2007;56:2997–3005 [DOI] [PubMed] [Google Scholar]
- 17.Obrosova IG, Stavniichuk R, Drel VR, et al. Different roles of 12/15-lipoxygenase in diabetic large and small fiber peripheral and autonomic neuropathies. Am J Pathol 2010;177:1436–1447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bianchi R, Buyukakilli B, Brines M, et al. Erythropoietin both protects from and reverses experimental diabetic neuropathy. Proc Natl Acad Sci USA 2004;101:823–828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eizirik DL, Cardozo AK, Cnop M. The role for endoplasmic reticulum stress in diabetes mellitus. Endocr Rev 2008;29:42–61 [DOI] [PubMed] [Google Scholar]
- 20.Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 2010;140:900–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hotamisligil GS. Endoplasmic reticulum stress and atherosclerosis. Nat Med 2010;16:396–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Minamino T, Komuro I, Kitakaze M. Endoplasmic reticulum stress as a therapeutic target in cardiovascular disease. Circ Res 2010;107:1071–1082 [DOI] [PubMed] [Google Scholar]
- 23.Tabas I. The role of endoplasmic reticulum stress in the progression of atherosclerosis. Circ Res 2010;107:839–850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saxena S, Cabuy E, Caroni P. A role for motoneuron subtype-selective ER stress in disease manifestations of FALS mice. Nat Neurosci 2009;12:627–636 [DOI] [PubMed] [Google Scholar]
- 25.Schonthal AH. Targeting endoplasmic reticulum stress for cancer therapy. Front Biosci (Schol Ed) 2012;4:412–431 [Schol Ed] [DOI] [PubMed] [Google Scholar]
- 26.Fonseca SG, Lipson KL, Urano F. Endoplasmic reticulum stress signaling in pancreatic beta-cells. Antioxid Redox Signal 2007;9:2335–2344 [DOI] [PubMed] [Google Scholar]
- 27.Kars M, Yang L, Gregor MF, et al. Tauroursodeoxycholic Acid may improve liver and muscle but not adipose tissue insulin sensitivity in obese men and women. Diabetes 2010;59:1899–1905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maris M, Overbergh L, Gysemans C, et al. Deletion of C/EBP homologous protein (Chop) in C57Bl/6 mice dissociates obesity from insulin resistance. Diabetologia 2012;55:1167–1178 [DOI] [PubMed] [Google Scholar]
- 29.Wu J, Zhang R, Torreggiani M, et al. Induction of diabetes in aged C57B6 mice results in severe nephropathy: an association with oxidative stress, endoplasmic reticulum stress, and inflammation. Am J Pathol 2010;176:2163–2176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhong Y, Li J, Chen Y, Wang JJ, Ratan R, Zhang SX. Activation of endoplasmic reticulum stress by hyperglycemia is essential for Müller cell-derived inflammatory cytokine production in diabetes. Diabetes 2012;61:492–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sims-Robinson C, Zhao S, Hur J, Feldman EL. Central nervous system endoplasmic reticulum stress in a murine model of type 2 diabetes. Diabetologia 2012;55:2276–2284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martin CL, Albers J, Herman WH, et al. DCCT/EDIC Research Group Neuropathy among the diabetes control and complications trial cohort 8 years after trial completion. Diabetes Care 2006;29:340–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kharroubi I, Ladrière L, Cardozo AK, Dogusan Z, Cnop M, Eizirik DL. Free fatty acids and cytokines induce pancreatic beta-cell apoptosis by different mechanisms: role of nuclear factor-kappaB and endoplasmic reticulum stress. Endocrinology 2004;145:5087–5096 [DOI] [PubMed] [Google Scholar]
- 34.Fu S, Yang L, Li P, et al. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature 2011;473:528–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dyck PJ, Norell JE, Tritschler H, et al. Challenges in design of multicenter trials: end points assessed longitudinally for change and monotonicity. Diabetes Care 2007;30:2619–2625 [DOI] [PubMed] [Google Scholar]
- 36.Pittenger GL, Ray M, Burcus NI, McNulty P, Basta B, Vinik AI. Intraepidermal nerve fibers are indicators of small-fiber neuropathy in both diabetic and nondiabetic patients. Diabetes Care 2004;27:1974–1979 [DOI] [PubMed] [Google Scholar]
- 37.Quattrini C, Tavakoli M, Jeziorska M, et al. Surrogate markers of small fiber damage in human diabetic neuropathy. Diabetes 2007;56:2148–2154 [DOI] [PubMed] [Google Scholar]
- 38.Smith AG, Ramachandran P, Tripp S, Singleton JR. Epidermal nerve innervation in impaired glucose tolerance and diabetes-associated neuropathy. Neurology 2001;57:1701–1704 [DOI] [PubMed] [Google Scholar]
- 39.Shevalye H, Watcho P, Stavniichuk R, Dyukova E, Lupachyk S, Obrosova IG. Metanx alleviates multiple manifestations of peripheral neuropathy and increases intraepidermal nerve fiber density in zucker diabetic Fatty rats. Diabetes 2012;61:2126–2133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Song B, Scheuner D, Ron D, Pennathur S, Kaufman RJ. Chop deletion reduces oxidative stress, improves beta cell function, and promotes cell survival in multiple mouse models of diabetes. J Clin Invest 2008;118:3378–3389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kaufman RJ, Back SH, Song B, Han J, Hassler J. The unfolded protein response is required to maintain the integrity of the endoplasmic reticulum, prevent oxidative stress and preserve differentiation in β-cells. Diabetes Obes Metab 2010;12(Suppl 2):99–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dickhout JG, Hossain GS, Pozza LM, Zhou J, Lhoták S, Austin RC. Peroxynitrite causes endoplasmic reticulum stress and apoptosis in human vascular endothelium: implications in atherogenesis. Arterioscler Thromb Vasc Biol 2005;25:2623–2629 [DOI] [PubMed] [Google Scholar]
- 43.Guo R, Ma H, Gao F, Zhong L, Ren J. Metallothionein alleviates oxidative stress-induced endoplasmic reticulum stress and myocardial dysfunction. J Mol Cell Cardiol 2009;47:228–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tu BP, Weissman JS. Oxidative protein folding in eukaryotes: mechanisms and consequences. J Cell Biol 2004;164:341–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li G, Scull C, Ozcan L, Tabas I. NADPH oxidase links endoplasmic reticulum stress, oxidative stress, and PKR activation to induce apoptosis. J Cell Biol 2010;191:1113–1125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am J Physiol Cell Physiol 2004;287:C817–C833 [DOI] [PubMed] [Google Scholar]
- 47.Simmen T, Lynes EM, Gesson K, Thomas G. Oxidative protein folding in the endoplasmic reticulum: tight links to the mitochondria-associated membrane (MAM). Biochim Biophys Acta 2010;1798:1465-147348. [DOI] [PMC free article] [PubMed]
- 48.Yu T, Jhun BS, Yoon Y. High-glucose stimulation increases reactive oxygen species production through the calcium and mitogen-activated protein kinase-mediated activation of mitochondrial fission. Antioxid Redox Signal 2011;14:425–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol 2001;21:1249–1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bek MF, Bayer M, Müller B, et al. Expression and function of C/EBP homology protein (GADD153) in podocytes. Am J Pathol 2006;168:20–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.