

Abstract
It is generally believed that inflammatory cues can attract noncognate, “bystander” T-cell specificities to sites of inflammation. We have shown that recruitment of naive and in vitro activated autoreactive CD8+ T cells into endogenous islets requires local autoantigen expression. Here, we demonstrate that absence of an autoantigen in syngeneic extrapancreatic islet grafts in diabetic hosts renders the grafts “invisible” to cognate memory (and naive) T cells. We monitored the recruitment of islet-specific glucose-6-phosphatase catalytic subunit-related protein (IGRP)206–214-reactive CD8+ T cells into IGRP206–214-competent and IGRP206–214-deficient islet grafts in diabetic wild-type or IGRP206–214−/− nonobese diabetic hosts (harboring either naive and memory T cells or only naive IGRP206–214-specific T-cells, respectively). All four host–donor combinations had development of recurrent diabetes within 2 weeks. Wild-type hosts recruited IGRP206–214-specific T cells into IGRP206–214+/+ but not IGRP206–214−/− grafts. In IGRP206–214−/− hosts, there was no recruitment of IGRP206–214-specific T cells, regardless of donor type. Graft-derived IGRP206–214 activated naive IGRP206–214-specific T cells, but graft destruction invariably predated their recruitment. These results indicate that recurrent diabetes is exclusively driven by autoreactive T cells primed during the primary autoimmune response, and demonstrate that local antigen expression is a sine qua non requirement for accumulation of memory T cells into islet grafts. These findings underscore the importance of tackling autoreactive T-cell memory after β-cell replacement therapy.
Nonobese diabetic (NOD) mice have development of a form of type 1 diabetes that results from destruction of β cells by CD4+ and CD8+ T cells recognizing many autoantigenic peptides (1). A significant fraction of islet-associated CD8+ cells recognize the mimotope NRP-V7 in the context of the major histocompatibility complex (MHC) molecule Kd (2). These cells are a significant component of the earliest NOD islet CD8+ infiltrates (2,3), are diabetogenic (4,5), and target residues 206–214 of islet-specific glucose-6-phosphatase catalytic subunit-related protein (IGRP) (6). The peripheral IGRP206–214-reactive CD8+ T-cell pool is sizeable (7) and, on recruitment into islets, undergoes a local avidity maturation process that contributes to disease progression (8).
Studies in infection and autoimmune disease models have suggested that recruitment of T cells into sites of extralymphoid inflammation does not require local expression of cognate peptide–MHC (pMHC) (9–11). However, we recently have shown that cues emanating from pancreatic islets undergoing spontaneous autoimmune inflammation in NOD mice cannot recruit naive or newly activated bystander T-cell specificities. This was established by monitoring the recruitment of naive or in vitro activated IGRP206–214-specific CD8+ T cells in gene-targeted NOD mice expressing a T-cell “invisible” IGRP206–214 sequence. These mice had development of diabetes with normal incidence, but their insulitic lesions could not recruit either cell type. These results indicated that recruitment of naive T cells or effector cytotoxic T lymphocytes to a site of autoimmune inflammation results from an active process that is strictly dependent on local display of cognate pMHC (12).
Here, we asked whether this revised paradigm also applies to recruitment of memory (autoantigen-experienced) autoreactive T cells and/or recruitment of naive and memory T cells to syngeneic islet grafts. We reasoned that the “nonphysiological” lymphatic and vascular anatomy of islets grafts transplanted under the kidney capsule (13–15), coupled with a high rate of graft cell death (16), should allow recruitment of “graft-irrelevant” (i.e., nonautoreactive) memory T cells to the site in response to local inflammatory cues, including those caused by grafting. We demonstrate that recruitment of CD8+ T cells to islet grafts during disease recurrence exclusively involves autoantigen-specific T cells from the memory pool, excluding a role for bystander T-cell specificities or graft antigen-activated autoreactive T cells.
RESEARCH DESIGN AND METHODS
Mice.
NOD.IGRPK209A/F213AKI/KI mice, encoding an immunologically silent IGRP206–214 epitope, have been described (12). These studies were approved by the local Animal Care Committee.
Diabetes.
Diabetes was monitored twice per week by measuring urine glucose levels and was confirmed by tail vein blood glucose measurements. All recipient mice had at least two successive blood glucose measurements >22.2 mmol/L and underwent transplantation within 1–2 weeks of diabetes onset.
Peptides and tetramers.
The peptides IGRP206–214, NRP-V7, and TUM, and the corresponding tetramers (phycoerythrin -labeled), were prepared as described (17).
Flow cytometry.
Cell suspensions were stained with pMHC tetramers and FITC-conjugated or peridinin chlorophyll protein (PerCP)-conjugated anti-CD8α and anti-CD4 mAbs (BD Pharmingen) for 60 min at 4°C, fixed in 1% paraformaldehyde/PBS, and analyzed by fluorescence-activated cell sorting.
Islet isolation.
Pancreatic islets were isolated by hand-picking after collagenase P digestion of the pancreas and cultured overnight at 37°C in 5% CO2.
Islet transplantation and graft harvest.
Five hundred islets were transplanted under the left kidney capsule. Successful engraftment was defined as restoration of glycemic control for >48 h. Graft failure was defined as nonfasting blood glucose >15 mmol/L.
Specificity of islet-associated CD8+ T cells.
The grafts of recurrent diabetic hosts were cut into ∼2-mm3 fragments and cultured for 1 week in 0.5 units/mL rIL-2. T cells were analyzed by fluorescence-activated cell sorter as described. Measurements of interferon-γ secretion by graft-associated T cells (2 × 104/well) in response to peptide-pulsed irradiated NOD splenocytes (105/well) were determined by enzyme-linked immunosorbent assay (R&D Systems) and normalized to values obtained with TUM.
Adoptive transfer.
Purified splenic CD8+ T cells were labeled with carboxyfluorescein succinimidyl ester (CFSE) (2.5 μmol/L) and injected intravenously (5 × 106) 24 h after transplantation. Mice were killed 7 days later, and the grafted and nongrafted kidney-draining lymph nodes, spleens, pancreatic lymph nodes, and mesenteric lymph nodes were examined for dilution of CFSE in the CFSE+CD8+ gate.
Statistical analyses.
Data were compared by Mann-Whitney U or χ2 log-rank tests. Statistical significance was assumed at P < 0.05.
Online supplementary materials.
The online supplementary materials provide supporting data on the recruitment of IGRP206–214-reactive or InsB15–23–reactive CD8+ T cells to islet grafts, as well as representative fluorescence-activated cell sorter profiles.
RESULTS
NOD.IGRPK209A/F213AKI/KI mice (referred to as IGRP206–214−/− or “epitope-deficient” mice) have development of diabetes with the same incidence and kinetics as wild-type NOD (“epitope-competent”) mice but cannot trigger the activation or recruitment of naive or in vitro activated IGRP206–214-reactive CD8+ T cells (12). Here, we investigated if the naive IGRP206–214-reactive CD8+ T cells of epitope-deficient hosts and/or their memory counterparts arising in epitope-expressing hosts are recruited into epitope-competent or epitope-deficient islet grafts (from NOD.scid and NOD.rag2−/−.IGRPK209A/F213AKI/KI donors, respectively).
We first tracked the recruitment of IGRP206–214-reactive CD8+ T cells from diabetic IGRP206–214+ hosts (harboring both naive and memory IGRP206–214-reactive CD8+ T cells) into IGRP206–214+ or IGRP206–214−/− grafts. The presence of IGRP206–214-reactive CD8+ T cells recruited into the graft was analyzed by flow cytometry using pMHC tetramers. We also measured the amount of interferon-γ that graft-infiltrating T cells secreted in response to peptide-pulsed irradiated splenocytes as an additional read-out of T-cell recruitment.
Diabetic IGRP206–214+ hosts receiving IGRP206–214−/− islets had development of recurrence of disease, but did so a few days later than those receiving IGRP206–214+ islets (12.7 ± 3.5 vs. 5.9 ± 0.7 days; Figs. 1A top, B, and C left). This indicated that recruitment of naive and/or preactivated IGRP206–214-reactive CD8+ T cells contributes to, but is dispensable for, graft destruction in diabetic IGRP206–214+ hosts. Importantly, however, whereas IGRP206–214-reactive T cells accounted for a significant fraction of IGRP206–214+ graft-associated CD8+ T cells (18.2% ± 4.7%), they were undetectable in IGRP206–214−/− grafts (Fig. 2A–C and Supplementary Fig. 1A). Furthermore, the lymph nodes draining the grafted (left) kidney in mice receiving IGRP-206–214–expressing islet grafts harbored more IGRP206–214-reactive CD8+ T cells than those draining the contralateral (nongrafted) kidney, and this was not seen in diabetic hosts grafted with IGRP206–214−/− islets (Fig. 3A). In addition, the pancreatic lymph nodes and the spleen, and to a lesser extent the mesenteric lymph nodes, of mice grafted with IGRP206–214+ islets contained more IGRP206–214-reactive CD8+ T cells than those from mice grafted with IGRP206–214−/− islets (Figs. 3B and C). This suggests that graft-derived IGRP206–214 induces the activation and retention of host naive and/or preactivated IGRP206–214-reactive CD8+ T cells in graft-proximal lymphoid organs.
FIG. 1.

Survival of islet grafts from IGRP206–214-competent or IGRP206–214-deficient donors in spontaneously diabetic IGRP206–214-competent or IGRP206–214-deficient NOD hosts. A: Individual blood glucose curves of diabetic NOD hosts receiving NOD.scid (n = 10) or NOD.rag2KI/KI.IGRPK209A/F213AKI/KI islets (n = 9), and diabetic NOD.IGRPK209A/F213AKI/KI hosts receiving NOD.scid (n = 9) or NOD.rag2−/−.IGRPK209A/F213AKI/KI islets (n = 5). B: Average onset of disease recurrence after transplantation (in days) in the four different donor/host combinations. P values were obtained by Mann-Whitney U test. C: Survival curves of grafts in diabetic NOD (left) or NOD.IGRPK209A/F213AKI/KI hosts (right). P values were calculated via log-rank test. For (B) and (C), differences between epitope-positive and epitope-negative grafts in epitope-positive hosts remained statistically significant upon exclusion of the epitope-negative graft that survived to 40 days (P = 0.0395 in B; and P = 0.0353 in C).
FIG. 2.

Recruitment of IGRP206–214-reactive CD8+ T cells from diabetic IGRP206–214-competent or IGRP206–214-deficient NOD hosts into islet grafts from IGRP206–214-competent or IGRP206–214-deficient donors. A: Percentages of NRP-V7/Kd tetramer-positive in islet-graft–associated CD8+ T cells. Data (average ± SEM) correspond, from left to right, to eight, four, four, and three grafts per group, respectively. B: Interferon-γ (IFN-γ) secretion by islet graft-associated CD8+ T cells in response to NRP-V7 peptide-pulsed NOD dendritic cells. Data correspond, from left to right, to five, four, four, and three grafts per group, respectively. C: Representative fluorescence-activated cell sorting staining profiles of CD8+ T cells isolated from islet grafts in the four different donor/host combinations. TUM/Kd was used as a negative control tetramer. P values in (A) and (B) were obtained with Mann-Whitney U test. Grafts were harvested immediately after the last blood glucose measurement in Fig. 1. (A high-quality color representation of this figure is available in the online issue.)
FIG. 3.

Frequencies of IGRP206–214-reactive CD8+ T cells in lymphoid organs and blood of diabetic IGRP206–214-competent or IGRP206–214-deficient hosts grafted with IGRP206–214-competent or IGRP206–214-deficient islets. Percentages of NRP-V7/Kd tetramer-positive cells in CD8+ T cells from lymph nodes (LNs) draining the grafted compared with contralateral kidneys (A), the pancreatic lymph nodes (PLNs) and the mesenteric lymph nodes (MLNs) (B), and the spleen (C). Data (average ± SEM) correspond to six and eight mice, respectively. D: Frequencies of IGRP206–214-reactive CD8+ T cells in lymphoid organs and blood of IGRP206–214-competent compared with IGRP206–214-deficient hosts grafted with IGRP206–214-competent (left) compared with IGRP206–214-deficient islets (right). Data (average ± SEM) correspond to six, six, eight, and five mice per group, respectively. Background staining with the negative control tetramer TUM/Kd was subtracted. P values were obtained with Mann-Whitney U test.
We next investigated whether the IGRP206–214-reactive CD8+ T cells recruited to the epitope-expressing grafts include naive T-cells primed by graft-derived IGRP206–214. We followed the fate of IGRP206–214+ and IGRP206–214−/− islet grafts and IGRP206–214-reactive CD8+ T cells in diabetic IGRP206–214−/− hosts, which are unable to generate antigen-experienced IGRP206–214-reactive CD8+ T cells from an otherwise normal pool of naive T-cell precursors. IGRP206–214−/− hosts rejected IGRP206–214+ and IGRP206–214−/− islets with kinetics similar to those seen in NOD mice (Figs. 1A–C). Yet, IGRP206–214-reactive CD8+ T cells were barely detectable in IGRP206–214+ and IGRP206–214−/− grafts implanted into IGRP206–214−/− hosts (Figs. 2A and B), indicating that the grafts do not recruit newly primed IGRP206–214-reactive CD8+ T cells, at least within the first 2 weeks after transplantation. Interestingly, IGRP206–214+ grafts in IGRP206–214−/− hosts recruited slightly more InsB15–23-reactive CD8+ T cells than in IGRP206–214+ hosts (Supplementary Figs. 1B and C). Although these differences were not statistically significant, they suggest that in these mice the IGRP206–214-reactive CD8+ T-cell niche is occupied by other memory T-cell specificities. In addition, the islet graft-associated CD8+ T cells express markers of memory (i.e., are CD44high, CD62L−, and CD127+; data not shown).
In agreement with these data, the proximal lymphoid organs (graft-draining lymph nodes, pancreatic lymph nodes, and spleen) and blood of IGRP206–214−/− hosts transplanted with antigen-expressing islets contained fewer IGRP206–214-reactive CD8+ T cells than their IGRP206–214+ host counterparts, suggesting that graft-derived antigen does not induce a detectable peripheral expansion of naive autoreactive T cells (Fig. 3D). In addition, because the percentages of IGRP206–214-reactive CD8+ T cells in the graft-draining lymph nodes, pancreatic lymph nodes, and spleen of IGRP206–214+ hosts grafted with IGRP206–214-deficient islets were also low (Fig. 3D, right panels), we conclude that the peripheral expansion of IGRP206–214-reactive CD8+ T cells seen in IGRP206–214+ islet-grafted IGRP206–214+ hosts (Figs. 3A–C) largely, if not exclusively, involves antigen-experienced T cells. Interestingly, NOD hosts grafted with IGRP206–214−/− islets accumulated IGRP206–214-reactive CD8+ T cells in the bloodstream (Fig. 3D), suggesting that, in the absence of antigen in the graft and graft-draining lymphoid organs, preactivated IGRP206–214-reactive CD8+ T cells are “trapped” in the bloodstream.
Finally, we asked whether absence of graft-antigen-primed naive IGRP206–214-reactive CD8+ T cells in the epitope-expressing grafts of IGRP206–214−/− hosts was caused by inability of graft-derived IGRP206–214 to activate cognate naive CD8+ T cells, or to protracted recruitment and/or accumulation of these T cells into the graft. This was performed by tracking the proliferation of naive splenic CFSE-labeled IGRP206–214-reactive CD8+ T cells from 8.3 T-cell receptor transgenic mice in NOD hosts grafted with IGRP206–214+ or IGRP206–214−/− islets the previous day. Various host lymphoid organs were examined for dilution of CFSE in the CFSE+CD8+ gate 7 days after T-cell transfer. Naive 8.3-CD8+ T cells proliferated vigorously in the lymph nodes draining IGRP206–214+ (but not IGRP206–214−/−) grafts and, to a lesser extent, in the pancreatic lymph node and spleen, where some of the proliferation appears to be induced by host-derived (residual) IGRP206–214 (Fig. 4A and B). There were very few donor 8.3-CD8+ T cells in IGRP206–214+ or IGRP206–214−/− grafts (0.06% ± 0.03% vs. 0.06% ± 0.016% of CD8+ cells, respectively) (Fig. 4C). These observations indicate that destruction of IGRP206–214+ and IGRP206–214−/− grafts in IGRP206–214+ hosts (Fig. 1A) predates recruitment of newly activated T cells.
FIG. 4.
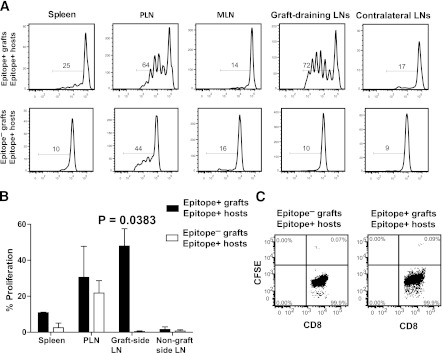
Proliferation of naive CFSE-labeled 8.3-CD8+ T cells in lymphoid organs of IGRP206–214-competent hosts grafted with IGRP206–214-competent compared with IGRP206–214-deficient islets. A: Representative CFSE dilution profiles. B: Average ± SEM of the percentage of proliferated cells. C: Representative flow profiles of graft-associated CD8+ T cells of these mice. Data in (A–C) correspond to three mice per host/donor combination. P values were obtained with Mann-Whitney U test. LN, lymph node; MLN, mesenteric lymph node; PLN, pancreatic lymph node.
DISCUSSION
The data presented herein challenge a current paradigm stating that nonantigen-specific inflammatory cues can attract and retain noncognate, bystander T-cell specificities to sites of inflammation, including syngeneic islet transplants in diabetic mice. We demonstrate that absence of the cognate autoantigen in a syngeneic extrapancreatic islet graft in a diabetic host renders the graft invisible to cognate memory (and naive) T cells. Local antigen expression, in addition to MHC class I expression (18), is thus a sine qua non requirement for accumulation of autoreactive CD8+ T cells into islet grafts.
The absolute need for local autoantigen expression is highlighted by two important considerations. First, IGRP206–214/Kd (NRP-V7)-reactive CD8+ T cells are among the most prevalent in NOD islet infiltration (8). Second, the vascular beds irrigating islet grafts, including the subcapsular kidney space, have a porous, fenestrated architecture (13–15) that could conceivably render them permeable to bystander T cells. Therefore, it is remarkable that autoantigen-experienced (i.e., memory) IGRP206–214-reactive T cells, despite their prevalence in the periphery, do not accumulate into IGRP206–214-deficient grafts. Recruitment of memory CD8+ T cells to islet grafts thus follows the same rules that we have described for the recruitment of naive and in vitro activated CD8+ T cells into endogenous islets (i.e., requiring a cognate pMHC interaction in situ). A recent report demonstrates a striking similarity in human insulitis: all the CD8+ T cells found in the inflamed islets of type 1 diabetic patients bound self-pMHC complexes (19).
Our findings further imply that individual autoantigenic specificities, even when prevalent, play relatively minor roles in the anamnestic autoimmune response contributing to graft destruction in autoimmune disease-affected hosts. Our results also clearly indicate that destruction of syngeneic islet grafts in diabetic NOD mice is largely, if not exclusively, effected by autoantigen-experienced T cells primed during the primary autoimmune response. Although graft antigen-loaded antigen-presenting cells residing in the graft-draining lymph nodes can readily induce the activation of naive autoreactive CD8+ T cells, graft destruction precedes recruitment of these T cells into the graft. The high physical and functional pMHC-binding avidities of antigen-experienced T cells coupled with their ability to mount rapid recall responses to limiting amounts of antigen (20) likely afford them a competitive advantage, particularly during the first 2 weeks after transplantation. Differences in the bio-distribution of memory versus naive T cells may be another contributing factor. These considerations, however, do not exclude the likely involvement of graft antigen-primed naive autoreactive T cells in chronic loss of graft function, such as, for example, in the context of partially matched islet allografts.
Recurrent autoimmunity in allogeneic islet cell transplantation has become a topic of growing interest. For example, the pretransplant peripheral frequencies of autoreactive T cells in diabetic recipients are predictive of islet allograft fate, and posttransplant increases are associated with loss of graft function (21–24), suggesting that recurrent autoimmunity may contribute to allograft destruction. Although clinical islet transplantation is a more complex situation, our model has allowed us to dissect the specific roles of bystander immunity versus anamnestic and naive autoimmunity to islet graft rejection. Our observations emphasize the importance of developing therapies capable of preventing priming of naive alloreactive T cells causing allograft rejection, recruitment of memory autoreactive T cells causing anamnestic autoimmunity in the immediate posttransplant period, and the priming of naive autoreactive T cells causing chronic loss of graft function, giving special attention to the pretransplant autoreactivity status of the diabetic host.
Supplementary Material
ACKNOWLEDGMENTS
The work described here was funded by grants from the Canadian Diabetes Association (CDA) and the Canadian Institutes of Health Research. G.M.A. and B.O.R. are supported by the Dutch Diabetes Research Foundation, the Juvenile Diabetes Research Foundation, and the European Union FP7 Program (BetaCellTherapy and NAIMIT). S.T. and X.C.-C. are supported by studentships from Alberta Innovates–Health Solutions (AIHS, formerly AHFMR) and the AXA Research Fund, respectively. J.R.W. was supported by a fellowship from the CDA. P.S. is a Scientist of the AIHS and a Scholar of the Juvenile Diabetes Research Foundation and Instituto de Investigaciones Sanitarias Carlos III. The Julia McFarlane Diabetes Research Centre is supported by the Diabetes Association (Foothills) and the CDA.
No potential conflicts of interest relevant to this article were reported.
G.M.A., X.C.-C., S.T., and J.R.W. researched data and contributed to the editing of the manuscript. G.M.A., X.C.-C., S.T., J.R.W., and B.O.R. contributed to the discussion. B.-Y.X. and J.W. assisted with experiments. J.W. and B.O.R. reviewed and edited the manuscript. P.S. designed and supervised the study. P.S. is the guarantor of this work and, as such, had full access to all the data and takes responsibility for the integrity of the data and the accuracy of data analysis.
The authors thank A. Shameli, S. Thiessen, J. Luces, and R. Barasi (University of Calgary) for technical assistance and L. Kennedy and L. Robertson (University of Calgary) for flow cytometry.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db12-0600/-/DC1.
G.M.A. is currently affiliated with the Department of Endocrinology, University Medical Center Groningen, Groningen, the Netherlands.
B.-Y.X. is currently affiliated with the Alberta Diabetes Institute, University of Alberta, Edmonton, Alberta, Canada.
REFERENCES
- 1.Babad J, Geliebter A, DiLorenzo TP. T-cell autoantigens in the non-obese diabetic mouse model of autoimmune diabetes. Immunology 2010;131:459–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anderson B, Park BJ, Verdaguer J, Amrani A, Santamaria P. Prevalent CD8(+) T cell response against one peptide/MHC complex in autoimmune diabetes. Proc Natl Acad Sci USA 1999;96:9311–9316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.DiLorenzo T, Graser T, Ono T, et al. Major histocompatibility complex class I-restricted T cells are required for all but end stages of diabetes development in nonobese diabetic mice and use a prevalent T cell receptor alpha chain gene rearrangement. Proc Natl Acad Sci USA 1998;95:12538–12543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Verdaguer J, Yoon JW, Anderson B, et al. Acceleration of spontaneous diabetes in TCR-beta-transgenic nonobese diabetic mice by beta-cell cytotoxic CD8+ T cells expressing identical endogenous TCR-alpha chains. J Immunol 1996;157:4726–4735 [PubMed] [Google Scholar]
- 5.Verdaguer J, Schmidt D, Amrani A, Anderson B, Averill N, Santamaria P. Spontaneous autoimmune diabetes in monoclonal T cell nonobese diabetic mice. J Exp Med 1997;186:1663–1676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lieberman SM, Evans AM, Han B, et al. Identification of the beta cell antigen targeted by a prevalent population of pathogenic CD8+ T cells in autoimmune diabetes. Proc Natl Acad Sci USA 2003;100:8384–8388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Trudeau J, Kelly-Smith C, Verchere B, Finegood D, Santamaria P, Tan R. Prediction of spontaneous autoimmune diabetes in NOD mice by quantification of autoreactive T cells in peripheral blood. J Clin Invest 2003;111:217–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Amrani A, Verdaguer J, Serra P, Tafuro S, Tan R, Santamaria P. Progression of autoimmune diabetes driven by avidity maturation of a T-cell population. Nature 2000;406:739–742 [DOI] [PubMed] [Google Scholar]
- 9.Stephens R, Randolph DA, Huang G, Holtzman MJ, Chaplin DD. Antigen-nonspecific recruitment of Th2 cells to the lung as a mechanism for viral infection-induced allergic asthma. J Immunol 2002;169:5458–5467 [DOI] [PubMed] [Google Scholar]
- 10.Ostler T, Pircher H, Ehl S. “Bystander” recruitment of systemic memory T cells delays the immune response to respiratory virus infection. Eur J Immunol 2003;33:1839–1848 [DOI] [PubMed] [Google Scholar]
- 11.Chen AM, Khanna N, Stohlman SA, Bergmann CC. Virus-specific and bystander CD8 T cells recruited during virus-induced encephalomyelitis. J Virol 2005;79:4700–4708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang J, Tsai S, Shameli A, Yamanouchi J, Alkemade G, Santamaria P. In situ recognition of autoantigen as an essential gatekeeper in autoimmune CD8+ T cell inflammation. Proc Natl Acad Sci USA 2010;107:9317–9322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lukinius A, Jansson L, Korsgren O. Ultrastructural evidence for blood microvessels devoid of an endothelial cell lining in transplanted pancreatic islets. Am J Pathol 1995;146:429–435 [PMC free article] [PubMed] [Google Scholar]
- 14.Vajkoczy P, Menger MD, Simpson E, Messmer K. Angiogenesis and vascularization of murine pancreatic islet isografts. Transplantation 1995;60:123–127 [PubMed] [Google Scholar]
- 15.Morini S, Brown ML, Cicalese L, et al. Revascularization and remodelling of pancreatic islets grafted under the kidney capsule. J Anat 2007;210:565–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Davalli AM, Scaglia L, Zangen DH, Hollister J, Bonner-Weir S, Weir GC. Vulnerability of islets in the immediate posttransplantation period. Dynamic changes in structure and function. Diabetes 1996;45:1161–1167 [DOI] [PubMed] [Google Scholar]
- 17.Tsai S, Shameli A, Yamanouchi J, et al. Reversal of autoimmunity by boosting memory-like autoregulatory T cells. Immunity 2010;32:568–580 [DOI] [PubMed] [Google Scholar]
- 18.Serreze DV, Chapman HD, Varnum DS, Gerling I, Leiter EH, Shultz LD. Initiation of autoimmune diabetes in NOD/Lt mice is MHC class I-dependent. J Immunol 1997;158:3978–3986 [PubMed] [Google Scholar]
- 19.Coppieters KT, Dotta F, Amirian N, et al. Demonstration of islet-autoreactive CD8 T cells in insulitic lesions from recent onset and long-term type 1 diabetes patients. J Exp Med 2012;209:51–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tailor P, Tsai S, Shameli A, et al. The proline-rich sequence of CD3epsilon as an amplifier of low-avidity TCR signaling. J Immunol 2008;181:243–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roep BO, Stobbe I, Duinkerken G, et al. Auto- and alloimmune reactivity to human islet allografts transplanted into type 1 diabetic patients. Diabetes 1999;48:484–490 [DOI] [PubMed] [Google Scholar]
- 22.Huurman VA, Hilbrands R, Pinkse GG, et al. Cellular islet autoimmunity associates with clinical outcome of islet cell transplantation. PLoS ONE 2008;3:e2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hilbrands R, Huurman VA, Gillard P, et al. Differences in baseline lymphocyte counts and autoreactivity are associated with differences in outcome of islet cell transplantation in type 1 diabetic patients. Diabetes 2009;58:2267–2276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pinkse GG, Tysma OH, Bergen CA, et al. Autoreactive CD8 T cells associated with beta cell destruction in type 1 diabetes. Proc Natl Acad Sci USA 2005;102:18425–18430 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.