

Abstract
Chronic endoplasmic reticulum (ER) stress is a major contributor to obesity-induced insulin resistance in the liver. However, the molecular link between obesity and ER stress remains to be identified. Proteasomes are important multicatalytic enzyme complexes that degrade misfolded and oxidized proteins. Here, we report that both mouse models of obesity and diabetes and proteasome activator (PA)28-null mice showed 30–40% reduction in proteasome activity and accumulation of polyubiquitinated proteins in the liver. PA28-null mice also showed hepatic steatosis, decreased hepatic insulin signaling, and increased hepatic glucose production. The link between proteasome dysfunction and hepatic insulin resistance involves ER stress leading to hyperactivation of c-Jun NH2-terminal kinase in the liver. Administration of a chemical chaperone, phenylbutyric acid (PBA), partially rescued the phenotypes of PA28-null mice. To confirm part of the results obtained from in vivo experiments, we pretreated rat hepatoma-derived H4IIEC3 cells with bortezomib, a selective inhibitor of the 26S proteasome. Bortezomib causes ER stress and insulin resistance in vitro—responses that are partly blocked by PBA. Taken together, our data suggest that proteasome dysfunction mediates obesity-induced ER stress, leading to insulin resistance in the liver.
Obesity is a major cause of insulin resistance and contributes to the development of type 2 diabetes (1). Growing evidence suggests that chronic endoplasmic reticulum (ER) stress in the liver is a major contributor to obesity-induced insulin resistance (2–4). However, the molecular mechanisms linking obesity and ER stress are not fully understood.
We previously identified metabolic pathways that are significantly altered by obesity in the livers of people with type 2 diabetes by analyzing comprehensive gene expression profiles using DNA chips (5). We found that genes involved in ubiquitin-proteasome pathways were coordinately upregulated in obese individuals. Proteasomes play fundamental roles in processes that are essential for cell viability (6).
Eukaryotic cells contain several types of proteasomes. Core 20S proteasomes (20S) have binding sites for the regulatory particles proteasome activator (PA)700 and PA28 (7). PA700–20S-PA700 complexes are known as 26S proteasomes and are ATP-dependent machines that degrade cell proteins (7). PA28 is found in both previously described PA28–20S-PA28 complexes and PA700–20S-PA28 complexes, which also contain PA700 (8). The PA28 family comprises three members: α, β, and γ. PA28α encoded by the PSME1 gene and PA28β encoded by the PSME2 gene form a heteropolymer, which is mainly located in the cytoplasm, whereas PA28γ encoded by the PSME3 gene forms a homopolymer that predominantly occurs in the nucleus (9). The association of the PA28 with the 20S may play a role in antigen processing by modulating peptide cleavage in the 20S (10,11), but it appears that the PA28 may play a greater role in intracellular protein degradation than in antigen processing (12). Recently, it was reported that PA28α overexpression enhances ubiquitin-proteasome system–mediated degradation of abnormal proteins (13).
It has been reported that fatty acids, insulin (14), and oxidative stress (15) inhibit proteasome activity in cultured hepatocellular carcinoma (Hep)G2 cells. However, it remains to be determined whether liver proteasome function is dysregulated in obesity and type 2 diabetes.
Based on these findings, we hypothesized that proteasome dysregulation in the liver is involved in the development of hepatic insulin resistance in obesity and type 2 diabetes. To test this hypothesis, we generated PA28α-PA28β-PA28γ triple-knockout (PA28 KO) mice as a model of impaired proteasome function and investigated their metabolic phenotypes.
RESEARCH DESIGN AND METHODS
Human studies.
This study was approved by the ethics committee of Kanazawa University. Liver biopsy specimens were obtained from 21 patients with type 2 diabetes (15 men and 6 women; mean age 53.0 ± 2.1 years, BMI 24.4 ± 0.9 kg/m2, fasting plasma glucose 7.94 ± 0.59 mmol/L, HbA1c 7.3 ± 0.3%, and alanine aminotransferase 34.4 ± 5.5 IU/L) admitted to Kanazawa University Hospital between 2000 and 2003 as previously described (5,16). Statistical analyses of DNA chip gene expression data were performed as previously described (5). To test the significance of expression ratios for individual genes or pathways, we performed a supervised analysis using a permutation-based method with BRB-ArrayTools software (17), developed for the statistical analysis of DNA chip gene expression data by the Biometric Research Branch of the U.S. National Cancer Institute.
Animal experiments.
PA28 KO mice were generated by cross-breeding PA28α-PA28β double-KO mice (18) and PA28γ KO mice (19). Genetic background of both lines of KO mice is almost completely homologous to that of C57BL/6J mice because they were backcrossed to C57BL/6J mice for at least eight generations. C57BL/6J mice and ob/ob mice were obtained from Sankyo Laboratory Service (Tokyo, Japan), and genetically diabetic model male db/db mice were obtained from Charles River (Tokyo, Japan). The study protocol was reviewed and approved by the animal care and use committee of Kanazawa University. All mice were housed in specific pathogen-free barrier facilities, maintained under a 12-h light/dark cycle, fed a standard rodent food diet (Oriental Yeast) (STD) or rodent food containing 60% fat (Research Diet) (HFD) for up to 28 weeks, and provided with water ad libitum. Sodium 4-phenylbutyrate (PBA, Enzo Life Sciences, Farmingdale, NY) was mixed in drinking water at a concentration of 4 mg/mL and administered ad libitum for 3 weeks as previously described (20).
Gene expression analyses in mice.
We used Genopal DNA chips (21) (Mitsubishi Rayon, Tokyo, Japan [http://www.mrc.co.jp/genome/e/index.html]) and RT-PCR to identify significant changes in hepatic gene transcription. Total RNA was isolated using an RNeasy Mini Kit (Qiagen, Valencia, CA) and converted into cDNA using a cDNA synthesis kit (Applied Biosystems, Tokyo, Japan) as previously described (22). Quantitative real-time PCR analysis was performed on an ABI Prism 7900HT Sequence Detection System (Invitrogen) using TaqMan gene expression assays (Applied Biosystems) or the SYBR Green Master Mix (Takara Bio, Otsu, Japan) as described previously (23). The primers for mouse acetyl-CoA carboxylase-1 (Acc1) were as follows: forward, 5′-TGGAGAGCC CCACACACA-3′; reverse, 5′-TGACAGACTGATCGCAGAGAAAG-3′. The primers for mouse X box-binding protein 1 (XBP-1) were as follows: forward, 5′-AAACAGAGTAGCAGCGCAGACTGC-3′; reverse, 5′-GGATCTCTAAAACTAGAGGCTTGGTG-3′. The primers for mouse C/EBP homologous protein (CHOP) were as follows: forward, 5′-TATCTCATCCCCAGGAAACG-3′; reverse, 5′-GGGCACTGACCACTCTGTTT-3′. TaqMan gene expression assays (Applied Biosystems) were used for PSME1 (Hs00389209_m1); PSME2 (Hs01581609_g1); PSME3 (Hs00195072_m1); Psme1 (Mm00650858_g1); Psme2 (Mm01702832_g1); Psme3 (Mm00839833_m1); insulin receptor substrate 2 (Irs2) (Mm03038438_m1); sterol regulatory element–binding factor 1 (Srebf1) (Mm00550338_m1); forkhead box O1 (Foxo1) (Mm00490672_m1); glucose-6-phosphatase, catalytic (G6pc) (Mm00839363_m1); phosphoenolpyruvate carboxykinase 1 (Pck1) (Mm01247058_m1); insulin-like growth factor binding protein 1 (Igfbp1) (Mm00515154_m1); and peroxisome proliferator–activated receptor γ coactivator 1α (Ppargc1a) (Mm01208835_m1).
Determination of proteasome activity.
Frozen livers were minced and homogenized in ice-cold buffer (25 mmol/L Tris-HCl [pH 7.5], containing 1 mmol/L dithiothreitol and 2 mmol/L ATP) using an homogenizer and then frozen and thawed three times. Extracts were centrifuged at 12,000 rpm for 10 min at 4–8°C. Protein concentrations were determined using the Lowry Protein Assay (Bio-Rad Laboratories, Richmond, CA). Proteasome activity was assayed as chymotrypsin-like activity in proteasome assay buffer containing 100 mmol/L Tris-HCl (pH 8.0), 2 mmol/L ATP, and fluorogenic peptide substrate (0.1 mmol/L) according to a procedure previously described (24). The assay for chymotrypsin-like activity is based on the detection of the fluorophore 7-amino-4-methylcoumarin (AMC) after its cleavage from the labeled substrate Suc-Leu-Leu-Val-Tyr-AMC (Peptide Institute, Osaka, Japan). Free AMC fluorescence was quantified using a 355/460 nm filter set in a luminescence spectrophotometer (Fluoroskan Ascent FL; ThermoLab Systems, Franklin, MA). Proteasome activity was calculated as the difference between the total activity in tissue extracts and the remaining activity in the presence of the proteasome inhibitor MG132 (20 μmol/L).
Immunoprecipitation and Western blotting.
Proteins were extracted from tissues and subjected to SDS-PAGE as previously described (25). Membranes were incubated overnight at 4°C with antibodies against ubiquitin (DAKO Japan, Kyoto, Japan), PA28α (Cell Signaling Technology, Danvers, MA), PA28β (Cell Signaling Technology), PA28γ (Cell Signaling Technology), Akt (Cell Signaling Technology), phosphorylated (p)–Ser473-Akt (p-Akt) (Cell Signaling), IRS-1 (Millipore, Billerica, MA), p–IRS-1 (Ser307) (Cell Signaling Technology), IRS-2 (Cell Signaling Technology), phosphatidylinositol 3-kinase p85 (PI3K p85; Millipore Corp.), SREBP-1 (Santa Cruz Biotechnology, Santa Cruz, CA), pan-cadherin (Cell Signaling), glucose-regulated protein 78 (GRP78) (Santa Cruz Biotechnology), immunoglobulin heavy-chain binding protein (BiP) (Cell Signaling), p–protein kinase R–like endoplasmic reticulum kinase (p-PERK) (Cell Signaling), inositol-requiring enzyme (IRE) 1α (Novus Biologicals, Littleton, CO), p-IRE1α (Novus), eukaryotic initiation factor 2α (eIF2α) (Cell Signaling), p-eIF2α (Cell Signaling), XBP-1 (Santa Cruz Biotechnology), c-Jun NH2-terminal kinase (JNK) (Cell Signaling), p-JNK (Cell Signaling), p–c-Jun (Santa Cruz Biotechnology), and CHOP (Cell Signaling). The membranes were incubated with secondary antibody conjugated with enhanced chemiluminescence Western Blotting Detection Reagents (GE Healthcare) and were visualized using an LAS-3000 luminescent image analyzer (FUJIFILM, Tokyo, Japan). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Santa Cruz Biotechnology) was used as a control for protein loading.
Measurement of biochemical parameters.
Plasma glucose levels, blood insulin levels, and triglyceride content in the liver were determined with a Glucocard Diameter, (ARKRAY, Kyoto City, Japan), an insulin kit (Morinaga, Yokohama, Japan), and TG-E Test Wako kits (Wako, Osaka, Japan), respectively.
Glucose and insulin tolerance tests and insulin infusion.
Glucose and insulin tolerance tests were performed through injection of glucose (1–1.5 g/kg i.p.) and insulin (2.0 IU/kg i.p.), respectively, after an overnight fast. After the withdrawal of food for 4 h, mice were anesthetized. Twenty minutes after an injection of insulin (10 IU/kg i.p.), tissues were removed, frozen in liquid nitrogen, and stored at −80°C until processing.
Hyperinsulinemic-euglycemic clamp studies in mice.
Clamp studies were performed after 2–3 days of recovering from cannulization as previously described (25). Clamp studies were performed on conscious and unrestrained animals. During clamp studies, insulin (Novolin R; Novo Nordisk, Denmark) was continuously infused at a rate of 5.0 mU/kg/min, and the blood glucose concentration (monitored every 5 min) was maintained at 100 mg/dL through the administration of glucose (50% enriched to ~20% with [6,6-2H2]glucose; Sigma) for 120 min. Blood was sampled through tail-tip bleeds at 90, 105, and 120 min for the purpose of determining the glucose Rd. Rd values were calculated according to non–steady-state equations, and hepatic glucose production was calculated as the difference between the Rd and the exogenous glucose infusion rates.
Histopathological examination.
Liver samples were fixed in 4% buffered formalin and embedded in Tissue-Tek OCT compound (Sakura Finetek USA) and paraffin for histological analysis. The formalin-fixed and paraffin-embedded section (5 mm) was processed routinely for hematoxylin-eosin staining. The OCT-embedded samples were serially sectioned at 4 mm. For the evaluation of fat deposition, the liver section was stained with Oil-red O.
Preparation for nuclear matrix and cell membrane fractions from whole cell lysates.
Nuclear matrix and cell membrane fractions were extracted from whole cell lysates by using the CNMCS compartmental protein extraction kit (BioChain Institute) according to the manufacturer's protocol.
Electron microscopy.
Small pieces of mouse liver were fixed through incubation with 2% paraformaldehyde and 2% glutaraldehyde in 0.1 mol/L phosphate buffer (pH 7.2) for 2 h at 4°C and then postfixed through incubation with 1% OsO4 for 2 h at 4°C. Specimens were stained with 1% uranium acetate for 30 min, dehydrated in a graded ethanol series, and embedded in an epoxy resin based on Glycidether (Selva Feinbiochemica, Heidelberg, Germany). Ultrathin sections were prepared using an ultramicrotome, stained with uranium acetate and lead citrate, and visualized using a JEM-1210 electron microscope (JEOL, Tokyo, Japan).
Cell culture.
Studies were performed using H4IIEC3 rat hepatoma cells that were purchased from the American Type Culture Collection (Manassas, VA). Cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) (Invitrogen) supplemented with 10% FBS (Invitrogen), penicillin (100 units/mL), and streptomycin (0.1 mg/mL; Invitrogen) at 37°C under an atmosphere of 5% CO2 in air in a humidified incubator.
Chemicals.
Bortezomib was purchased from LC laboratories (Woburn, MA). PBA for in vitro experiments was purchased from Calbiochem (San Diego, CA).
Statistical analysis.
All data are expressed as means ± SE and were analyzed by the Mann-Whitney U test with the level of statistical significance set at P < 0.05. All calculations were performed using SPSS, version 11.0, software for Windows (SPSS, Chicago, IL).
RESULTS
Expression of genes involved in proteasome pathways is altered in the livers of patients with type 2 diabetes and obesity.
We previously identified metabolic pathways that are altered in association with obesity in the livers of patients with type 2 diabetes (5,16). Clinical characteristics of study obese and nonobese subjects are shown in Supplementary Table 1. The hepatic expression of genes involved in a proteasome pathway, the gene encoding PA28α (P < 0.05), was coordinately upregulated in diabetic patients who were obese compared with those who were not obese (Fig. 1A). To further strengthen the significance of the hepatic expression of the genes for PA28s, we analyzed an independent new sample set of the human liver using RT-PCR. Clinical information of the subjects is described in Supplementary Table 2. As shown in Fig. 1B, mRNA levels of genes encoding PA28α, -β, and -γ in the human liver correlated positively with an insulin resistance index (homeostasis model assessment of insulin resistance). Therefore, hepatic expression of PA28s was associated with insulin resistance in humans. We next analyzed hepatic gene expression in a mouse model of diet-induced obesity (HFD mice) using a custom-made array, Metabolic Chip (21). Consistent with human data, the hepatic expression of genes involved in the proteasome pathway was coordinately upregulated in C57BL/6 mice fed the HFD compared with those fed the STD. Of these, the gene encoding PA28β (Psme2) was significantly upregulated in mice fed the HFD compared with those fed the STD (Supplementary Table 3). These findings prompted us to test the hypothesis that proteasome degradation pathways are involved in the development of obesity-induced insulin resistance in the liver.
FIG. 1.

Proteasome dysfunction in mouse models of obesity. A: Gene expression and proteasome activity in the livers of patients with type 2 diabetes and obesity. Coordinate upregulation of genes involved in proteasome degradation pathways in the livers of type 2 diabetic patients with obesity compared with those without obesity. GenMAPP (Gene MicroArray Pathway Profiler [http://www.genmapp.org]) was used to annotate the pathway with expression ratios for the genes involved. The fold changes presented beside the gene names are for obese versus nonobese patients. Genes significantly upregulated in obesity (P < 0.05) are shown in red; genes analyzed whose expression was not significantly altered in obesity are shown in gray. B: Graphs showing associations between homeostasis model assessment of insulin resistance (HOMA-IR) and the mRNA expression of subunits of PA28: PA28α (PSME1), PA28β (PSME2), and PA28γ (PSME3). Data were normalized according to 18S mRNA level. C: Comparison of liver proteasome activity in 32-week-old C57BL6 mice fed STD and HFD for 28 weeks (from 4 to 32 weeks of age), in WT and db/db mice (20 weeks old), and in WT mice and ob/ob mice (20 weeks old). Proteasome activity was estimated by measuring chymotrypsin-like activity in liver tissues as described in research design and methods. Data represent means ± SE (n = 4 per group). *P < 0.05, **P < 0.01. D: Accumulation of polyubiquitinated proteins was assessed by Western blot in the livers isolated from C57BL6 mice fed the STD and the HFD, db/db mice, and ob/ob mice. GAPDH served as internal control.
Liver proteasome activity is reduced in animal models of type 2 diabetes and obesity.
Next, we examined liver proteasome activity in animal models of type 2 diabetes and obesity. Proteasome activity was measured using a substrate for chymotrypsin-like activity. Unexpectedly, liver proteasome activity was reduced by ~30–40% in genetically obese ob/ob mice, diabetic db/db mice, and C57BL/6 mice fed the HFD (Fig. 1C). As a consequence, accumulation of ubiquitinated proteins was increased in the liver of C57BL/6 mice fed the HFD, db/db mice, and ob/ob mice (Fig. 1D). These results suggest that liver proteasome activity is reduced in animal models of type 2 diabetes and obesity. Coordinate upregulation of genes involved in the ubiquitin-proteasome pathway in obese patients and mouse models of type 2 diabetes and obesity may compensate for impaired proteasome function.
PA28 KO mice show impaired proteasome function and accumulation of polyubiquitinated proteins in the liver.
To test the hypothesis that proteasome dysregulation in the liver is involved in the development of hepatic insulin resistance in obesity and type 2 diabetes, we generated a mouse model of impaired proteasome function: PA28 KO mice. We first performed a genomic DNA PCR analysis to confirm that the mutant mice expressed the messages of neither the PA28α nor the PA28γ gene (Fig. 2A). Knockout of the Psme1, Psme2, and Psme3 in the liver of PA28 KO mice was confirmed both by RT-PCR and by Western blotting analyses (Fig. 2B and C). Up to 30 weeks after birth, PA28 KO mice were indistinguishable in appearance and growth from age-matched wild-type (WT) mice (Fig. 2D).
FIG. 2.

PA28 KO mice show impaired proteasome function and accumulation of polyubiquitinated proteins in the liver. A: Mice were genotyped by PCR analysis as previously described (18,19). Genomic DNA extracted from mouse tails was analyzed by PCR. Primers used for genotyping were 5'-TTTCCTGTACGTGACTTCCATCCTGTTG-3' (primer 1), 5'-GGTCCACATACAATAAAGACATGGGCTG-3' (primer 2), and 5'-GATTGTGGTCCTCCTGCAACGCCTAAA-3' (primer 3). Primers 1 and 3 amplified 1.2-kb fragments from the wild-type PA28α allele. Primers 1 and 2 amplified 2.2-kb fragments from the mutant allele. Primers used for genotyping were 5'-CCGGGACAATAAGACACATCACTC-3' (primer 1), 5'-TTGTCCCTCCCTCCAGTTGTCTAA-3' (primer 2), and 5'-GATCCCCTCAGAAGAACTCGTCAA-3' (primer 3). Primers 2 and 3 amplified 0.9-kb fragments from the wild-type PA28γ allele. Primers 1 and 3 amplified 1.9-kb fragments from the mutant allele. B: RT-PCR analysis of total RNA prepared from liver of PA28 KO mice (n = 7). N.D., not determined. Transcripts of mouse PA28α, -β, and -γ were examined by quantitative RT-PCR. Data were normalized according to GAPDH mRNA level and presented as a value relative to that for WT mice. C: Western blot analysis of extracts prepared from the liver of PA28 KO mice. The blot was probed with the anti-PA28α, anti-PA28β, and anti-PA28γ antibodies. D: Body weights of WT and PA28 KO mice fed an STD or the HFD. ○, WT mice fed the STD (n = 5); ●, WT mice fed the HFD (n = 4); □, PA28 KO mice fed the STD (n = 4); ■, PA28 KO mice fed the HFD (n = 4). Data represent means ± SE. E: Comparison of liver proteasome activity in WT (n = 13, STD; n = 4, HFD) and PA28 KO (n = 11, STD; n = 4, HFD) mice. Data represent means ± SE. *P < 0.05. F: Proteasome activity was evaluated in the muscles isolated from 32-week-old WT mice (n = 5) and PA28 KO mice (n = 4). The activity was normalized to the total protein content. Data represent means ± SE. *P < 0.05. G: Western blot analyses of total ubiquitinated proteins in the livers of WT and KO mice. Livers were isolated from 32-week-old WT and PA28 KO mice fed the STD or the HFD for 28 weeks (from 4 to 32 weeks of age). Quantitation of ubiquitinated proteins levels is normalized to GAPDH and is represented as means ± SE (n = 3 per group). *P < 0.05.
When fed the STD, PA28 KO mice had 35% reduced hepatic proteasome activity compared with WT mice (Fig. 2E). Proteasome activity was also reduced by ~50% in the skeletal muscle of PA28 KO mice compared with that of WT mice (Fig. 2F). These results are consistent with previous observations in mouse embryonic fibroblasts from PA28α−/−PA28β−/− mice (18). The degree of proteasome inactivation in the livers of PA28 KO mice corresponded with that in the livers of db/db mice and C57BL mice fed the HFD (Fig. 1C and Fig. 2E). Thus, the PA28 KO mouse appears to be an appropriate animal model that mimics the proteasome dysfunction observed in obesity and type 2 diabetes. When fed the HFD, PA28 KO mice tended to have further reduced hepatic proteasome activity compared with WT mice (P = 0.086) (Fig. 2E).
For assessment of the impact of proteasome dysfunction in the liver, polyubiquitinated proteins were detected by Western blotting (Fig. 2G). The HFD increased the accumulation of ubiquitinated proteins in the liver in both WT and PA28 KO mice. As expected, more polyubiquitinated proteins accumulated in the livers of PA28 KO mice compared with those of WT mice for both STD-fed and HFD-fed mice. Accumulation of the ubiquitinated proteins in the liver of PA28 KO mice fed HFD was significantly increased compared with those fed STD, suggesting that HFD further impairs proteasome function in PA28 KO mice.
PA28 KO mice show glucose intolerance and attenuated insulin signaling in the liver.
To examine the metabolic effects of proteasome dysfunction in vivo, we performed glucose and insulin loading tests. Fasting glucose levels were similar in PA28 KO mice and WT mice (Fig. 3A). However, PA28 KO mice showed glucose intolerance after glucose loading (Fig. 3B), which was further exacerbated by the HFD (Fig. 3B). While serum insulin levels were significantly higher in PA28 KO mice compared with WT mice (Fig. 3A), there was no significant difference in insulin tolerance test results (Fig. 3C), suggesting the existence of hepatic insulin resistance in the PA28 KO mice.
FIG. 3.

PA28 KO mice show proteasome dysfunction, glucose intolerance, and attenuated insulin signaling in the liver. A: Blood glucose concentrations and serum insulin levels measured in an overnight fasting state at 20 weeks of age. WT (n = 9, STD; n = 5, HFD) and PA28 KO (n = 6, STD; n = 4, HFD) mice. Data represent means ± SE. *P < 0.05. B: Intraperitoneal glucose tolerance tests (IPGTT). C: Intraperitoneal insulin tolerance tests (IPITT). Blood glucose level measured during the intraperitoneal glucose tolerance tests and intraperitoneal insulin tolerance tests at 20 weeks of age. WT mice fed the STD, n = 5; WT mice fed the HFD, n = 4; PA28 KO mice fed the STD, n = 4; PA28 KO mice fed the HFD, n = 4. Data represent means ± SE. *P < 0.05. **P < 0.01. D: Insulin sensitivity was assayed by using a hyperinsulinemic-euglycemic clamp study in WT (n = 8) and PA28 KO (n = 10) mice. Left panel: Glucose infusion ratio. Middle panel: HGP before and after insulin clamp. Right panel: Glucose disposal rate. Data represent means ± SE. *P < 0.05. E: Equal amounts of protein in total lysates of liver and muscle were immunoblotted (IB) with anti–p-Akt (Ser473) and anti-Akt antibodies. p-Akt values of insulin-injected fasted mice were displayed relative to those of saline-injected mice (WT mice fed the STD, n = 7; PA28 KO mice fed the STD, n = 7). Data represent means ± SE. *P < 0.05. F: Expression of mRNAs encoding Irs2 in the livers of 12-week-old WT and PA28 KO mice analyzed by quantitative real-time RT-PCR. Expression values were normalized to 18S mRNA. Data represent means ± SE (n = 7 per group). *P < 0.05. Liver lysates from mice were immunoblotted with anti–IRS-2 antibody. G: Liver lysates from mice were immunoprecipitated (IP) using anti–IRS-1 antibody bound to protein A agarose. The immunoprecipitates were immunoblotted with anti–p–IRS-1 (Ser307) and p85. Representative results from five mice of each genotype are shown. Right panel: densitometry quantitation of IRS-1 to p85 signal ratio is shown. Data represent means ± SE. *P < 0.05.
Because the PA28 KO mouse appears to be a model that mimics the proteasome dysfunction observed in HFD-fed mice, subsequent experiments were performed only in mice fed the STD. To identify the responsible organ that contributes to insulin resistance in PA28 KO mice, we next performed hyperinsulinemic-euglycemic clamp experiments and Western blot analysis of the insulin-signaling pathway. As shown in Fig. 3D, endogenous glucose production was significantly increased in PA28 KO mice, whereas peripheral glucose disposal tended to be increased. In the liver of PA28 KO mice, insulin-induced phosphorylation of Akt at Ser473 was impaired (Fig. 3E) in association with marked reduction in IRS-2 protein levels (Fig. 3F) and enhanced serine phosphorylation of IRS-1 compared with WT mice (Fig. 3G). Insulin-induced increment in IRS-1–associated p85 subunit of PI3K was slightly impaired in the liver of PA28 KO mice compared with that of WT mice (Fig. 3G). On the other hand, in the skeletal muscle of the PA28 KO mice, insulin-induced phosphorylation of Akt at Ser473 was rather enhanced compared with that of WT mice (Fig. 3E). These findings indicate that PA28 deficiency impairs insulin signaling mainly in the liver and thereby induces systemic glucose intolerance in vivo.
PA28 KO mice show hepatic steatosis.
In insulin-resistant states, the hyperinsulinemia drives hepatic lipogenesis via a SREBP-1c pathway (26). Oil-red O staining of liver tissue sections revealed a slight hepatic steatosis in the PA28 KO mice fed STD (Fig. 4A). Triglyceride content was significantly increased in the liver of PA28 KO mice (Fig. 4B). To understand the molecular basis underlying the enhanced hepatic steatosis in PA28-null livers, we analyzed gene expression of SREBP-1c that regulates lipid biosynthesis. As shown in Fig. 4C, expression of Srebf1 was modestly increased in the PA28-deleted livers compared with control livers. Furthermore, the levels of cleaved/active SREBP1c were much more increased in the liver of the PA28 KO mice compared with that of WT mice (Fig. 4D). Indeed, mRNA expression of Acc1, a target of SREBP-1c, was upregulated in the liver of the PA28 KO mice compared with that of WT mice (Fig. 4C). Nuclear SREBP-1c, but not membrane-bound precursor of SREBP-1c, was significantly increased in the livers of PA28 KO mice compared with those of WT mice (Fig. 4D).
FIG. 4.

PA28 KO mice show hepatic SREBP-1c activation and steatosis. A: Oil-red O staining of lipid droplets in the livers of 12-week-old WT and PA28 KO mice. The highlighted region of the upper panel is shown at a higher magnification in the lower panel. Scale bars, 100 μm. B: Triglyceride contents in the livers of WT and PA28 KO mice. Data are expressed as milligrams per gram of liver tissue. Data represent means ± SE (n = 4–5 per group). *P < 0.05. C: Expression of mRNAs encoding Srebf1 and Acc1 in the livers of 12-week-old WT and PA28 KO mice analyzed by quantitative real-time RT-PCR. Expression values were normalized to GAPDH mRNA. Data represent means ± SE (n = 7–9 per group). D: Whole cell, membrane, and nuclear fractions in liver extracts were subjected to SDS-PAGE and blotted using an anti–SREBP-1 antibody. GAPDH, pan-cadherin, and lamin A/C served as internal controls. Quantitation of SREBP-1 68-kDa protein levels is normalized to GAPDH and is represented as means ± SE. *P < 0.05.
PA28 deficiency–induced proteasome dysfunction activates an unfolded protein response and increases ER stress in the liver.
To investigate the mechanisms underlying proteasome dysfunction–induced insulin resistance in the liver, we evaluated morphological changes in the liver through histological examination. Hematoxylin-eosin staining of liver tissues showed that hepatocytes were indistinguishable in the two groups (Fig. 5A). Next, we examined morphological changes in organelles within hepatocytes by electron microscopy. Electron micrographs revealed massive expansion of the ER in the livers of PA28 KO mice (Fig. 5B), suggestive of an unfolded protein response (UPR) (27). ER stress is caused by the accumulation of unfolded and misfolded proteins in the ER lumen and is associated with several human diseases (28,29). In addition, proteasome inhibitors have been reported to induce ER stress in cultured primary rat hepatocytes (30). Therefore, to examine whether PA28 deficiency–induced proteasome dysfunction causes ER stress in the liver, we analyzed the expression patterns of several molecular indicators of ER stress in liver extracts from 12-week-old WT and PA28 KO mice fed the STD (Fig. 5C and D). Proteasome dysfunction induced by PA28 gene deletion resulted in ER stress in the liver tissues of lean mice, as evidenced by increased levels of Grp78, CHOP, p-PERK, p-eIF2α, and p-IRE1α, as well as ER stress–inducible mRNAs encoding CHOP and the spliced form of XBP-1 (XBP-1s), compared with WT mice (Fig. 5C and D). XBP-1s protein amounts in nuclear fractions increased significantly in the liver tissue of PA28 KO mice compared with that of WT mice (Fig. 5C). Hyperactivation of JNK through phosphorylation is another marker of ER stress (31) and plays a role in linking ER stress and insulin resistance (32). Phosphorylation of JNK and its downstream target c-Jun was significantly increased in the livers of PA28 KO mice compared with those of WT mice (Fig. 5). Based on these findings, it might be possible that general accumulation of polyubiquitinated proteins or accumulation of some specific substrate for PA28s promotes a UPR and ER stress in the livers of PA28 KO mice.
FIG. 5.

Deletion of PA28 genes causes ER stress in the liver. A: Hematoxylin-eosin–stained liver sections from PA28 KO and WT mice. Scale bar, 200 μm. B: Electron microscopic analyses of the ER in livers from WT and PA28 KO mice. The highlighted region of the upper panel is shown at a higher magnification in the lower panel. Scale bars: 10 μm (upper panel) and 1 μm (lower panel). C: Western blotting for the ER stress–associated markers GRP78, CHOP, p-PERK, p-eIF2α, and p-IRE1α in the livers of 12-week-old male WT and PA28 KO mice. Upper right panel: Each expression level was quantified (n = 3 per group). Data represent means ± SE. *P < 0.05. Lower right panel: Western blotting for nuclear spliced form of XBP-1s protein amounts in the livers of 12-week-old male WT (n = 5) and PA28 KO (n = 5) mice. Lamin A/C served as internal control. Data represent means ± SE. *P < 0.05. D: Expression of mRNAs encoding CHOP and XBP-1s in the livers of WT (□) and PA28 KO (■) mice. Expression values were normalized to Actb mRNA. Data represent means ± SE (n = 4–7 per group). *P < 0.05. E: Western blotting for p-JNK, p–c-Jun, and total JNK in the livers of WT and PA28 KO mice.
Proteasome dysfunction–induced ER stress and insulin resistance are partly blocked by an orally active chemical chaperone, PBA, in the liver of PA28 KO mice.
We next investigated the effects of PBA administration on proteasome dysfunction–induced ER stress and insulin resistance in PA28 KO mice. Intraperitoneal glucose tolerance test was performed to further evaluate the effect of PBA on whole-body glucose metabolism. After glucose loading, blood glucose levels tended to decrease in PBA-administered PA28 KO mice compared with untreated PA28 KO mice (Supplementary Fig. 1). The plasma insulin levels after glucose loading significantly increased in PBA-administered PA28 KO mice compared with untreated PA28 KO mice (Supplementary Fig. 1). Administration of PBA ameliorated massive expansion of the ER in the livers of PA28 KO mice (Fig. 6A) and decreased hepatic CHOP expression and IRE1a phosphorylation, suggesting that PBA administration alleviates hepatic ER stress in PA28 KO mice (Fig. 6B). As a consequence, PBA administration improved impaired insulin-induced phosphorylation of Akt at Ser473 in liver tissue of PA28 KO mice (Fig. 6C). These findings confirm the critical role of ER stress in the proteasome dysfunction–mediated insulin resistance.
FIG. 6.
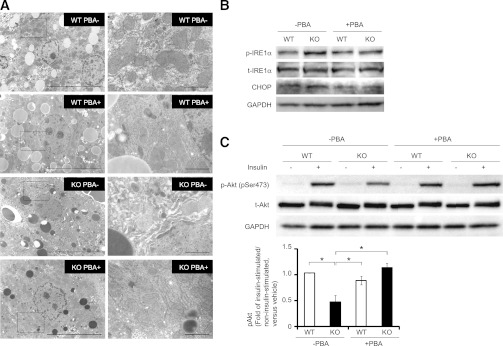
Effects of a chemical chaperone, PBA, administration on proteasome dysfunction–induced ER stress and insulin resistance in PA28 KO mice. A–C: WT and PA28 KO mice were administered mixed PBA (4 mg/mL) through drinking water for 3 weeks. A: Electron microscopic analyses of the ER in livers from WT and PA28 KO mice administered orally with or without PBA. The highlighted region of the left panel (magnification ×5,000) is shown at a higher magnification in the right panel (magnification ×20,000). Scale bars: 10 μm (left panel) and 1 μm (right panel). B: IRE1α phosphorylation and CHOP levels in the livers of WT and PA28 KO mice. C: WT and PA28 KO mice fed STD were starved overnight and injected with insulin (10 IU/kg i.p.). Equal amounts of protein in total lysates of liver and muscle were immunoblotted with anti–p-Akt (Ser473) and anti-Akt antibodies. p-Akt values of insulin-injected fasted mice values were displayed relative to those of saline-injected mice. Data represent means ± SE (n = 3 per group). *P < 0.05. t-, total.
PA28 deficiency–induced proteasome dysfunction increases FoxO1 protein amounts and gluconeogenic genes in the liver.
It has been shown that the FoxO1 protein is targeted for proteasomal degradation (33–35). FoxO1 protein amounts dramatically increased in total cell lysates as well as in nuclear fractions of PA28 KO mice compared with WT mice (Fig. 7A), whereas Foxo1 mRNA levels were unaltered (Fig. 7B). In addition, FoxO1 phosphorylation was decreased in the liver tissue of PA28 KO mice compared with that of WT mice (Fig. 7A). Gluconeogenic genes, such as Pck1 and Igfbp1, which are targets of FoxO1 were upregulated in the livers of PA28 KO mice compared with those of WT mice (Fig. 7C). These findings suggest that impaired degradation of FoxO1 is one of the candidate pathways leading to impaired suppression of gluconeogenic genes and increased hepatic glucose production in PA28 KO mice.
FIG. 7.
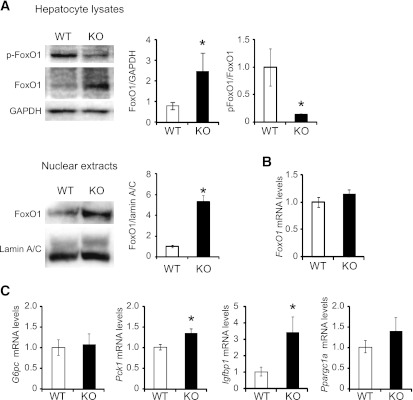
Proteasome dysfunction upregulates FoxO1 protein amounts and gluconeogenic gene expression in the liver of PA28 KO mice. A: Total liver extracts and nuclear fractions from the livers of WT and PA28 KO mice were analyzed by Western blotting for phosphorylated and total FoxO1. GAPDH served as internal control. Data represent means ± SE (n = 4–5 per group). *P < 0.05. Nuclear fractions from in the livers of WT and PA28 KO mice were analyzed by Western blotting for total FoxO1. Lamin A/C GAPDH served as internal control. Data represent means ± SE (n = 5 per group). *P < 0.05. B and C: Relative mRNA levels of FoxO1, G6pc, Pck1, Igfbp1, and Ppargc1a in the liver of WT and PA28 KO mice were analyzed by RT-PCR. Data were normalized according to GAPDH levels. Data represent means ± SE (n = 7 per group). *P < 0.05.
Proteasome inhibition causes ER stress and insulin resistance in vitro—responses that are partly blocked by a chemical chaperone.
To confirm that proteasome dysfunction causes ER stress and insulin resistance at the cellular level, we pretreated rat hepatoma-derived H4IIEC3 cells with bortezomib, a selective inhibitor of the 26S proteasome. Bortezomib concentration-dependently increased levels of UPR molecular markers, such as BiP, CHOP, and phosphorylated forms of IRE1α and JNK (Fig. 8A). Bortezomib also reduced insulin-stimulated serine phosphorylation of Akt at Ser473 in H4IIEC3 hepatocytes in a concentration-dependent manner (Fig. 8B). To address whether ER stress is responsible for proteasome dysfunction–induced insulin resistance, we tested the effect of the chemical chaperone PBA on H4IIEC3 cells treated with bortezomib. PBA partly prevented the accumulation of BiP and CHOP, the phosphorylation of JNK (Fig. 8C), and the impaired phosphorylation of Akt at Ser473 (Fig. 8D) induced by bortezomib. These findings indicate that proteasome dysfunction at least partly accelerates insulin resistance via ER stress in hepatocytes.
FIG. 8.

The proteasome inhibitor bortezomib (BZ) induces ER stress and insulin resistance in H4IIEC3 cells. A and B: H4IIEC3 cells were treated with the indicated concentrations of bortezomib (in DMEM supplemented with 10% FBS) for 24 h. After washing, cells were serum starved for 16 h and then treated with insulin (1 nmol/L) or phosphate-buffered saline for 15 min. Cells were solubilized, and equal amounts of proteins were analyzed by Western blotting using BiP-, p-IRE1α–, CHOP-, p-JNK–, total JNK (t-JNK)-, p-Akt–, and total Akt (t-Akt)-specific antibodies. C and D: H4IIEC3 cells were pretreated or not for 24 h with 2 mmol/L PBA and then treated with the indicated concentrations of bortezomib (in DMEM supplemented with 10% FBS) for 24 h. Cells were washed, serum starved for 16 h, and treated with insulin (1 nmol/L) or phosphate-buffered saline for 15 min. Cells were solubilized, and equal amounts of proteins were analyzed by Western blotting using BiP-, CHOP-, p-JNK–, total JNK–, p-Akt–, and total Akt–specific antibodies. Blots of p-Akt were quantitated densitometrically and expressed as ratios to total Akt (n = 4 for each condition). Relative density is mean ± SE fold increase over control. *P = 0.05 vs. treatment with insulin alone.
DISCUSSION
Our study shows that liver proteasome activity is reduced by ~30–40% in genetic and dietary models of obesity and diabetes with coordinate upregulation of genes involved in the ubiquitin-proteasome pathway. In concert with our findings, recent systematic analyses of liver tissue in obese mice also revealed increased proteasome components (36,37), which may compensate for impaired proteasome function. To test a hypothesis that proteasome dysfunction may be a primary event that links obesity and insulin resistance in the liver, we established a mouse model of impaired proteasome function by deleting PA28 genes. The livers of PA28 KO mice showed impaired proteasome function similar to that observed in mouse models of obesity and type 2 diabetes, accumulation of ubiquitinated proteins and damaged organelles, ER stress, JNK activation, and insulin resistance (Supplementary Fig. 2). A chemical chaperone PBA administration almost completely alleviated proteasome dysfunction–mediated insulin resistance, confirming the critical role of ER stress in the development of insulin resistance under proteasome dysfunction.
Accumulating evidence suggests that obesity promotes ER stress, which is detected as enhanced UPR signaling, that activates JNK and impairs insulin signaling at the level of IRSs in adipose tissue and the liver (2,4). However, the link between obesity and ER stress has remained unclear. We propose that obesity-associated proteasome dysfunction induces ER stress in the liver, as PA28 KO mice showed accumulation of polyubiquitinated proteins and massive expansion of the ER in the liver, probably due to a reduced capacity for proteasome-mediated degradation of ubiquitinated proteins. This is the first in vivo evidence of proteasome dysfunction–induced insulin resistance mediated by ER stress in the liver. Furthermore, we showed that the selective proteasome inhibitor bortezomib increases ER stress and thereby activates JNK in a cultured hepatocyte cell line.
Yang et al. (38) recently reported that hepatic autophagy is downregulated in the livers of ob/ob mice and that defective autophagy in Atg7 KO mice causes ER stress and hepatic insulin resistance. Therefore, it is possible that both proteasome-mediated protein degradation and autophagy-mediated protein degradation are impaired in the livers of obese individuals, further exacerbating ER stress.
Proteasome function seems to be altered differently in different tissues. Streptozotocin-induced hyperglycemia impairs proteasome activity in the liver and kidney (39,40), whereas proteasome activity is enhanced in the wasted muscle of obese diabetic db/db mice (41). Taken together, these results indicate that obesity predominantly induces proteasome dysfunction in the liver. This clarifies the previous finding that ER stress causes insulin resistance in the liver together with the adipose tissue (2) and brain (42,43). Mechanisms underlying enhanced insulin sensitivity in the skeletal muscle of PA28 KO mice should be investigated in the future.
Why obesity impairs proteasome function in the liver remains unclear. One possible link may be overnutrition-induced oxidative stress. Visceral adiposity in obesity causes excessive flux of free fatty acids into the liver via the portal vein, resulting in oxidative stress in the liver (44). The saturated fatty acid palmitate induces excessive production of reactive oxygen species in mitochondria, activates JNK, and causes insulin resistance at the level of IRSs in hepatocytes (44,45). In addition, genes involved in oxidative phosphorylation are upregulated in parallel with insulin resistance in patients with type 2 diabetes who are obese compared with those who are not obese (5,16). Severe oxidative stress causes the covalent modification of 20S proteasome subunits, thereby reducing proteasome activity in the liver and in cultured hepatocytes (15). On the other hand, PA28α overexpression protects against oxidative stress in cultured cardiomyocytes, likely through enhancing the removal of oxidized proteins (46). PA28α and PA28β proteins interact with each other. The degradation rate of PA28β was also significantly decreased by PA28α overexpression in cultured cardiomyocytes (46). These findings suggest that obesity-associated mitochondrial reactive oxygen species and oxidative stress may impair proteasome function. Interestingly, expression of genes involved in the proteasome pathway, including PA28 genes, is increased in the liver tissues of obese mice compared with controls. A recent report compellingly demonstrated that upregulation of the immunoproteasome by interferons not only plays a previously recognized role in helping antigen presentation but also facilitates the removal of damaged proteins generated by interferon-induced oxidative stress (47). We speculate that these are compensatory mechanisms because the hybrid proteasome is better equipped to degrade misfolded proteins than is the conventional 26S proteasome. It was previously shown that the association of the 11S, also known as PA28 or REG, increases the peptidase activities of the 20S (48). PA28 KO mice may therefore be a suitable model for the development of therapies for proteasome dysfunction–associated diseases and metabolic abnormalities.
In the current study, PA28 KO mice showed hepatic steatosis associated with upregulated Srebf1 and Acc1 and increased cleaved/active SREBP-1c (Supplementary Fig. 2). There may be a cross-talk between ER stress pathways and hepatic lipogenesis. Lee et al. (49) reported that the IRE1/XBP1 pathway induces expression of critical genes involved in fatty acid synthesis, such as Acc1. In addition, the PERK/eIF2α pathway decreases Insig1 protein translation, which increases cleaved/active SREBP-1c (50). In concert with these reports, we observed enhanced phosphorylation of IRE1α, PERK, and eIF2α and increased protein level of XBP-1s in the liver of PA28 KO mice. Indeed, our results are compatible with a model in which the ubiquitin-proteasome system degrades the amount of the endogenous nuclear SREBPs but not the precursors (51).
XBP-1s directly binds FoxO1 and promotes its protein degradation via the proteasome (52). In the current study, XBP-1s protein was increased in the liver of PA28 KO mice, probably owing to increased phosphorylation of IRE1α, an endonuclease for XBP1 gene. Even in such condition, FoxO1 protein amounts dramatically increased in total cell lysates as well as in cytoplasmic and nuclear fractions, probably owing to proteasome dysfunction in the liver of PA28 KO mice. In addition, hepatic insulin resistance caused by ER stress/JNK pathway and increased SREBP-1c that downregulates IRS-2 further accumulates FoxO1 in the nucleus, leading to induction of genes involved in gluconeogenesis such as Pepck1 (Supplementary Fig. 2).
A limitation of the current study is the fact that we cannot rule out the possibility of altered insulin secretion in the PA28 KO mice. In vitro studies suggest that the ubiquitin-proteasome system plays a role in insulin secretion by maintaining the normal function of voltage-dependent calcium channels (53). Recently, the ER-associated degradation (ERAD)/ubiquitin/proteasome system was reported to be compromised in β cells of obese patients with type 2 diabetes (54), which is compatible with our observation in the liver. Therefore, it might be possible that PBA ameliorated proteasome dysfunction–induced inhibition of glucose-stimulated insulin secretion in the current study (Supplementary Fig. 1). Indeed, PBA was reported to ameliorate β-cell dysfunction in nondiabetic obese humans infused with fatty acids (55). On the other hand, proteasome activity is rather enhanced in the skeletal muscle of obese diabetic db/db mice (41). Therefore, future research should involve the tissue-specific regulation of proteasome function in obesity and diabetes.
In conclusion, proteasome function is impaired in obesity, which contributes to the development of hepatic insulin resistance and steatosis via activating JNK and SREBP-1c by ER stress. Proteasome dysfunction also increases total and nuclear FoxO1 that enhances hepatic gluconeogenesis (Supplementary Fig. 2). Therefore, proteasome dysfunction may be a primary event linking obesity and ER stress–induced insulin resistance in the liver.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by Grants-in-Aid from the Ministry of Education, Culture, Sports, Science, and Technology, Tokyo, Japan (to T.Taka. and S.K.). S.K. has received research grants from Takeda Pharmaceutical Company. T.Taka. and S.K. have a collaborative association with Mitsubishi Rayon developing Genopal DNA chips. No other potential conflicts of interest relevant to this article were reported.
T.Oto. researched the data, contributed to discussion, and wrote the manuscript. T.Taka. conceived and designed the experiments, researched the data, contributed to discussion, wrote the manuscript, and reviewed and edited the manuscript. H.M. and T. Ota contributed to discussion. S.M. contributed to discussion and reviewed and edited manuscript. H.H., H.T., A.K., T.K., K.R.S., F.L., T.Take., S.K., K.Is., Y.K., K.Iw., K.-i.K., M.U., Y.T., M.Y., and K.To. researched data. S.I. contributed to discussion. K.Ta. and S.K. contributed to discussion and reviewed and edited manuscript. T.Taka. is the guarantor of this work, had full access to all the data in the study, and takes responsibility for the integrity of the data and the accuracy of the data analysis.
The authors thank Maki Wakabayashi, Yoko Hashimoto, and Shima Kitakata of the Department of Disease Control and Homeostasis, Kanazawa University Graduate School of Medical Science, for technical assistance.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db11-1652/-/DC1.
See accompanying commentary, p. 691.
REFERENCES
- 1.de Luca C, Olefsky JM. Stressed out about obesity and insulin resistance. Nat Med 2006;12:41–42 [DOI] [PubMed] [Google Scholar]
- 2.Ozcan U, Cao Q, Yilmaz E, et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 2004;306:457–461 [DOI] [PubMed] [Google Scholar]
- 3.Ozcan U, Yilmaz E, Ozcan L, et al. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 2006;313:1137–1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nakatani Y, Kaneto H, Kawamori D, et al. Involvement of endoplasmic reticulum stress in insulin resistance and diabetes. J Biol Chem 2005;280:847–851 [DOI] [PubMed] [Google Scholar]
- 5.Takamura T, Misu H, Matsuzawa-Nagata N, et al. Obesity upregulates genes involved in oxidative phosphorylation in livers of diabetic patients. Obesity (Silver Spring) 2008;16:2601–2609 [DOI] [PubMed] [Google Scholar]
- 6.Voges D, Zwickl P, Baumeister W. The 26S proteasome: a molecular machine designed for controlled proteolysis. Annu Rev Biochem 1999;68:1015–1068 [DOI] [PubMed] [Google Scholar]
- 7.DeMartino GN, Slaughter CA. The proteasome, a novel protease regulated by multiple mechanisms. J Biol Chem 1999;274:22123–22126 [DOI] [PubMed] [Google Scholar]
- 8.Tanahashi N, Murakami Y, Minami Y, Shimbara N, Hendil KB, Tanaka K. Hybrid proteasomes. Induction by interferon-gamma and contribution to ATP-dependent proteolysis. J Biol Chem 2000;275:14336–14345 [DOI] [PubMed] [Google Scholar]
- 9.Soza A, Knuehl C, Groettrup M, Henklein P, Tanaka K, Kloetzel PM. Expression and subcellular localization of mouse 20S proteasome activator complex PA28. FEBS Lett 1997;413:27–34 [DOI] [PubMed] [Google Scholar]
- 10.Kloetzel PM. Generation of major histocompatibility complex class I antigens: functional interplay between proteasomes and TPPII. Nat Immunol 2004;5:661–669 [DOI] [PubMed] [Google Scholar]
- 11.Preckel T, Fung-Leung WP, Cai Z, et al. Impaired immunoproteasome assembly and immune responses in PA28-/- mice. Science 1999;286:2162–2165 [DOI] [PubMed] [Google Scholar]
- 12.Chen X, Barton LF, Chi Y, Clurman BE, Roberts JM. Ubiquitin-independent degradation of cell-cycle inhibitors by the REGgamma proteasome. Mol Cell 2007;26:843–852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li J, Horak KM, Su H, Sanbe A, Robbins J, Wang X. Enhancement of proteasomal function protects against cardiac proteinopathy and ischemia/reperfusion injury in mice. J Clin Invest 2011;121:3689–3700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hamel FG. Preliminary report: inhibition of cellular proteasome activity by free fatty acids. Metabolism 2009;58:1047–1049 [DOI] [PubMed] [Google Scholar]
- 15.Osna NA, Haorah J, Krutik VM, Donohue TM., Jr Peroxynitrite alters the catalytic activity of rodent liver proteasome in vitro and in vivo. Hepatology 2004;40:574–582 [DOI] [PubMed] [Google Scholar]
- 16.Misu H, Takamura T, Matsuzawa N, et al. Genes involved in oxidative phosphorylation are coordinately upregulated with fasting hyperglycaemia in livers of patients with type 2 diabetes. Diabetologia 2007;50:268–277 [DOI] [PubMed] [Google Scholar]
- 17.Ye QH, Qin LX, Forgues M, et al. Predicting hepatitis B virus-positive metastatic hepatocellular carcinomas using gene expression profiling and supervised machine learning. Nat Med 2003;9:416–423 [DOI] [PubMed] [Google Scholar]
- 18.Murata S, Udono H, Tanahashi N, et al. Immunoproteasome assembly and antigen presentation in mice lacking both PA28alpha and PA28beta. EMBO J 2001;20:5898–5907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Murata S, Kawahara H, Tohma S, et al. Growth retardation in mice lacking the proteasome activator PA28gamma. J Biol Chem 1999;274:38211–38215 [DOI] [PubMed] [Google Scholar]
- 20.Kimura K, Yamada T, Matsumoto M, et al. Endoplasmic reticulum stress inhibits STAT3-dependent suppression of hepatic gluconeogenesis via dephosphorylation and deacetylation. Diabetes 2012;61:61–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ando H, Takamura T, Matsuzawa-Nagata N, et al. The hepatic circadian clock is preserved in a lipid-induced mouse model of non-alcoholic steatohepatitis. Biochem Biophys Res Commun 2009;380:684–688 [DOI] [PubMed] [Google Scholar]
- 22.Takamura T, Sakurai M, Ota T, Ando H, Honda M, Kaneko S. Genes for systemic vascular complications are differentially expressed in the livers of type 2 diabetic patients. Diabetologia 2004;47:638–647 [DOI] [PubMed] [Google Scholar]
- 23.Ota T, Gayet C, Ginsberg HN. Inhibition of apolipoprotein B100 secretion by lipid-induced hepatic endoplasmic reticulum stress in rodents. J Clin Invest 2008;118:316–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bonelli MA, Alfieri RR, Desenzani S, Petronini PG, Borghetti AF. Proteasome inhibition increases HuR level, restores heat-inducible HSP72 expression and thermotolerance in WI-38 senescent human fibroblasts. Exp Gerontol 2004;39:423–432 [DOI] [PubMed] [Google Scholar]
- 25.Misu H, Takamura T, Takayama H, et al. A liver-derived secretory protein, selenoprotein P, causes insulin resistance. Cell Metab 2010;12:483–495 [DOI] [PubMed] [Google Scholar]
- 26.Brown MS, Goldstein JL. Selective versus total insulin resistance: a pathogenic paradox. Cell Metab 2008;7:95–96 [DOI] [PubMed] [Google Scholar]
- 27.Bernales S, McDonald KL, Walter P. Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol 2006;4:e423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Harding HP, Calfon M, Urano F, Novoa I, Ron D. Transcriptional and translational control in the Mammalian unfolded protein response. Annu Rev Cell Dev Biol 2002;18:575–599 [DOI] [PubMed] [Google Scholar]
- 29.Kaufman RJ, Scheuner D, Schröder M, et al. The unfolded protein response in nutrient sensing and differentiation. Nat Rev Mol Cell Biol 2002;3:411–421 [DOI] [PubMed] [Google Scholar]
- 30.Acharya P, Engel JC, Correia MA. Hepatic CYP3A suppression by high concentrations of proteasomal inhibitors: a consequence of endoplasmic reticulum (ER) stress induction, activation of RNA-dependent protein kinase-like ER-bound eukaryotic initiation factor 2alpha (eIF2alpha)-kinase (PERK) and general control nonderepressible-2 eIF2alpha kinase (GCN2), and global translational shutoff. Mol Pharmacol 2009;76:503–515 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 31.Urano F, Wang X, Bertolotti A, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000;287:664–666 [DOI] [PubMed] [Google Scholar]
- 32.Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. J Clin Invest 2005;115:1111–1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Matsuzaki H, Daitoku H, Hatta M, Tanaka K, Fukamizu A. Insulin-induced phosphorylation of FKHR (Foxo1) targets to proteasomal degradation. Proc Natl Acad Sci USA 2003;100:11285–11290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aoki M, Jiang H, Vogt PK. Proteasomal degradation of the FoxO1 transcriptional regulator in cells transformed by the P3k and Akt oncoproteins. Proc Natl Acad Sci USA 2004;101:13613–13617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang H, Regan KM, Wang F, et al. Skp2 inhibits FOXO1 in tumor suppression through ubiquitin-mediated degradation. Proc Natl Acad Sci USA 2005;102:1649–1654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fu S, Yang L, Li P, et al. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature 2011;473:528–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fu S, Fan J, Blanco J, et al. Polysome profiling in liver identifies dynamic regulation of endoplasmic reticulum translatome by obesity and fasting. PLoS Genet 2012;8:e1002902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang L, Li P, Fu S, Calay ES, Hotamisligil GS. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab 2010;11:467–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Queisser MA, Yao D, Geisler S, et al. Hyperglycemia impairs proteasome function by methylglyoxal. Diabetes 2010;59:670–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Portero-Otín M, Pamplona R, Ruiz MC, Cabiscol E, Prat J, Bellmunt MJ. Diabetes induces an impairment in the proteolytic activity against oxidized proteins and a heterogeneous effect in nonenzymatic protein modifications in the cytosol of rat liver and kidney. Diabetes 1999;48:2215–2220 [DOI] [PubMed] [Google Scholar]
- 41.Wang X, Hu Z, Hu J, Du J, Mitch WE. Insulin resistance accelerates muscle protein degradation: Activation of the ubiquitin-proteasome pathway by defects in muscle cell signaling. Endocrinology 2006;147:4160–4168 [DOI] [PubMed] [Google Scholar]
- 42.Ozcan L, Ergin AS, Lu A, et al. Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell Metab 2009;9:35–51 [DOI] [PubMed] [Google Scholar]
- 43.Kozuka C, Yabiku K, Sunagawa S, et al. Brown rice and its component, γ-oryzanol, attenuate the preference for high-fat diet by decreasing hypothalamic endoplasmic reticulum stress in mice. Diabetes 2012;61:3084–3093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Matsuzawa-Nagata N, Takamura T, Ando H, et al. Increased oxidative stress precedes the onset of high-fat diet-induced insulin resistance and obesity. Metabolism 2008;57:1071–1077 [DOI] [PubMed] [Google Scholar]
- 45.Nakamura S, Takamura T, Matsuzawa-Nagata N, et al. Palmitate induces insulin resistance in H4IIEC3 hepatocytes through reactive oxygen species produced by mitochondria. J Biol Chem 2009;284:14809–14818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li J, Powell SR, Wang X. Enhancement of proteasome function by PA28α overexpression protects against oxidative stress. FASEB J 2011;25:883–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Seifert U, Bialy LP, Ebstein F, et al. Immunoproteasomes preserve protein homeostasis upon interferon-induced oxidative stress. Cell 2010;142:613–624 [DOI] [PubMed] [Google Scholar]
- 48.Stohwasser R, Salzmann U, Giesebrecht J, Kloetzel PM, Holzhütter HG. Kinetic evidences for facilitation of peptide channelling by the proteasome activator PA28. Eur J Biochem 2000;267:6221–6230 [DOI] [PubMed] [Google Scholar]
- 49.Lee AH, Scapa EF, Cohen DE, Glimcher LH. Regulation of hepatic lipogenesis by the transcription factor XBP1. Science 2008;320:1492–1496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rutkowski DT, Wu J, Back SH, et al. UPR pathways combine to prevent hepatic steatosis caused by ER stress-mediated suppression of transcriptional master regulators. Dev Cell 2008;15:829–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hirano Y, Yoshida M, Shimizu M, Sato R. Direct demonstration of rapid degradation of nuclear sterol regulatory element-binding proteins by the ubiquitin-proteasome pathway. J Biol Chem 2001;276:36431–36437 [DOI] [PubMed] [Google Scholar]
- 52.Zhou Y, Lee J, Reno CM, et al. Regulation of glucose homeostasis through a XBP-1-FoxO1 interaction. Nat Med 2011;17:356–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kawaguchi M, Minami K, Nagashima K, Seino S. Essential role of ubiquitin-proteasome system in normal regulation of insulin secretion. J Biol Chem 2006;281:13015–13020 [DOI] [PubMed] [Google Scholar]
- 54.Costes S, Huang CJ, Gurlo T, et al. β-cell dysfunctional ERAD/ubiquitin/proteasome system in type 2 diabetes mediated by islet amyloid polypeptide-induced UCH-L1 deficiency. Diabetes 2011;60:227–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xiao C, Giacca A, Lewis GF. Sodium phenylbutyrate, a drug with known capacity to reduce endoplasmic reticulum stress, partially alleviates lipid-induced insulin resistance and beta-cell dysfunction in humans. Diabetes 2011;60:918–924 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.