

Abstract
A−β+ ketosis-prone diabetes (KPD) is an emerging syndrome of obesity, unprovoked ketoacidosis, reversible β-cell dysfunction, and near-normoglycemic remission. We combined metabolomics with targeted kinetic measurements to investigate its pathophysiology. Fasting plasma fatty acids, acylcarnitines, and amino acids were quantified in 20 KPD patients compared with 19 nondiabetic control subjects. Unique signatures in KPD—higher glutamate but lower glutamine and citrulline concentrations, increased β-hydroxybutyryl-carnitine, decreased isovaleryl-carnitine (a leucine catabolite), and decreased tricarboxylic acid (TCA) cycle intermediates—generated hypotheses that were tested through stable isotope/mass spectrometry protocols in nine new-onset, stable KPD patients compared with seven nondiabetic control subjects. Free fatty acid flux and acetyl CoA flux and oxidation were similar, but KPD had slower acetyl CoA conversion to β-hydroxybutyrate; higher fasting β-hydroxybutyrate concentration; slower β-hydroxybutyrate oxidation; faster leucine oxidative decarboxylation; accelerated glutamine conversion to glutamate without increase in glutamate carbon oxidation; and slower citrulline flux, with diminished glutamine amide–nitrogen transfer to citrulline. The confluence of metabolomic and kinetic data indicate a distinctive pathogenic sequence: impaired ketone oxidation and fatty acid utilization for energy, leading to accelerated leucine catabolism and transamination of α-ketoglutarate to glutamate, with impaired TCA anaplerosis of glutamate carbon. They highlight a novel process of defective energy production and ketosis in A−β+ KPD.
Ketosis-prone diabetes (KPD) is characterized by presentation with diabetic ketoacidosis (DKA) in persons who do not fit traditional categories of types 1 or 2 diabetes (1–5). We have defined four subgroups of KPD based on presence or absence of β-cell autoantibodies (A+ or A−), and recovery or lack of recovery of β-cell functional reserve following the index episode of DKA (β+ or β−) (1,6,7).
The A−β+ KPD subgroup represents a novel syndrome of severe but reversible β-cell dysfunction (1,3,5,8,9). Approximately 50% of these patients develop DKA without a precipitating factor at diagnosis of diabetes. These new-onset, unprovoked A−β+ KPD patients display male predominance (10) and low frequencies of human leukocyte antigen class II susceptibility alleles for autoimmune diabetes (11). β-Cell function increases markedly within 1–3 months after the index DKA, with sustained glycemic improvement and insulin independence (1,9,11,12). The cause of the unprovoked ketoacidosis is unknown. Over 5–10 years, patients may relapse to unprovoked ketosis (3,5).
This syndrome provides a model to identify novel mechanisms of obesity, ketosis, and reversible β-cell dysfunction. We used a metabolomics approach to identify unique alterations in A−β+ KPD patients, with a kinetics approach to specify the pathophysiology.
RESEARCH DESIGN AND METHODS
Metabolomic analysis
Human subjects.
Protocols were approved by the Human Studies Institutional Review Boards of Baylor College of Medicine and Duke University (13). Written informed consent was obtained. New-onset, unprovoked male A−β+ KPD patients (n = 20) were identified by absence of GAD65/67, IA-2, or ZnT8 autoantibodies and presence of β-cell functional reserve 4–8 weeks after the index DKA (1,6). Men (n = 19) from the Study of the Effects of Diet on Metabolism and Nutrition study (13) were nondiabetic obese control subjects. KPD patients were selected from a longitudinal database using the MatchIT program (15) to minimize intragroup differences in initial HbA1c and intergroup differences in age, ethnicity, BMI, waist circumference, fasting C-peptide, glucose, and lipids. At the time of blood sampling, all patients were clinically stable, taking twice-daily neutral protamine Hagedorn insulin (± short-acting insulin); three patients were also taking metformin (1 g daily). Five additional, newly diagnosed, unprovoked A−β+ KPD patients were subsequently recruited for measurements of glutamine, glutamate, isoleucine, and leucine.
Plasma collection.
The KPD samples were collected in acid citrate dextrose (#364606, BD Biosciences) after an overnight fast before the morning dose of insulin, 20 days (median) after the index DKA (5). Blood was kept on ice for 2–4 h, centrifuged at 4°C, and the plasma stored at −20°C. Control subjects’ samples were collected after an overnight fast in serum separator tubes (#366510, BD Biosciences), centrifuged, and the serum stored at −80°C. Blood samples were also taken from five fresh A−β+ KPD patients 1 month after their index DKA, collected in serum separator tubes, processed immediately, and stored.
Metabolite analysis.
To measure 15 amino acids and 45 acylcarnitines, samples were deproteinized by methanol precipitation, esterified with hot, acidic methanol or n-butanol, and then analyzed by tandem mass spectrometry (MS/MS) with a Quattro Micro instrument (Waters Corporation, Milford, MA) (13). Leucine and isoleucine are reported as a single analyte (Leu/Ile) because they were not resolved by this method. These conditions partially hydrolyze glutamine to glutamate and asparagine to aspartate, so “glx” (Glu/Gln) or “asx” (Asp/Asn) signify glutamate or aspartate with contributions from hydrolysis reactions of glutamine and asparagine. Final concentrations of isoleucine, leucine, glutamine, and glutamate were measured by reverse-phase high-performance liquid chromatography (HPLC; Hewlett-Packard 1090; Hewlett-Packard, Avondale, PA); frozen samples were thawed on ice prior to HPLC and glutamine standards prepared fresh in ice-cold water.
To measure seven nonesterified free fatty acids (FFAs), samples were methylated and purified by solid-phase extraction (13). Derivatized fatty acids were analyzed by gas chromatography/mass spectrometry (GC/MS; Trace DSQ; Thermo Electron Corporation, Austin, TX).
All MS analyses used stable isotope dilution to quantify metabolites (13). Because acid citrate dextrose tubes were used for the original KPD patients, a dilution correction factor (1.31) was used for analytes measured in those samples (16).
Plasma insulin, C-peptide, glucagon, cortisol, and free metanephrines were measured by radioimmunoassay (Linco, St. Louis, MO), and β-hydroxybutyrate (BOHB) by spectrophotometry (Wako, Richmond, VA).
Statistical analysis.
Data were inspected for normality using the Shapiro-Wilk tests. The t tests were used to compare levels of hormones, baseline clinical parameters, and metabolites. BOHB values were not normally distributed, so median levels were compared using the Mann-Whitney U test. The Fisher exact test was used to assess deviations from the order of metabolites as an indicator of substrate preference due to altered consumption or production. Significance was defined at P < 0.05.
Kinetic analyses
Human subjects.
The study was approved by the Baylor institutional review board and written informed consent obtained. Nine new-onset, unprovoked A−β+ KPD patients (six men, three women), all clinically stable, were recruited 6–8 weeks after the index DKA episode. Seven nondiabetic control subjects (six men, one woman) were matched for BMI, age, and ethnicity, with the six men and one woman among the KPD patients. Exclusions were: serum creatinine >1.2 mg/day, retinopathy, neuropathy, or heart failure; history of alcohol abuse; alanine aminotransferase/aspartate aminotransferase (AST)/alkaline phosphatase >2× upper limit of normal; medications likely to affect fat, glucose, or protein metabolism; and other chronic illnesses. Fasting plasma C-peptide and glucose, peak and area under the curve of C-peptide response to glucagon, fasting lipids, and liver and thyroid functions were measured. Details of the glucagon stimulation test and cutoffs to classify β-cell reserve have been reported (1).
Subjects underwent four stable isotope protocols in the General Clinical Research Center. For 2 days prior to each study, they consumed an isocaloric, standardized diet (30% calories from fat, 65% from carbohydrates, 1 g/kg protein) prepared by the General Clinical Research Center kitchen. KPD patients were treated with low doses of neutral protamine Hagedorn insulin for 8 weeks prior to the studies. The last dose was administered at 8–10 p.m., and each study commenced the following morning after a 10–14-h fast, with no insulin administered until study completion. One venous catheter was placed for infusions, with another in the opposite hand, heated for arterialized blood sampling.
Protocol 1.
After baseline blood and breath sampling, an oral 50 mg/kg bolus of 2H2O was administered. After an intravenous priming dose of NaH13CO3 (1.2 µmol/kg), 2H5-glycerol (prime 4.5 µmol/kg, constant 9.0 µmol/kg/h), 2H2-potassium palmitate (prime 3 µmol/kg, constant 4 µmol/kg/h), and 13C1-acetate (prime 62.5 µmol/kg, constant 150 µmol/kg/h) were infused for 4 h. Blood was collected hourly for the first 3 h and every 15 min during the fourth hour. Indirect calorimetry was performed during the last 30 min of the fourth hour. At hour 4, glycerol and palmitate infusions ceased, but the acetate infusion continued for another 2 h. Blood and breath samples were drawn every 15 min between hours 5 and 6.
Tracer-to-tracee ratios of plasma free palmitate, acetate, and BOHB were determined by negative chemical ionization GC/MS of its pentafluorobenzyl derivative (17). Plasma palmitate and BOHB concentrations were measured by in vitro isotope dilution with U-13C-palmitate or 13C2-BOHB (Cambridge Isotope Laboratories, Andover, MA) as internal standards, using negative chemical ionization-GC/MS (17).
Tracer-to-tracee ratio of plasma glycerol was determined by electron impact ionization GC/MS of its tripropionate derivative (18). Breath 13CO2 content was determined by GC/gas isotope ratio MS. Zinc was used to reduce water in 10-µl plasma in quartz vessels and 2H2 abundance of the resulting hydrogen determined by GC/gas isotope ratio MS.
Protocol 2.
After baseline blood sampling, 2H2-citrulline (prime 1.0 µmol/kg, constant 1.0 µmol/kg/h), 15N1-amide-glutamine (prime 13.6 µmol/kg, constant 13.6 µmol/kg/h), 2H8-valine (prime 1 µmol/kg, constant 1 µmol/kg/h), and NaH13CO3 (prime 4 µmol/kg, constant 4 µmol/kg/h) were infused. A 0.5-µmol/kg prime of 15N1-citrulline was also given. Citrulline, glutamine, and valine were infused for 6 h. NaH13CO3 infusion was infused for 2 h; at the second hour, 13C1-leucine (prime 4 µmol/kg, constant 4 µmol/kg/h) was started and infused until hour 6. Blood was collected hourly during the first 4 h and every 15 min during the fifth hour. Breath was collected at baseline and every 15 min between hours 1 and 2 and 5 and 6.
Tracer-to-tracee ratio of plasma α-ketoisocaproic acid (α-KICA) from leucine was measured by negative chemical ionization-GC/MS of its pentafluorobenzyl derivative (17).
Plasma citrulline, glutamine, and valine isotopic enrichment (IE) were measured after conversion to their 5-(dimethylamino)-1-napthalene sulfonamide derivatives by liquid chromatography-MS/MS (Synergi MAX-RP column; Phenomenex, Torrance, CA) (19).
Protocol 3.
After baseline blood and breath sampling, 13C1-glutamine (prime 13.6 µmol/kg, constant 13.6 µmol/kg/h) was infused for 6 h. A priming dose of NaH13CO3 (4 µmol/kg) was given at hour 0. Indirect calorimetry was performed during the last 30 min of the fourth hour. Blood and breath were collected hourly for 5 h and then every 15 min between hours 5 and 6.
Plasma glutamine and glutamate IE were measured by liquid chromatography-MS/MS as in protocol 2.
Protocol 4.
After baseline blood and breath sampling, a priming dose of NaH13CO3 (4 µmol/kg) was given, and then 2,4-13C-β-hydroxybutyrate (prime 20 µmol/kg, constant 12 µmol/kg/h) was infused for 3 h. Blood and breath were collected hourly for the first 2 h and every 15 min during the third hour. Indirect calorimetry was performed during the last 30 min of the second hour. Blood was collected in K-EDTA tubes and spun immediately and one aliquot deproteinized with perchloric acid and frozen in liquid nitrogen.
IE of BOHB was measured as in protocol 1.
Plasma glucose was measured with a glucose analyzer (YSI, Yellow Springs, OH), insulin by radioimmunoassay (Linco) and FFAs by spectrophotometry (Wako). Amino acids were measured by HPLC.
Calculations.
Ra (palmitate, glycerol, acetate, or BOHB) (in µmol/kg/h):
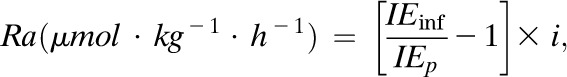 |
Eq. (1) |
where IEinf is the IE (mole %) in the infusate, IEp is IE (mole %) in the plasma at steady state, and i is the tracer infusion rate.
Rate of appearance of FFAs (µmol · kg−1 · h−1):
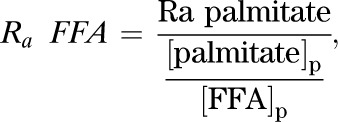 |
Eq. (2) |
where Ra palmitate is the rate of appearance of palmitate (Eq. 1), [palmitate]p is the concentration of plasma palmitate, and [FFA]p is the concentration of plasma FFA.
Whole-body fatty acid oxidation is calculated from RQ, VO2, and 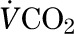 (20).
(20).
Rate of acetate oxidation (µmol · kg−1 · h−1) in the tricarboxylic acid (TCA) cycle:
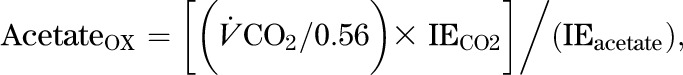 |
Eq. (3) |
where 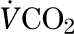 is the CO2 excretion rate measured by indirect calorimetry, IECO2 is the IE of CO2 in the breath, IEacetate is the IE of plasma acetate, and 0.56 is the acetate retention factor (21).
is the CO2 excretion rate measured by indirect calorimetry, IECO2 is the IE of CO2 in the breath, IEacetate is the IE of plasma acetate, and 0.56 is the acetate retention factor (21).
Acetate oxidized in the TCA cycle (%):
| Eq. (4) |
BOHB from acetate (µmol/kg/h):
| Eq. (5) |
where RaBOHB is the value from Eq. 1, IEBOHB is the M+1 isotopomer enrichment of BOHB derived from the 13C1-acetate infusate, and IEacetate is the IE of acetate.
Total body water (TBW) (from oral deuterated water):
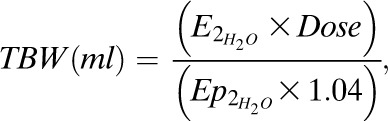 |
Eq. (6) |
where 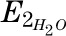 is the enrichment of deuterated water consumed,
is the enrichment of deuterated water consumed, 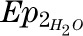 is the enrichment of plasma H2O at steady state, and the constant 1.04 corrects for deuterium’s volume of distribution. Fat-free mass (FFM) is TBW/0.72, where 0.72 is the hydration constant of fat-free tissue. Fat mass (kg) = body weight (kg) − FFM.
is the enrichment of plasma H2O at steady state, and the constant 1.04 corrects for deuterium’s volume of distribution. Fat-free mass (FFM) is TBW/0.72, where 0.72 is the hydration constant of fat-free tissue. Fat mass (kg) = body weight (kg) − FFM.
Ra (citrulline or glutamine amide-N) (μmol/kg/h) was calculated as in Eq. (1).
Rate of transfer of glutamine amide 15N to citrulline:
| Eq. (7) |
where Ra citrulline is the value from Eq. 7, IE15N-citrulline is the isotopic (M+1) enrichment of citrulline from 15N-glutamine, and IE15N-glutamine is the isotopic (M+1) enrichment of glutamine.
Flux of glutamine carbon is calculated as in Eq. (7).
13C-glutamine reaches steady state quickly, but 13C-glutamate formed from the glutamine does not, because of the large whole-body free glutamate pool. Hence, the fraction of the glutamate pool derived from glutamine (fractional synthesis rate [FSR] of glutamate from glutamine) is calculated using the precursor-product equation:
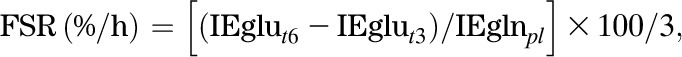 |
Eq. (8) |
where IEglut6 − IEglut3 is the increase in glutamate IE from 3–6 h of the infusion, and IEglnpl is the plateau IE of glutamine.
Absolute synthesis rate (ASR) of glutamate from glutamine:
| Eq. (9) |
The rate of formation of 13CO2 from 13C1-glutamine over the 6-h infusion, an index of glutamate dehydrogenase activity, is calculated as follows:
13C1-glutamine oxidation (mmol/6 h) = area under the curve of breath 13CO2 = AUC0–6 h of 13CO2:
| Eq. (10) |
where VCO2/0.78 is the production rate of CO2, assuming 22% of CO2 is retained in the bicarbonate pool (22), and IECO2 is the IE of CO2 (atom percent excess).
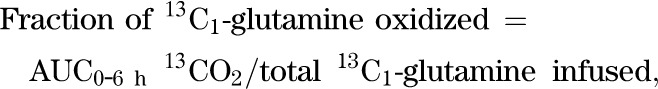 |
Eq. (11) |
a relative measure of glutamate entry in the TCA cycle at α-ketoglutarate and an estimate of glutaminase plus glutamate dehydrogenase activity.
RESULTS
Metabolomics
Clinical and biochemical parameters.
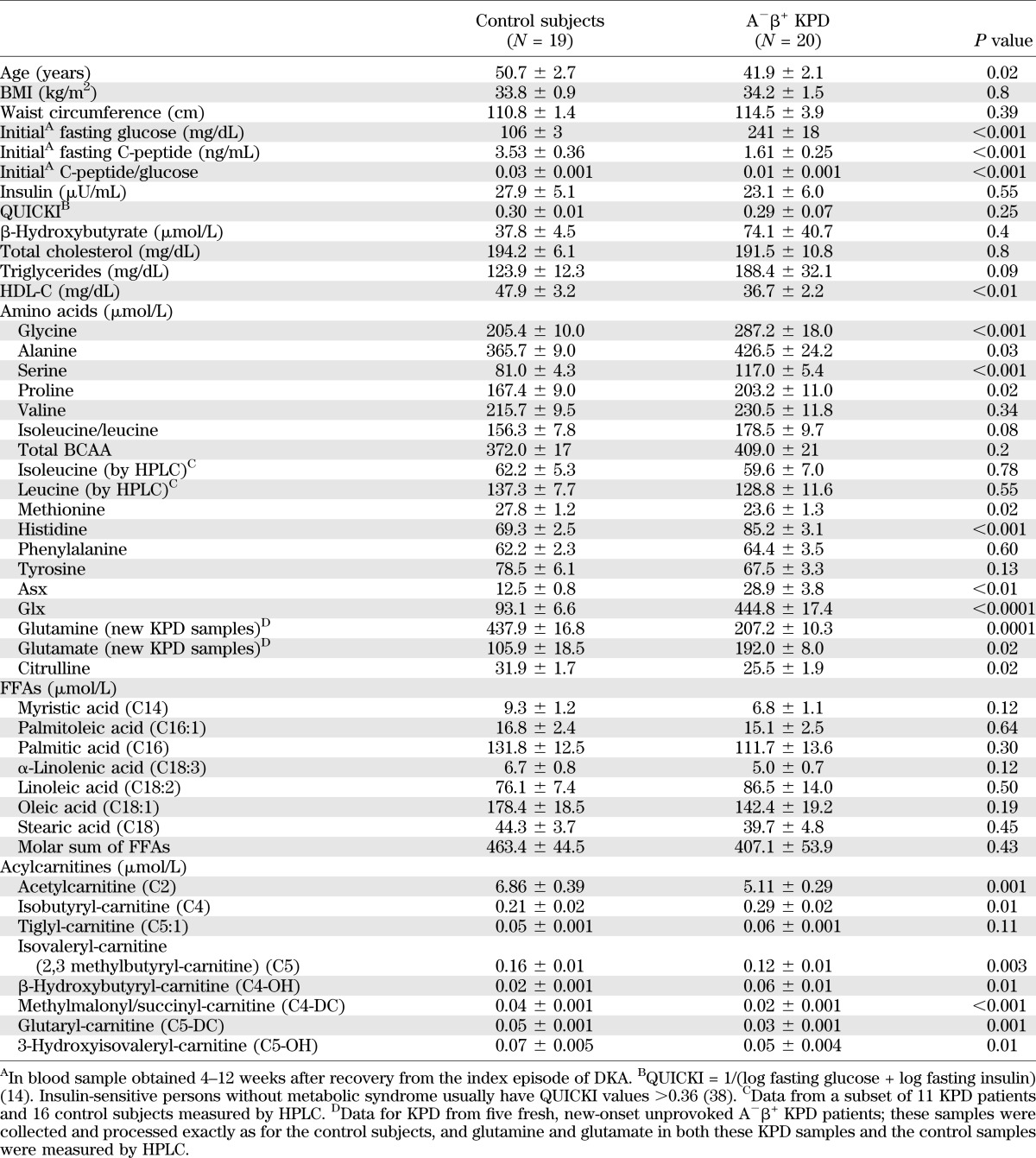
BMI and waist circumference were similar in KPD and control subjects (Table 1). KPD subjects had elevated initial HbA1c (13.5 ± 0.5 %) and fasting plasma glucose levels 4–12 weeks after the index DKA episode. Characteristically, HbA1c was markedly improved 3–6 months after the index DKA (7.2 ± 0.5), when the patients were on low doses of insulin (N = 13) or off insulin (N = 7). Fasting C-peptide was higher in control subjects. The C-peptide/glucose ratio was one-third of the control level in the KPD group. Fasting insulin levels were not different, and quantitative insulin sensitivity check index (QUICKI), an index of insulin sensitivity, demonstrated similar levels of insulin resistance in the two groups. Triglycerides showed a trend to be higher, and HDL-cholesterol (HDL-C) was ∼30% lower in KPD. The mean BOHB level was twice as high in KPD as in control subjects, but the difference did not attain statistical significance due to individual variability. KPD patients had normal fasting metanephrines and cortisol and normal thyroid, renal, and liver functions. The KPD group included 1 white, 7 African American, and 11 Hispanic subjects, whereas the control subjects included 14 white, 4 African American, and no Hispanic subjects; ethnicity was not correlated with any inter- or intragroup differences in metabolite or hormone levels.
TABLE 1.
Metabolomic survey: demographic and biochemical data and fasting plasma concentrations of amino acids, acylcarnitines, and FFAs
Amino acids and related acylcarnitines.
“Asx” was 2.3-fold (P < 0.01) and “glx” 4.8-fold (P < 0.0001) higher in the KPD group (Table 1). Smaller elevations were observed in KPD for glycine (1.4-fold; P < 0.001), serine (1.4-fold; P < 0.001), alanine (1.2-fold; P = 0.03), proline (1.2-fold; P = 0.02), and histidine (1.2-fold; P < 0.001). Methionine (0.85-fold; P = 0.02) and citrulline (0.75-fold; P = 0.02) were lower in KPD.
The unusually high levels of glx in KPD suggested partial ex vivo conversion of glutamine to glutamate or pyroglutamine (23–25) in those samples. HPLC also demonstrated very low glutamine and very high glutamate levels in the KPD samples. To avoid alterations of glutamine and glutamate due to collection methods or storage, five additional, newly diagnosed A−β+ KPD patients were recruited and their plasma collected using stringent collection and processing methods (26). The mean glutamine concentration in these new KPD samples was 207.20 ± 10.29 μmol/L (0.56-fold less than control subjects; P = 0.0001) and the mean glutamate concentration 192.03 ± 7.98 μmol/L (1.75-fold more than control subjects; P = 0.02).
Concentrations of total branched chain amino acids (BCAA) were similar in KPD and control subjects, with no significant differences in valine or isoleucine/leucine. Because MS/MS cannot distinguish isoleucine from leucine, HPLC was used to separate them in samples from 11 KPD patients and 16 control subjects. Leucine and isoleucine concentrations were not different between KPD and control subjects (128.8 ± 11.6 vs. 137.3 ± 7.7 and 59.6 ± 7.0 vs. 62.2 ± 5.3 μmol/L, respectively; P = NS for both). This was surprising because fasting plasma concentrations of leucine and isoleucine are usually elevated in diabetic patients with imperfect glycemic control (27–30). Hence, we tracked the catabolic pathways of BCAA through acylcarnitine esters of their distinct ketoacid metabolites (Table 1). Isobutyryl-carnitine (C4-AC), from decarboxylation of the valine ketoacid, was 1.5-fold higher in KPD (P < 0.01). In contrast, C5-AC, representing acylcarnitine esters of both isovaleryl-CoA and α-isomethylbutyryl-CoA (from decarboxylation of the leucine and isoleucine ketoacids, respectively), was 1.3-fold lower in KPD (P < 0.01), but tiglyl-carnitine (C5:1-AC), an isoleucine-specific catabolite, was 1.2-fold higher in KPD (P = 0.1). In sum, these data indicated that the leucine ketoacid metabolite was decreased in KPD patients.
BCAA are deaminated by branch chain aminotransferase (BCAT) to produce KICA from leucine, α-keto-β-methyvaleric acid (KMVA) from isoleucine and α-ketoisovaleric acid (KIVA) from valine, and the NH2 group is transferred to α-ketoglutarate to generate glutamate. The ketoacids are processed by branch chain ketoacid dehydrogenase (BCKDH) and subsequently follow distinct pathways to either generate ketone bodies (KICA, KMVA) or enter the TCA cycle for oxidation (KIVA, KMVA). Glutamate is exported from mitochondria with aspartate and the remaining ketoacids. Glutamate, aspartate, KMVA, and KIVA are precursors for gluconeogenesis; in contrast, KICA is a primary amino acid source of ketones. The metabolomic data suggested acceleration of the initial steps of BCAA catabolism in muscle as an explanation for key metabolic signatures in KPD patients: lack of elevated BCAA, low C5-AC, elevated glutamate, and elevated ketones.
TCA/anaplerotic metabolites.
Acetylcarnitine (C2, acylcarnitine ester of acetyl CoA) was lower in KPD (1.3-fold; P = 0.001), suggesting either decreased production of acetyl CoA or increased shunting of acetyl CoA into oxidative or synthetic pathways (Table 1). β-hydroxybutyryl-carnitine (C4-OH), representing intracellular BOHB, was threefold higher in KPD (P = 0.01), suggesting increased ketone synthesis or diminished ketone oxidation. Other short-chain acylcarnitines (C4-DC, C5-DC), which include metabolites arising distal to α-ketoglutarate in the TCA cycle, such as methylmalonyl/succinyl-carnitine and glutaryl-carnitine, were diminished in KPD.
Fatty acids and ketones.
The molar sum of the seven most abundant fatty acids was similar in KPD and control subjects (Table 1). Persons consuming American diets have the following rank order of fasting fatty acid concentrations: oleate > palmitate ≥ linoleate (31); this order was preserved in both groups, and levels of palmitate and oleate were similar, making it unlikely that there were group differences in habitual dietary fat composition. These data suggested that the proclivity for ketosis in KPD patients is not due to excessive fatty acid supply.
Kinetics.
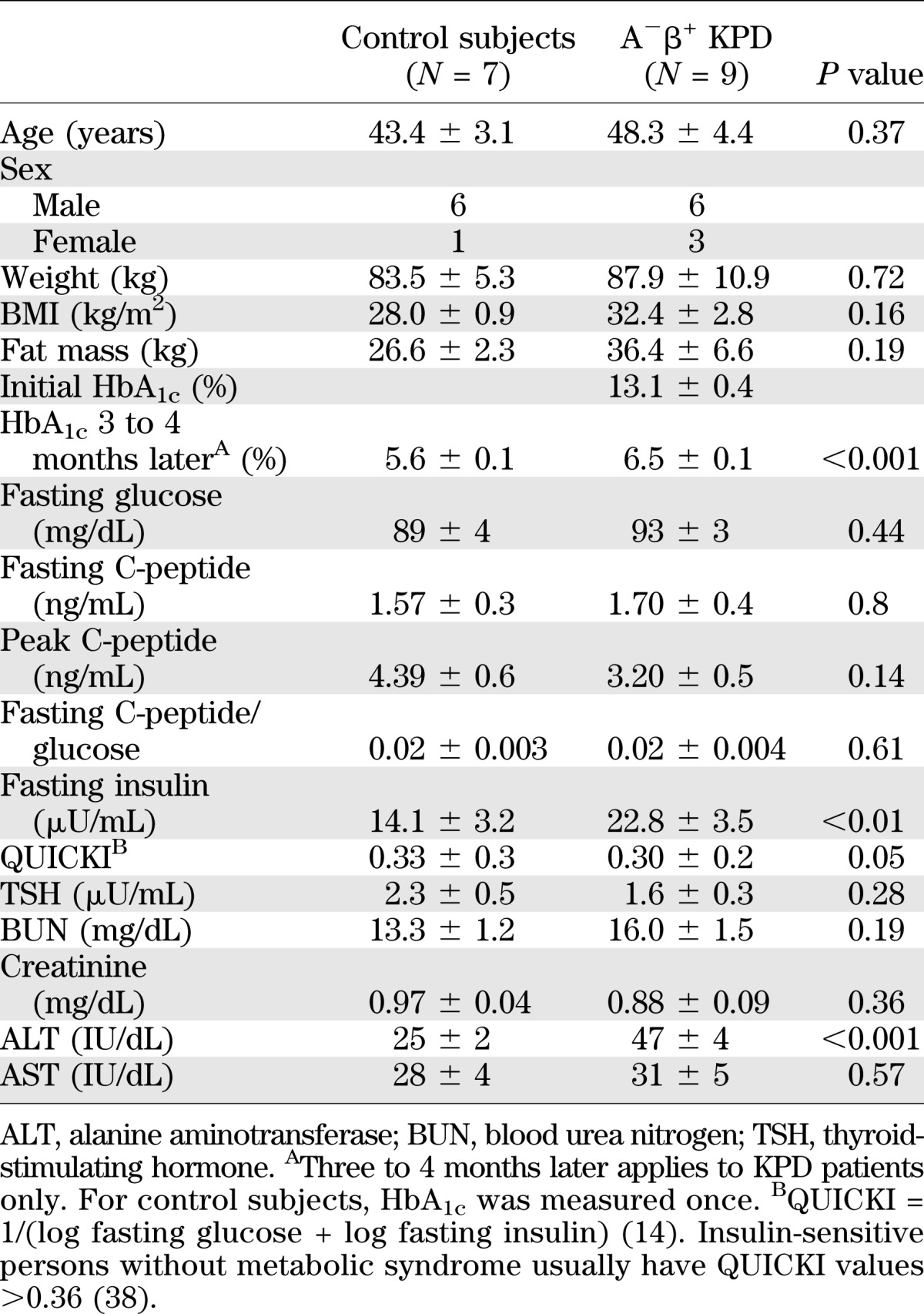
In the kinetic study cohort, age, weight, and BMI were similar in the two groups (Table 2). HbA1c was very high in the KPD patients at the time of the index DKA; 3 to 4 months later, mean HbA1c and fasting glucose were markedly improved, but still higher than in the control subjects. At that time, fasting and glucagon-stimulated peak C-peptide and C-peptide/glucose ratio were similar in KPD and control subjects. Fasting insulin levels were higher in KPD; however, they were very similar to those of the KPD patients in the metabolomics study (Table 1), and QUICKI demonstrated a significantly higher degree of insulin resistance in KPD. Thyroid-stimulating hormone, blood urea nitrogen, creatinine, and AST were normal and similar in both groups. Triglycerides (163 ± 9 mg/dL) were elevated, whereas HDL-C (40.6 ± 3.9 mg/dL) was low, and LDL-cholesterol level was 85 ± 9 mg/dL in the KPD group.
TABLE 2.
Kinetic study: demographic, hormonal, and biochemical data
1. Fasting rates of FFA release and acetyl CoA flux are similar in KPD and control subjects. BOHB oxidation and fatty acid disposal are slowed, leading to elevated plasma concentrations of BOHB and FFA.
Ra FFA (total FFA flux) was not different, whereas Ra glycerol (index of triglyceride hydrolysis) was significantly lower, and Ra palmitate (palmitate flux) trended lower in KPD (Fig. 1). Furthermore, Ra acetate (index of whole-body acetyl CoA flux) was similar in KPD and control subjects (1,344 ± 260 vs. 1,570 ± 141 µmol/kgFM/h; P = 0.50), and acetate oxidation (breath 13CO2 liberated by isocitrate dehydrogenase activity, reflecting acetyl CoA flux through the TCA cycle) was also similar (861 ± 175 vs. 1,007 ± 87 µmol/kgFM/h; P = 0.49). Despite the similar FFA flux, plasma concentration of FFA was higher in KPD (587 ± 51 vs. 412 ± 8 µmol/L; P = 0.04) (Fig. 1), suggesting slower FFA disposal (corroborated by the slower palmitate clearance). Plasma concentration of BOHB was also higher in the KPD patients (97 ± 17 vs. 43 ± 10 μmol/L; P = 0.02) in the face of slower conversion of acetyl CoA to BOHB (Fig. 2). Collectively, these findings indicated defective BOHB and fatty acid disposal, which, after a 10–14-h fast, is predominantly oxidative. To test BOHB oxidation directly, protocol 4 was performed in three control subjects and seven KPD patients. KPD had slower BOHB flux (330 ± 80 vs. 696 ± 103 µmol/kgFM/h; P = 0.03) but also slower oxidative BOHB disposal (191 ± 47 vs. 427 ± 84 μmol/kgFM/h; P = 0.03), a combination that would increase BOHB concentration. (Of note, plasma BOHB enrichment was used in this study as a proxy for intracellular acetate or acetyl CoA enrichment; in reality, it is acetyl CoA that is oxidized. The IE of the acetyl CoA would be lower than the plasma BOHB enrichment, because there are additional sources of unlabeled acetyl CoA in the intracellular pool. Because the more highly enriched BOHB is the denominator used in the calculations, the actual oxidation rate of BOHB [which must first be converted to acetyl CoA to be oxidized via intermediates in the TCA cycle] is probably underestimated in all subjects.)
FIG. 1.

Lipid kinetics (mean ± SE) in A−β+ KPD patients and control subjects. *P ≤ 0.05; †P < 0.1 (N = 9 for KPD, N = 7 for control subjects).
FIG. 2.

BOHB concentration, rate of conversion of acetate to BOHB, BOHB flux, and BOHB oxidation (mean ± SE) in A−β+ KPD patients and control subjects (for BOHB concentration: N = 9 for KPD, N = 7 for control subjects; for all other measurements N = 7 for KPD, N = 3 for control subjects). *P < 0.05; †P = 0.09.
2. Oxidative decarboxylation of leucine is accelerated in KPD.
There was no difference between KPD and control subjects in leucine flux and a trend toward higher valine flux in KPD (Fig. 3). The rate of oxidative decarboxylation, or removal of the #1 carbon of leucine by BCKDH, showed a strong trend to be faster in KPD (60.0 ± 2.8 vs. 51.7 ± 2.9 μmol/kgFFM/h; P = 0.06). Increased BCKDH activity requires increased flux through the earlier BCAT-catalyzed step that drives transfer of NH2 from leucine to α-ketoglutarate to generate glutamate (32). Unless matched by acceleration of the opposing reaction that oxidatively deaminates glutamate to α-ketoglutarate, this would promote accumulation of glutamate.
FIG. 3.

Leucine flux, oxidative decarboxylation, and nonoxidative disposal and valine flux (mean ± SE) in A−β+ KPD patients and control subjects. †P = 0.06 (N = 9 for KPD, N = 7 for control subjects).
3. Conversion of glutamine to glutamate is accelerated in KPD, without a parallel increase in glutamate carbon oxidation.
There was no group difference in glutamine flux, but plasma glutamine concentration trended lower in KPD (557 ± 29 vs. 655 ± 36 μmol/L; P = 0.06) (Fig. 4A). The rate of glutamine carbon transfer to glutamate was faster in KPD by several measures: slope of change, absolute synthesis rate of glutamate from glutamine (Fig. 4A), and fractional synthesis rate of glutamate from glutamine (Fig. 4B). However, the rate of oxidation of the same glutamate carbon in the TCA cycle was not different between the groups (Fig. 4B). Thus, increased glutamate production from glutamine in KPD is not matched by a parallel increase in glutamate carbon anaplerosis into the TCA cycle, as predicted from the results of the leucine infusion study.
FIG. 4.

A: Glutamine flux, glutamine plasma concentration, and absolute conversion rate of glutamine to glutamate (mean ± SE) in A−β+ KPD patients and control subjects (N = 9 for KPD, N = 7 for control subjects). B: Fractional synthesis rate of glutamate from glutamine, oxidation of glutamate carbon (derived from glutamine), and glutamate plasma concentration (mean ± SE) in A−β+ KPD patients and control subjects. *P < 0.05; †P = 0.06 (N = 9 for KPD, N = 7 for control subjects).
4. Transfer of amide nitrogen from glutamine to citrulline is slower in KPD.
Citrulline flux was slower in KPD than in control subjects (Fig. 5), with a trend toward slower transfer of glutamine amide nitrogen to ornithine (7.8 ± 0.4 vs. 8.6 ± 0.3 μmol/kgFFM/h; P = 0.1) for citrulline synthesis.
FIG. 5.

Citrulline flux and rate of transfer of glutamine amide-N to citrulline (mean ± SE) in A−β+ KPD patients and control subjects. *P < 0.05; †P = 0.10 (N = 9 for KPD, N = 7 for control subjects).
DISCUSSION
In this study, the pathophysiology of A−β+ KPD was investigated comprehensively using a two-step discovery method. First, fasting plasma metabolomics compared KPD patients to nondiabetic obese subjects. Hypotheses generated from key differences were tested by kinetic tracer studies targeting the suspected metabolic pathways in a fresh cohort of patients and control subjects.
The metabolomic analysis revealed signals of altered BCAA catabolism in KPD associated with changes in glutamine/glutamate. We hypothesized that the lack of elevated leucine in KPD was due to accelerated catabolism of this amino acid, indicated by decreased C5-AC and elevated glutamate (33,34). Evidence that accelerated leucine catabolism extended beyond isovaleryl CoA to β-methylcrotonyl CoA and ketogenesis was provided by the lower level of 3-hydroxyisovaleryl-carnitine (C5-OH). Concomitantly, glutamine was 50% lower, whereas glutamate was 175% higher, in KPD. Glutamine and glutamate are nitrogen sinks for catabolism of other amino acids, including BCAA, in the fasted state. Elevated glutamate and decreased glutamine levels could reflect absence of upregulation of glutamate oxidation rates in KPD subjects in the face of faster conversion of glutamine to glutamate and accelerated leucine deamidation.
Changes in small-chain acylcarnitines reflecting TCA cycle intermediates and anaplerosis broadened the pathophysiologic hypothesis. Acetylcarnitine was significantly lower in KPD, suggesting decreased production or increased consumption of acetyl CoA. Acylcarnitines that include glutaryl CoA and succinyl CoA were lower and β-hydroxybutyryl-carnitine was threefold higher in KPD, suggesting diversion of acetyl CoA disposal toward ketogenesis. Finally, fasting FFA levels and composition were similar in KPD and control subjects, suggesting that excessive fatty acid flux from lipolysis is not the driver of ketogenesis in KPD.
In sum, the metabolomic data suggested that patients with unprovoked A−β+ KPD have accelerated leucine catabolism toward ketogenesis. This would expand the nitrogen sinks of transamination reactions, resulting in elevated levels of glutamate and aspartate. The data also indicated that KPD patients have defects in transferring carbon from glutamine/glutamate to the TCA cycle and nitrogen from glutamine/glutamate to the urea cycle.
The results of the kinetic studies confirmed the hypothesis and revealed also that KPD patients have altered ketone oxidation. Clinically stable KPD patients with excellent glycemic control have elevated plasma BOHB. The cause is not excessive fatty acid–derived ketone production—the conversion of acetyl CoA to BOHB is slower, and rates of acetyl CoA flux and isocitrate decarboxylation are unaltered. However, the rate of ketone oxidation is significantly slowed in KPD, leading to elevated concentrations of BOHB and its intracellular metabolites, and this likely plays a prominent role in the most unique characteristic of the phenotype, proclivity for ketosis. Fatty acid disposal is also impaired.
Unable to maximize rates of oxidation of fat-derived sources for energy, KPD patients after an overnight fast draw upon amino acids for energy. There is increased flux through the leucine catabolic pathway toward ketone production. Acceleration of leucine catabolism also leads, through reductive deamination of α-ketoglutarate, to glutamate accumulation (exacerbated by increased conversion from glutamine) and blunted glucose carbon anaplerosis in the TCA cycle as manifested by an unchanged rate of glutamate carbon oxidation. A similar cascade occurs in McArdle syndrome, in which accelerated BCAA catabolism eventually reduces flux into the TCA cycle through increased conversion of α-ketoglutarate to glutamate (35).
Peripheral oxidation of ketone bodies is a TCA cycle-dependent process; in this regard, an apparent conflict arises between the finding of slowed BOHB oxidation in KPD and the finding that there is no difference in the rate of oxidation in the TCA cycle when the groups are challenged with a similar flux of oxidative substrate (13C1-acetate). This apparent paradox may be explained because our measurements specifically demonstrate that KPD show no defect in the rate of removal of CO2 from isocitrate (at the step of isocitrate dehydrogenase) in the TCA cycle, and this leaves open the possibility of kinetic defects distal to that step in the TCA cycle and therefore in the rates at which reduced energy intermediates are delivered for oxidative phosphorylation. The data suggest that there may be such a kinetic defect at the level of α-ketoglutarate dehydrogenase associated with more rapid decarboxylation of leucine, as evidenced by the accumulation of glutamate and reduced levels of acylcarnitines of TCA intermediates beyond that step.
This study combines for the first time an unbiased metabolomics approach and a targeted kinetics approach to specify the pathophysiology of a novel diabetic syndrome (Fig. 6). Umpierrez et al. (36) previously explored the pathophysiology of ketosis-prone type 2 diabetes, a condition identical to unprovoked A−β+ KPD. They reported no ketosis or β-cell decompensation following prolonged infusion of fatty acids, supporting the concept that proneness to ketosis in KPD is not due to excessive fatty acid flux to the liver.
FIG. 6.

Schematic representation of metabolic defects in KPD as revealed by a combination of metabolomics and kinetics. Short black arrows represent increase or decrease in a metabolite measured in the metabolomics survey, whereas short white arrows represent increase, decrease, or no change in a metabolite measured in the kinetic studies. Lipid bilayer indicates the boundary of a liver cell. Dashed long arrows indicate decreased flux, and solid long arrows indicate increased flux. See text for details. alpha-KG, alpha-ketoglutarate; CPS, carbamoyl phosphate synthetase; GDH, glutamate dehydrogenase.
Defective oxidation of ketones and accelerated leucine catabolism are key defects of fasting energy metabolism that underlie the distinctive syndrome of unprovoked A−β+ KPD. The findings indicate the need to investigate mitochondrial function and BCAT and BCKDH regulation in the skeletal muscle of KPD patients. These defects are associated with pronounced changes in glutamine/glutamate metabolism; interestingly, elevated glutamate predicted type 1 diabetes in a longitudinal metabolomics study (37). Clinically and metabolically, patients with A−β+ KPD manifest features of both type 1 and type 2 diabetes. The present data explain how overweight patients with a type 2 diabetes phenotype and adequate β-cell reserve are prone to developing unprovoked ketoacidosis. The coupled metabolomics-kinetics approach should be useful in elucidating the pathophysiology of other phenotypes of diabetes.
ACKNOWLEDGMENTS
This study was supported by National Institutes of Health (NIH) R21-DK-082827 (to A.B.), the Diabetes and Endocrinology Research Center (P30-DK-079638) at Baylor College of Medicine, NIH RO1-DK-056689 (to F.J.), funds from the U.S. Department of Agriculture, Agricultural Research Service under Cooperative Agreement Number 58-6250-6001, and NIH PO1-DK-58398 (to C.B.N.).
No potential conflicts of interest relevant to this article were reported.
S.G.P. and F.J. helped to design the kinetic studies, analyzed data, and edited the manuscript. J.W.H. performed mass spectrometric analyses and analyzed data. I.C. recruited subjects and implemented the stable isotope protocols. J.R.B. and R.D.S. performed metabolomic analyses and reviewed the manuscript. D.I. performed biochemical assays. R.N. and K.O. maintained clinical care of the study subjects and edited the manuscript. C.S.H. performed autoantibody testing of the study subjects and reviewed the manuscript. C.B.N. helped with study design, supervised the metabolomic analyses, and edited the manuscript. A.B. designed and obtained funding for the study, implemented the kinetic protocols, analyzed data, and wrote the manuscript. A.B. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Parts of this study were presented in poster form at the 71st Scientific Sessions of the American Diabetes Association, San Diego, California, 24–28 June 2011.
The authors thank Elizabeth Fraser, Vy Pham, and Toni Oplt (all from Baylor College of Medicine) for technical assistance; Resa Labbe-Morris, RN, and the nursing staff of the Baylor College of Medicine General Clinical Research Center for meticulous attention to protocol and care of the study subjects; Varsha Patel, RPh, and the Investigational Pharmacy (The Methodist Hospital Research Institute, Houston, TX) for stable isotope preparation; Drs. Heinrich Taegtmeyer (University of Texas Health Sciences Center, Houston, TX), William Mitch, and Arun Sreekumar (both from Baylor College of Medicine) for helpful discussions; and all of the study subjects.
Footnotes
S.G.P. is currently affiliated with the Department of Surgery, UCLA Medical Center, Los Angeles, California.
See accompanying commentary, p. 682.
REFERENCES
- 1.Maldonado M, Hampe CS, Gaur LK, et al. Ketosis-prone diabetes: dissection of a heterogeneous syndrome using an immunogenetic and beta-cell functional classification, prospective analysis, and clinical outcomes. J Clin Endocrinol Metab 2003;88:5090–5098 [DOI] [PubMed] [Google Scholar]
- 2.Kitabchi AE. Ketosis-prone diabetes—a new subgroup of patients with atypical type 1 and type 2 diabetes? J Clin Endocrinol Metab 2003;88:5087–5089 [DOI] [PubMed] [Google Scholar]
- 3.Mauvais-Jarvis F, Sobngwi E, Porcher R, et al. Ketosis-prone type 2 diabetes in patients of sub-Saharan African origin: clinical pathophysiology and natural history of beta-cell dysfunction and insulin resistance. Diabetes 2004;53:645–653 [DOI] [PubMed] [Google Scholar]
- 4.Umpierrez GE, Smiley D, Kitabchi AE. Narrative review: ketosis-prone type 2 diabetes mellitus. Ann Intern Med 2006;144:350–357 [DOI] [PubMed] [Google Scholar]
- 5.Balasubramanyam A, Nalini R, Hampe CS, Maldonado M. Syndromes of ketosis-prone diabetes mellitus. Endocr Rev 2008;29:292–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Balasubramanyam A, Garza G, Rodriguez L, et al. Accuracy and predictive value of classification schemes for ketosis-prone diabetes. Diabetes Care 2006;29:2575–2579 [DOI] [PubMed] [Google Scholar]
- 7.Banerji MA, Dham S. A comparison of classification schemes for ketosis-prone diabetes. Nat Clin Pract Endocrinol Metab 2007;3:506–507 [DOI] [PubMed] [Google Scholar]
- 8.Umpierrez GE, Casals MM, Gebhart SP, Mixon PS, Clark WS, Phillips LS. Diabetic ketoacidosis in obese African-Americans. Diabetes 1995;44:790–795 [DOI] [PubMed] [Google Scholar]
- 9.Maldonado MR, Otiniano ME, Cheema F, Rodriguez L, Balasubramanyam A. Factors associated with insulin discontinuation in subjects with ketosis-prone diabetes but preserved beta-cell function. Diabet Med 2005;22:1744–1750 [DOI] [PubMed] [Google Scholar]
- 10.Nalini R, Ozer K, Maldonado M, et al. Presence or absence of a known diabetic ketoacidosis precipitant defines distinct syndromes of “A-β+” ketosis-prone diabetes based on long-term β-cell function, human leukocyte antigen class II alleles, and sex predilection. Metabolism 2010;59:1448–1455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nalini R, Gaur LK, Maldonado M, et al. HLA class II alleles specify phenotypes of ketosis-prone diabetes. Diabetes Care 2008;31:1195–1200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rasouli N, Elbein SC. Improved glycemic control in subjects with atypical diabetes results from restored insulin secretion, but not improved insulin sensitivity. J Clin Endocrinol Metab 2004;89:6331–6335 [DOI] [PubMed] [Google Scholar]
- 13.Newgard CB, An J, Bain JR, et al. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab 2009;9:311–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen H, Sullivan G, Quon MJ. Assessing the predictive accuracy of QUICKI as a surrogate index for insulin sensitivity using a calibration model. Diabetes 2005;54:1914–1925 [DOI] [PubMed] [Google Scholar]
- 15.Ho DE, Imai K, King G, Stuart EA. MatchIt: Nonparametric preprocessing for parametric causal inference [Internet], 2011. Vol. 42, Issue 8. Journal of Statistical Software. Available from http://www.jstatsoft.org/v42/i08 [Google Scholar]
- 16.Duarte NL, Wang XL, Wilcken DE. Effects of anticoagulant and time of plasma separation on measurement of homocysteine. Clin Chem 2002;48:665–668 [PubMed] [Google Scholar]
- 17.Hachey DL, Patterson BW, Reeds PJ, Elsas LJ. Isotopic determination of organic keto acid pentafluorobenzyl esters in biological fluids by negative chemical ionization gas chromatography/mass spectrometry. Anal Chem 1991;63:919–923 [DOI] [PubMed] [Google Scholar]
- 18.Mohammad MA, Sunehag AL, Rodriguez LA, Haymond MW. Galactose promotes fat mobilization in obese lactating and nonlactating women. Am J Clin Nutr 2011;93:374–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kao CC, Bandi V, Guntupalli KK, Wu M, Castillo L, Jahoor F. Arginine, citrulline and nitric oxide metabolism in sepsis. Clin Sci (Lond) 2009;117:23–30 [DOI] [PubMed] [Google Scholar]
- 20.Frayn KN. Calculation of substrate oxidation rates in vivo from gaseous exchange. J Appl Physiol 1983;55:628–634 [DOI] [PubMed] [Google Scholar]
- 21.Sidossis LS, Coggan AR, Gastaldelli A, Wolfe RR. A new correction factor for use in tracer estimations of plasma fatty acid oxidation. Am J Physiol 1995;269:E649–E656 [DOI] [PubMed] [Google Scholar]
- 22.Wolfe RR, Jahoor F. Recovery of labeled CO2 during the infusion of C-1- vs C-2-labeled acetate: implications for tracer studies of substrate oxidation. Am J Clin Nutr 1990;51:248–252 [DOI] [PubMed] [Google Scholar]
- 23.Schaefer A, Piquard F, Haberey P. Plasma amino-acids analysis: effects of delayed samples preparation and of storage. Clin Chim Acta 1987;164:163–169 [DOI] [PubMed] [Google Scholar]
- 24.Reichert MC. Innovations in sterilization technology for instrument processing. Med Instrum 1983;17:89–90 [PubMed] [Google Scholar]
- 25.Ukida M, Schäfer K, Bode JC. Effect of storage at 20° C on the concentration of amino acids in plasma. J Clin Chem Clin Biochem 1981;19:1193–1195 [DOI] [PubMed] [Google Scholar]
- 26.Scriver CR, Lamm P, Clow CL. Plasma amino acids: screening, quantitation, and interpretation. Am J Clin Nutr 1971;24:876–890 [DOI] [PubMed] [Google Scholar]
- 27.Felig P, Marliss E, Ohman JL, Cahill CF., Jr Plasma amino acid levels in diabetic ketoacidosis. Diabetes 1970;19:727–728 [DOI] [PubMed] [Google Scholar]
- 28.Wahren J, Felig P, Cerasi E, Luft R. Splanchnic and peripheral glucose and amino acid metabolism in diabetes mellitus. J Clin Invest 1972;51:1870–1878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carlsten A, Hallgren B, Jagenburg R, Svanborg A, Werkö L. Amino acids and free fatty acids in plasma in diabetes. I. The effect of insulin on the arterial levels. Acta Med Scand 1966;179:361–370 [DOI] [PubMed] [Google Scholar]
- 30.Nair KS, Garrow JS, Ford C, Mahler RF, Halliday D. Effect of poor diabetic control and obesity on whole body protein metabolism in man. Diabetologia 1983;25:400–403 [DOI] [PubMed] [Google Scholar]
- 31.Stefan N, Wahl HG, Fritsche A, Häring H, Stumvoll M. Effect of the pattern of elevated free fatty acids on insulin sensitivity and insulin secretion in healthy humans. Horm Metab Res 2001;33:432–438 [DOI] [PubMed] [Google Scholar]
- 32.Hutson SM. The case for regulating indispensable amino acid metabolism: the branched-chain alpha-keto acid dehydrogenase kinase-knockout mouse. Biochem J 2006;400:e1–e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Darmaun D, Déchelotte P. Role of leucine as a precursor of glutamine alpha-amino nitrogen in vivo in humans. Am J Physiol 1991;260:E326–E329 [DOI] [PubMed] [Google Scholar]
- 34.Darmaun D, Matthews DE, Bier DM. Physiological hypercortisolemia increases proteolysis, glutamine, and alanine production. Am J Physiol 1988;255:E366–E373 [DOI] [PubMed] [Google Scholar]
- 35.Wagenmakers AJ, Coakley JH, Edwards RH. Metabolism of branched-chain amino acids and ammonia during exercise: clues from McArdle’s disease. Int J Sports Med 1990;11(Suppl. 2):S101–S113 [DOI] [PubMed] [Google Scholar]
- 36.Umpierrez GE, Smiley D, Robalino G, Peng L, Gosmanov AR, Kitabchi AE. Lack of lipotoxicity effect on beta-cell dysfunction in ketosis-prone type 2 diabetes. Diabetes Care 2010;33:626–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Oresic M, Simell S, Sysi-Aho M, et al. Dysregulation of lipid and amino acid metabolism precedes islet autoimmunity in children who later progress to type 1 diabetes. J Exp Med 2008;205:2975–2984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hrebícek J, Janout V, Malincíková J, Horáková D, Cízek L. Detection of insulin resistance by simple quantitative insulin sensitivity check index QUICKI for epidemiological assessment and prevention. J Clin Endocrinol Metab 2002;87:144–147 [DOI] [PubMed] [Google Scholar]