

Abstract
Obesity is associated with insulin resistance, a major risk factor for type 2 diabetes and cardiovascular disease. However, not all obese individuals are insulin resistant, which confounds our understanding of the mechanistic link between these conditions. We conducted transcriptome analyses on 835 obese subjects with mean BMI of 48.8, on which we have previously reported genetic associations of gene expression. Here, we selected ∼320 nondiabetic (HbA1c <7.0) subjects and further stratified the cohort into insulin-resistant versus insulin-sensitive subgroups based on homeostasis model assessment–insulin resistance. An unsupervised informatics analysis revealed that immune response and inflammation-related genes were significantly downregulated in the omental adipose tissue of obese individuals with extreme insulin sensitivity and, to a much lesser extent, in subcutaneous adipose tissue. In contrast, genes related to β-oxidation and the citric acid cycle were relatively overexpressed in adipose of insulin-sensitive patients. These observations were verified by querying an independent cohort of our published dataset of 37 subjects whose subcutaneous adipose tissue was sampled before and after treatment with thiazolidinediones. Whereas the immune response and inflammation pathway genes were downregulated by thiazolidinedione treatment, β-oxidation and citric acid cycle genes were upregulated. This work highlights the critical role that omental adipose inflammatory pathways might play in the pathophysiology of insulin resistance, independent of body weight.
The number of obese individuals worldwide has reached two billion, leading to an explosion of obesity-related health problems associated with increased morbidity and mortality (1,2). The increase in the prevalence of obesity is strongly correlated with an increase in type 2 diabetes mellitus, reaching epidemic proportions in the United States (3,4). A key etiological factor linking obesity to type 2 diabetes mellitus is insulin resistance, characterized by decreased response in the cellular actions of insulin, leading to an impaired ability of insulin to inhibit glucose output from the liver and to promote glucose uptake in fat and muscle (5,6). The physiological mechanisms connecting obesity to insulin resistance have received intense investigation in recent years and several hypotheses have emerged, such as ectopic lipid accumulation in liver and muscle secondary to obesity-associated increase in serum free fatty acids, altered production of various adipocyte-derived factors (collectively known as adipokines), and low-grade inflammation of white adipose tissue resulting from chronic activation of the innate immune system (7,8).
The association between obesity and insulin resistance is likely a cause-and-effect relationship because human and animal studies indicate that weight loss and weight gain correlate closely with increasing and decreasing insulin sensitivity, respectively (9–11). However, not all obese individuals are insulin-resistant. In fact, insulin sensitivity can vary up to six-fold in this population, which highlights the importance of identifying genetic and environmental factors that place obese individuals at the greatest risk for obesity-related complications (12–14). It has been recognized that the adipose tissue, in addition to its role as an energy storage depot, is a bona fide endocrine organ with a key role in controlling whole-body metabolism (7,15). Adipose tissue actively secretes cytokines and hormones that regulate food intake, glucose metabolism, and whole-body nutrient homeostasis (16). The expansion of adipose tissue in obesity is associated with the activation of chronic proinflammatory pathways and macrophage infiltration, which ultimately impairs its function as an energy depot as well as an endocrine gland with detrimental consequences for the whole body (7,17–19). However, despite increasing awareness of the role inflamed adipose tissue plays in obesity-related insulin resistance, there is limited understanding of the molecular signals that differentiate insulin-resistant from insulin-sensitive obese individuals. This is because the majority of studies in this area have focused on comparisons of lean and obese individuals, obviating the potential causal factors of body weight per se on insulin sensitivity (20,21). These studies, however, could not reveal the gene expression signature responsible for the metabolically compromised status of obese individuals independent of their weight. A few recent studies of very small cohorts have been reported that point to a role for adipocyte size and differentiation potential, as well as to modest increases in a limited set of inflammatory genes in the development of insulin resistance, independent of obesity (22–27).
Because equally obese individuals can differ dramatically in their overall sensitivity to insulin, we performed a highly powered transcriptome study of our previously published transcription dataset from >800 obese individuals to identify the molecular pathways associated with insulin sensitivity, independent of body mass (28). Specifically, we conducted a comprehensive transcription profiling analysis on subcutaneous and omental adipose tissue samples collected from this entire cohort of obese subjects during bariatric surgery procedures. Our results emphasize the role of the immune system and mitochondrial function in the etiology of insulin resistance, independent of obesity.
RESEARCH DESIGN AND METHODS
Roux-en-Y gastric bypass (RYGB) profiling study.
Omental and subcutaneous adipose tissues were collected between 2000 and 2007 from patients before undergoing gastric bypass surgery at Massachusetts General Hospital. Demographic data including age, race, and gender for both the entire cohort and the selected subpopulation in our analysis are shown in Supplementary Fig. 1. Additional metabolic traits are summarized in Supplementary Fig. 3, which shows the distribution of BMI, white blood cell (WBC) count, fasting glucose, HbA1c, log10(Insulin), log10(HOMA-IR), LDL, and triglyceride, as well as a table summary of the minimum, maximum, median, and 25% and 75% percentiles for these traits. Samples were collected in RNAlater (Ambion/Applied Biosystems) stored at −80°C and shipped to Rosetta Inpharmatics Gene Expression Laboratory, Seattle, Washington, for extraction, amplification, labeling, and microarray processing (28). RNA was converted to fluorescently labeled cRNA that was then hybridized to custom 44 K DNA oligonucleotide microarrays manufactured by Agilent Technologies as described previously (29). Gene expression data were analyzed using Rosetta Resolver gene expression analysis software (version 7.0; Rosetta Biosoftware, Seattle, WA) and MATLAB (The MathWorks, Natick, MA). The expression data are available at GEO super series (Accession ID GSE24335). ANOVA analysis and Pearson correlation analysis were performed to select gene signatures that were differentially expressed between high HOMA-IR and low HOMA-IR subgroups from nondiabetic patients with HbA1c <7%. Monte Carlo simulation from a randomly permuted dataset was used to estimate and control the false discovery rate. In general, the omental and subcutaneous fat samples were taken from the same individual, but a small portion of fat tissue profiles were missing because of sample quality and array profiling quality issues. Table 1 shows the overlapping samples (i.e., samples from the same individual).
TABLE 1.
Patient pool samples used in this study

Peroxisome proliferator–activated receptor γ drug treatment profiling study.
Microarray data from a previously published study (30) were analyzed using the tools described. Adipose tissue biopsies were performed in normal lean subjects, obese insulin-resistant nondiabetic subjects, and obese insulin-resistant type 2 diabetic subjects (n = 8, 9, and 20, respectively) before and after 3-month treatment with either pioglitazone or rosiglitazone. RNA from the adipose tissue samples was profiled on Affymetrix Human Genome U133 Plus 2.0 Arrays. The array contains a total of 54,675 probe sets. A total of 74 arrays were available and used for the analysis. The dataset includes 38 profiles obtained from 19 subjects before and after pioglitazone treatment, and 36 profiles obtained from 18 subjects before and after rosiglitazone treatment. The log ratio of gene expression is calculated by subtracting the log intensity of baseline study from the log intensity of the compound treatment.
National Health and Nutrition Examination Survey data analysis.
National Health and Nutrition Examination Survey III Survey data were downloaded from the Centers for Disease Control and Prevention website http://www.cdc.gov/nchs/nhanes.htm. This survey includes >31,311 subjects and was conducted from 1988 to 1994. BMI data for subjects older than 20 years were extracted and plotted along with BMI data obtained from the obese subjects in the RYGB profiling study. HOMA-IR was calculated as fasting plasma glucose multiplied by fasting plasma insulin, with the units [(mg/dL)×(μU/mL)]/405, according to the protocol described previously (31).
Pathway enrichment analysis.
Enrichment of GO Biological Process terms in the annotation of sets of probes was tested using Gene Set Annotator in the Merck Target and Gene Information (TGI) System, which provides pathway enrichment analysis to finds gene sets in which a user-supplied set of identifiers is significantly enriched. The P value was calculated using the hypergeometric distribution, and the E value was calculated using the Bonferroni adjusted P value. Top 20 significantly enriched pathways with E value <0.005 were listed for the selected omental tissue signature genes; for the side-by-side comparison purpose, top 20 pathways ranked by E value also were listed for the selected subcutaneous tissue signature genes. Gene overlap was based on human database identification numbers. The allowed maximum and minimum gene set sizes for the pathway terms were 500 and 5, respectively.
Method and statistical algorithm used to select significantly differentially expressed genes in low and HOMA-IR analysis.
Within nondiabetic patients and patients not using medicine, we selected subjects with HbA1c ≤7. The following steps were taken. For step 1, subjects were ranked by HOMA-IR trait, then ANOVA was conducted (parametric) and Kruskal-Wallis (nonparametric) analyses were performed to select genes that were differentially expressed between the top 10% and bottom 10% of patient groups. Then, we identified the intersect genes that have P≤0.01 in both ANOVA and Kruskal-Wallis analyses. For step 2, we repeated step 1 for the top 15% and bottom 15% of patient groups. For step 3, we repeated step 1 for the top 20% and bottom 20% of patient groups. For step 4, we performed a union of the selected genes from steps 1–3. For step 5, we conducted Pearson (parametric) and Spearman (nonparametric) correlation analyses between the log ratio data of gene expression and log10 (HOMA-IR) trait, and selected genes that have correlation P≤0.01 in both Pearson and Spearman analyses. For step 6, we intersected selected genes between steps 4 and 5. The resulting differentially expressed genes for HOMA-IR trait in omental and subcutaneous tissues were 968 and 546, respectively. For step 7, we conducted Monte Carlo simulation, randomly permuted HOMA-IR trait, and then repeated steps 1–6 to determine the number of selected false-positive genes using the same statistical approaches. Finally, for step 8, we repeated step 7 250 times using the median of the number of selected false-positive genes to calculate the false discovery rate.
The numbers of false-positive genes selected by the described Monte Carlo process from the omental and subcutaneous tissue profiles were 94 and 68, respectively; therefore, the false discovery rates for these high and low HOMA-IR gene selections were 9.7% and 12.5% for the omental and subcutaneous tissues, respectively.
We used the Rosetta custom Agilent 44 K array. The array contains multiple probes for some genes. In the gene selection step, we treated each probe independently and did not intend to combine different probes for the same gene because we believed that different probe design for the same gene constitutes a systemic, instead of random, bias in detecting the gene expression level. Only in the pathway analysis step did we combine multiple probes for the same genes and we counted the gene only once in the enrichment test.
Algorithm of unsupervised clustering.
We used the hierarchical agglomerative clustering for the algorithm of unsupervised clustering figure. The basic algorithm was performed as follows: 1) we computed the distance matrix of the genes by using log10 ratio data, and we computed the similarity function of 1 − cosine correlation; 2) we amalgamated the data in the distance matrix by fusing a gene or groups of genes that were most similar, and the average link was used to calculate the distance from a cluster to all remaining unclustered points; and 3) we computed the dendrogram (returning a tree structure). The tree is a top-down collection of tree nodes such that the root node has a distance of 0.0. We believe that this unsupervised clustering algorithm is ideal to build a cluster hierarchy among the hidden data structure, and thus reveal the patterns of coexpression among selected signature genes.
RESULTS
A total of 835 omental and 697 subcutaneous fat samples were profiled using whole-genome microarrays to determine gene transcription levels in samples from obese individuals undergoing bariatric surgery. To test hypotheses regarding insulin sensitivity in this obese cohort, we excluded those patients with type 2 diabetes mellitus, as defined by their physician’s diagnosis, those with plasma HbA1c levels >7.0, or those who were using diabetes medication. We also excluded patients using long-term medications such as statins to minimize pharmacological effects on gene expression. As a result of this filtering process, the final sample set consisted of 387 omental and 323 subcutaneous fat expression profiles (Table 1). The distribution of the BMI of the studied cohort compared with the BMI of National Health and Nutrition Examination Survey III survey is shown in Fig. 1A. In general, a wide distribution of HOMA-IR and HbA1c was observed across the entire cohort, as shown in Fig. 1B, and particularly within the nondiabetic subgroup not using medication there was a broad range of HOMA-IR, highlighting the existence of both extremely insulin-sensitive and insulin-resistant obese individuals. Therefore, we aimed to determine whether gene expression patterns could be identified that correlate with the profound variability in insulin sensitivity in obese individuals, independent of BMI.
FIG. 1.

A: The BMI distribution curve (magenta) for patients who were involved in the RYGB profiling experiments, and the BMI distribution curve (gray) for the general population in National Health and Nutrition Examination Survey (NHANES) III survey. B: A dot plot comparing HOMA-IR trait vs. HbA1c trait for the patients who were involved in our profiling studies (nondiabetic, did not use any medicine, and had HbA1c <7%; gray). The red dots represent patients who had higher HOMA-IR level (top 15%), the blue dots represent patients who had lower HOMA-IR level (bottom 15%) among the patients included in the profiling analysis. C: A heat map showing the one-dimensional histogram of the 968 selected omental genes that differentiate between high and low HOMA-IR levels in the unsupervised analysis. Magenta in the heat map indicates upregulation and cyan indicates downregulation for the individual patient sample compared with the pool of all patient samples. D: A heat map showing the one-dimensional histogram of the 546 selected subcutaneous genes that differentiate between high and low HOMA-IR levels in the unsupervised analysis. The methodology used for calculating the algorithms for unsupervised clustering can be found in the Supplementary Materials. (A high-quality digital representation of this figure is available in the online issue.)
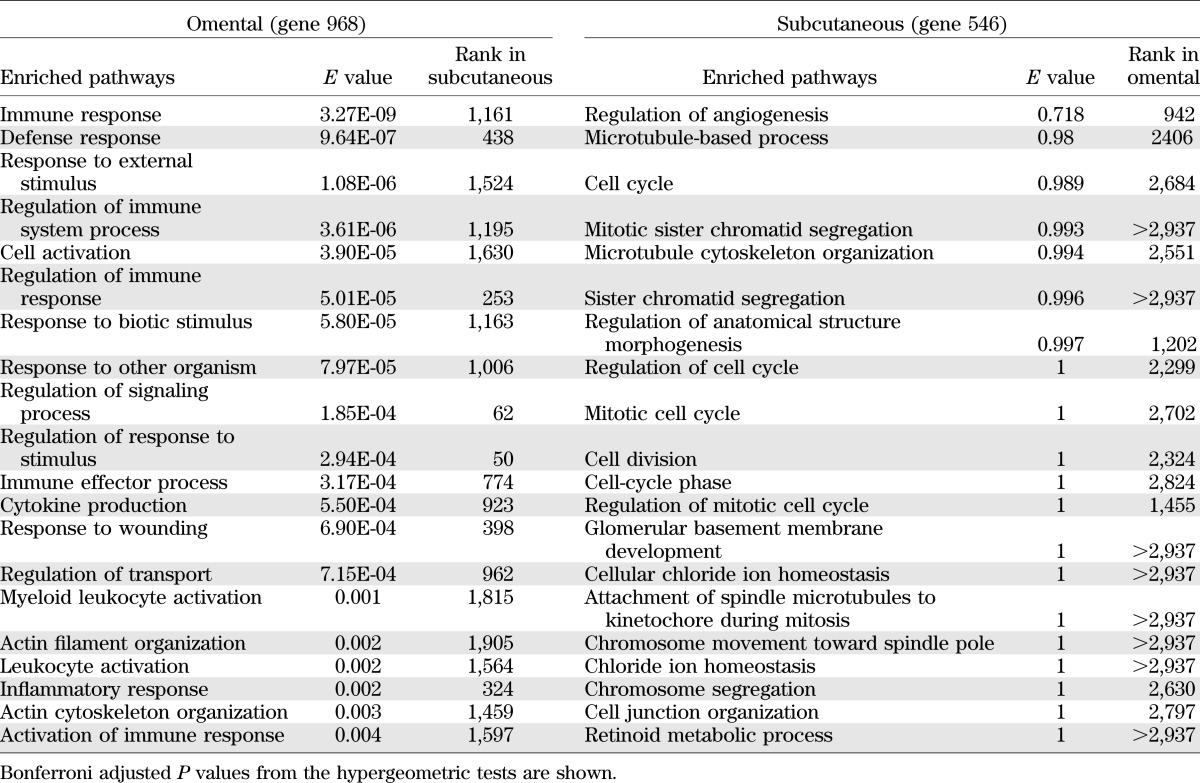
We conducted correlation analyses in the nondiabetic nonmedicated subgroup to identify genes displaying significant association with HOMA-IR. In addition, we conducted ANOVA analyses in which we selected the top and bottom 15 percentiles based on HOMA-IR and directly compared these two groups for differentiated expression patterns (Fig. 1C, D). It is worth noting that the HOMA-IR of the bottom 15 percentile in our obese insulin-sensitive subpopulation is similar to the HOMA-IR in a lean nondiabetic general population cohort (HOMA-IR, 2.8; interquartile range, 1.55) (32). The heat maps revealed hierarchical clustering and significant differentiation in gene expression across the HOMA-IR spectrum in both subcutaneous tissue and omental tissue, implicating both adipose depots as key phenotypic effectors of insulin sensitivity in obese individuals. Next, we conducted pathway enrichment analyses comprising the subgroup significantly associated with insulin sensitivity in omental (968 genes) and subcutaneous (546 genes) tissue samples. Description of these genes is presented in Supplementary Table 1. The rank order of pathways that were most significantly represented by the differential gene expression data are listed in Table 2. In omental tissue, inflammatory and immune-related pathway annotations were highly enriched in the gene set that showed significant regulation between extremes of insulin sensitivity. In contrast, in subcutaneous fat, we did not identify any significant pathway enrichment based on the 546 differentially expressed genes. Correlation analysis of HOMA-IR and the demographic traits, including age, race, and gender, showed no significant correlation (Supplementary Fig. 2). We realize that the lack of correlation between these factors and HOMA-IR does not preclude a correlation in expression of some HOMA-IR correlated transcripts. Hence, to strengthen our analysis, our gene expression data have been normalized by multiple steps, including the gender, age, and ethnic groups as described (28).
TABLE 2.
Listing of the top-ranked GO Biological Process terms from the enrichment analysis of the selected 968 omental genes and for the 546 selected subcutaneous genes
Progression from the unsupervised analysis to a semi-supervised approach enabled better resolution of the role of inflammatory and immune pathway regulation in adipose tissue with respect to whole-body insulin sensitivity. A set of 191 unique genes (270 probes) that have been associated with immune function were obtained from Ingenuity (Ingenuity Systems, www.ingenuity.com) and were examined by comparing relative gene expression levels in omental or subcutaneous fat between those individuals with high HOMA-IR and those with low HOMA-IR. In querying this integrated gene set, it was clear that the immune response pathway was downregulated in both fat depots of insulin-sensitive compared with insulin-resistant obese individuals, but that the signal was significantly more associated with insulin sensitivity in the omental versus subcutaneous fat (Fig. 2A). To better-qualify this observation, we leveraged a second gene expression data set from a separate cohort that provided relative gene transcription levels in subcutaneous fat from individuals who had undergone thiazoladinedione (TZD) treatment for 3 months (30). TZDs are a class of insulin-sensitizer drug that have been shown to robustly improve HOMA-IR in humans (33). As expected, the immune response signature showed an overall reduction in expression level, consistent with the inverse response observed in the insulin resistant state. When broken-down to individual genes, the immune response gene set showed a highly significant inverse correlation between the two cohorts, consistent with the directionality of HOMA-IR response in these separate studies (parametric Pearson correlation; correlation degree, −0.60071; P = 6.4e-20) (Fig. 2B). This degree of negative correlation at the level of individual genes indicated that immune response was oppositely regulated between insulin-resistant patients with high HOMA-IR level and subjects with improved insulin sensitivity after TZD treatment, further validating the observation that the majority of known immune genes were highly associated with the level of insulin resistance.
FIG. 2.

A: Heat map showing the expression of the known immune function genes in the omental and subcutaneous tissues of patients with high vs. low HOMA-IR levels in the RYGB profiling experiments and in the subcutaneous tissues of a different cohort of diabetic and nondiabetic subjects who received pioglitazone and rosiglitazone treatments in the peroxisome proliferator–activated receptor γ (PPARγ) profiling experiment. The data shown are the average of log10 of the ratio of gene expression between the high HOMA-IR patient group and the low HOMA-IR patient group for RYGB cohort, and the average of log10 of the ratio of gene expression for patients receiving PPARγ treatments and without PPARγ treatment for the PPARγ study cohort. B: Dot plot analysis showing the comparison of correlation coefficients between gene expression and HOMA-IR level in the RYGB profiling experiments vs. the correlation coefficients between gene expression and HOMA-IR level in the PPARγ profiling experiment for the known immune function genes. The magenta line is the least-squares fitted line for the comparison dot plot. C: Bar charts showing the evenness of the distribution of correlation coefficients for the expression of immune genes and WBCs in omental tissue (left, green) and in subcutaneous tissue (right, blue). The number of genes in each bin in the raw histogram is divided by the total number of genes in all bins to give the normalized histogram. The red curves are the distribution of correlation coefficients for all 44 K array genes with WBCs. (A high-quality digital representation of this figure is available in the online issue.)
Elevated WBC count is a marker of inflammation and has been reported to associate with insulin resistance and development of type 2 diabetes mellitus (34). We assessed whether specific adipose depots expressed an association between WBC levels and expression of genes known to be involved in immune response. Shown in Fig. 2C are normalized distribution charts for the correlation coefficients between expression of the known immune genes and WBC count. In omental tissue, 225 out of 270 probes for the known immune genes had a positive correlation with the WBC count, whereas 45 out of 270 probes had a negative correlation. The P value for this uneven distribution from the sign test was 1e−27, indicating that the majority of the known immune genes were associated with the high WBC in obese individuals with elevated HOMA-IR. In subcutaneous tissue, 185 out of 270 probes had a positive correlation with the WBC and 85 out of 270 probes had a negative correlation, and the degree of unevenness was significant with P = 1e−9. In conclusion, the majority of the known immune genes in both omental and subcutaneous tissues were overexpressed and positively associated with WBC count, although the degree of association between gene expression and WBC counts was much stronger in omental tissue than in subcutaneous tissue. However, we observed little correlation between WBC and HOMA-IR traits among the entire cohort in this dataset (Supplementary Fig. 4). This highlights the complex relationships among WBC counts, HOMA-IR, and immune genes, and supports the possibility that the changes in gene expression are not a reflection of changes in cell number but are more likely a reflection of changes in gene expression within each cell.
Finally, we defined small gene sets that relate to specific cell functions or cell phenotypes involved in the adipose inflammation response triggered by obesity (35). These subcellular signatures then were examined for association with insulin sensitivity. Notably, signatures related to macrophage cells were clearly upregulated as a function of high HOMA-IR, whereas there was no clear trend observed in mast cell signatures in the omental or subcutaneous adipose depots, but T-cell signatures were upregulated mainly in the omental depot (Fig. 3A). Concordantly, the macrophage signatures trended in the opposite direction in subcutaneous adipose tissue from subjects treated with TZDs, consistent with improved insulin sensitivity and lower HOMA-IR. Gene sets related to mitochondrial functions of β-oxidation and the tricarboxylic acid (TCA) cycle were of specific interest with respect to implications of metabolic status of adipose tissue on insulin sensitivity. In both omental and subcutaneous fat samples, there was a clear trend toward upregulation of mitochondrial function in individuals with low HOMA-IR, and the same trend was observed in individuals after treatment with TZDs. These data indicate that insulin sensitivity associates with increased levels of cellular metabolism in adipose tissue in obese individuals and suggest that increased mitochondrial function in adipocytes, along with reduced macrophage levels in fat depots, is tightly associated with improved insulin sensitivity. This potential link was further supported by distribution plots that revealed a positive correlation of inflammatory genes with WBC count and a significantly negative correlation of mitochondrial gene expression with WBC count (Fig. 3B). The fact that we did not identify the genes in mitochondrial function in our pathway analysis in Supplementary Table 1 might highlight one of the intrinsic limitations in the methodologies used in pathway analysis. These pathway methodologies do not allow differentiation of each pathway based on individual genes in the pathway (i.e., treatment of all genes equally without regard to how important each gene is in a particular pathway or where it is on the hierarchy). Moreover, pathway analysis depends on certain genes in a pathway and not on the magnitude of change in gene expression within that pathway. This is the reason we decided to use a combination of unsupervised, semisupervised, and targeted analyses in our analysis to better reveal the important relationships between inflammation and mitochondrial function and insulin resistance.
FIG. 3.

A: Heat map showing the expression of the literature-curated inflammation and mitochondrial function genes in the RYGB and peroxisome proliferator–activated receptor γ (PPARγ) profiling experiments. As in Fig. 2, the data shown are the average of log10 of the ratio of gene expression between the high HOMA-IR patient group and the low HOMA-IR patient group for RYGB cohort, and the average of log10 of the ratio of gene expression for patients receiving PPARγ treatments and those without PPARγ treatment for the PPARγ study cohort. B: Bar charts showing the evenness of the distribution of correlation coefficients for the relationships between expression of inflammation genes (left) and WBC count, and for the relationships between expression of mitochondrial function genes (right) and WBC count. The number of genes in each bin in the raw histogram is divided by the total number of genes in all bins to give the normalized histogram. The red curves are the distribution of correlation coefficients for the relationships between expression of all 44 K array genes and WBC count. (A high-quality digital representation of this figure is available in the online issue.)
DISCUSSION
Our findings demonstrate that inflammation of omental adipose tissue is strongly associated with insulin resistance, in the context of obesity, in a morbidly obese population. In contrast to the upregulation of the inflammation signature, genes related to β-oxidation and the citric acid cycle were underexpressed in the insulin-resistant state. In a separate population, these gene expression changes were reversed after TZD treatment, implicating these pathways in the mechanism of action for these insulin-sensitizing antidiabetic drugs. Our findings are consistent with a recently published report showing an increase in TCA cycle and β-oxidation gene signatures and a decrease in inflammation signatures correlating with insulin sensitivity before and after TZD treatments, which adds further validation to our findings (30).
Obesity is now recognized as a state of chronic low-grade systemic inflammation that may play a role in the development of insulin resistance with a central role for adipose tissue macrophages (18,19,36). Here, we confirm that macrophage gene signature is upregulated in the insulin-resistant state. Interestingly, signatures of both proinflammatory M1 as well as anti-inflammatory M2 macrophages are upregulated in contrast to what has been observed in animal studies (37). Moreover, gene signatures of T cells were upregulated with no difference in mast cell signature (38,39). It is not clear whether the difference in the inflammatory signature profile is attributable to changes in gene expression or attributable to a difference in immune cell recruitment to the adipose tissue, which will be the subject of a future study. Moreover, the divergence in the gene signature changes between pioglitazone and rosiglitazone, particularly in the genes representing the TCA cycle, is worth noting. This cannot be explained completely by the fact that that pioglitazone is a weaker peroxisome proliferator–activated receptor γ agonist, given that this phenomenon is not obvious in the other gene signatures (40). Deeper analysis of these differences might help us understand the triggers for the adverse events associated with the different TZD treatments.
It is difficult to determine a causal relationship between adipose tissue inflammation and insulin resistance from the current study. However, it is known that higher total leukocyte counts precede and predict the incident risk of type 2 diabetes mellitus (34). More recently, studies have shown that insulin resistance in mice with differential susceptibility to diabetes is preceded by differences in the inflammatory response of adipose tissue, emphasizing the notion that inflammation of adipose tissue might play a role in the etiology of insulin resistance independent of body weight (41). This suggests that targeting inflammatory pathways might prevent the development of insulin resistance and diabetes. A recent study shows that patients using anti-inflammatory drugs against rheumatoid arthritis or psoriasis had a significantly lower risk of development of diabetes (42). Rodent models also have shown that diet-induced insulin resistance is blunted when adipose tissue inflammation is blocked (36,43). Our data implicate a stronger role for visceral fat inflammation versus subcutaneous fat inflammation in the etiology of insulin resistance, which is consistent with studies that show that these adipose tissue depots have unique inflammatory patterns, and which supports the notion that the omental depot is a stronger contributor to insulin resistance (44). Finally, several studies have suggested that depot-specific adipocyte size may be linked to inflammation and can be a major factor driving insulin resistance (22,25,45,46). Increased adipocyte size of omental adipose tissue was associated with insulin resistance and, among moderately obese individuals, an increased proportion of small adipose cells is associated with inflammation in subcutaneous adipose tissue, which may reflect impaired adipogenesis or terminal differentiation (22,25). The latter notion was elegantly demonstrated in a recent article that shows that genetic predisposition for type 2 diabetes mellitus, but not obesity, is associated with an impaired ability to recruit new adipose cells to store excess lipids in the subcutaneous adipose tissue, thereby promoting ectopic lipid deposition (47). Studying the relationship among insulin resistance, inflammation, and adipocyte size in the obese subjects in our study will help highlight the interplay among these factors and will be the topic of future studies.
The results of our study highlight a different subpopulation of obese individuals who display an inflammatory signature in their omental adipose tissue, which might contribute to their state of insulin resistance. It is possible that individuals with higher basal inflammatory signature, before becoming obese, are more at risk for development of insulin resistance, diabetes, and other metabolic diseases. Understanding the mechanisms and pathways that lead to these differences will equip us with important targets to help stem the tide of such a debilitating disease.
Supplementary Material
ACKNOWLEDGMENTS
M.Q., Y.T., R.D., D.M.G., G.H., W.Z., and D.M.K. are associated with Merck Research Laboratories. No other potential conflicts of interest relevant to this article were reported.
M.Q., Y.T., R.D., D.M.G., W.Z., and D.D.S. researched the data. M.Q., Y.T., and D.M.K. wrote the manuscript. G.H., J.M.O., and L.M.K. reviewed and edited the manuscript and contributed to the discussion. D.M.K. is the guarantor of this work, had full access to all the data, and takes full responsibility for the integrity of data and the accuracy of data analysis.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db12-0399/-/DC1.
REFERENCES
- 1.Li Z, Bowerman S, Heber D. Health ramifications of the obesity epidemic. Surg Clin North Am 2005;85:681–701 [DOI] [PubMed] [Google Scholar]
- 2.Olshansky SJ. Projecting the future of U.S. health and longevity. Health Aff (Millwood) 2005;24(Suppl 2):W5R86–89 [DOI] [PubMed]
- 3.American Diabetes Association. Diabetes statistics. 2011. Available from http://www.diabetes.org/diabetes-statistics/prevalence.jsp Accessed 21 August 2011
- 4.Zimmet P, Alberti KG, Shaw J. Global and societal implications of the diabetes epidemic. Nature 2001;414:782–787 [DOI] [PubMed] [Google Scholar]
- 5.Flier JS. Obesity wars: molecular progress confronts an expanding epidemic. Cell 2004;116:337–350 [DOI] [PubMed] [Google Scholar]
- 6.Kahn BB, Flier JS. Obesity and insulin resistance. J Clin Invest 2000;106:473–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qatanani M, Lazar MA. Mechanisms of obesity-associated insulin resistance: many choices on the menu. Genes Dev 2007;21:1443–1455 [DOI] [PubMed] [Google Scholar]
- 8.Goldfine AB, Fonseca V, Shoelson SE. Therapeutic approaches to target inflammation in type 2 diabetes. Clin Chem 2011;57:162–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sims EA, Danforth E, Jr, Horton ES, Bray GA, Glennon JA, Salans LB. Endocrine and metabolic effects of experimental obesity in man. Recent Prog Horm Res 1973;29:457–496 [DOI] [PubMed] [Google Scholar]
- 10.Freidenberg GR, Reichart D, Olefsky JM, Henry RR. Reversibility of defective adipocyte insulin receptor kinase activity in non-insulin-dependent diabetes mellitus. Effect of weight loss. J Clin Invest 1988;82:1398–1406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bak JF, Møller N, Schmitz O, Saaek A, Pedersen O. In vivo insulin action and muscle glycogen synthase activity in type 2 (non-insulin-dependent) diabetes mellitus: effects of diet treatment. Diabetologia 1992;35:777–784 [DOI] [PubMed] [Google Scholar]
- 12.McLaughlin T, Abbasi F, Lamendola C, Reaven G. Heterogeneity in the prevalence of risk factors for cardiovascular disease and type 2 diabetes mellitus in obese individuals: effect of differences in insulin sensitivity. Arch Intern Med 2007;167:642–648 [DOI] [PubMed] [Google Scholar]
- 13.Reaven G. All obese individuals are not created equal: insulin resistance is the major determinant of cardiovascular disease in overweight/obese individuals. Diab Vasc Dis Res 2005;2:105–112 [DOI] [PubMed] [Google Scholar]
- 14.Sims EA. Are there persons who are obese, but metabolically healthy? Metabolism 2001;50:1499–1504 [DOI] [PubMed] [Google Scholar]
- 15.Hotamisligil GS. Inflammation and metabolic disorders. Nature 2006;444:860–867 [DOI] [PubMed] [Google Scholar]
- 16.Rosen ED, Spiegelman BM. Adipocytes as regulators of energy balance and glucose homeostasis. Nature 2006;444:847–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shoelson SE, Herrero L, Naaz A. Obesity, inflammation, and insulin resistance. Gastroenterology 2007;132:2169–2180 [DOI] [PubMed] [Google Scholar]
- 18.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW., Jr Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 2003;112:1796–1808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu H, Barnes GT, Yang Q, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest 2003;112:1821–1830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dahlman I, Kaaman M, Olsson T, et al. A unique role of monocyte chemoattractant protein 1 among chemokines in adipose tissue of obese subjects. J Clin Endocrinol Metab 2005;90:5834–5840 [DOI] [PubMed] [Google Scholar]
- 21.Dolinková M, Dostálová I, Lacinová Z, et al. The endocrine profile of subcutaneous and visceral adipose tissue of obese patients. Mol Cell Endocrinol 2008;291:63–70 [DOI] [PubMed] [Google Scholar]
- 22.McLaughlin T, Sherman A, Tsao P, et al. Enhanced proportion of small adipose cells in insulin-resistant vs insulin-sensitive obese individuals implicates impaired adipogenesis. Diabetologia 2007;50:1707–1715 [DOI] [PubMed] [Google Scholar]
- 23.McLaughlin T, Deng A, Gonzales O, et al. Insulin resistance is associated with a modest increase in inflammation in subcutaneous adipose tissue of moderately obese women. Diabetologia 2008;51:2303–2308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu A, McLaughlin T, Liu T, et al. Differential intra-abdominal adipose tissue profiling in obese, insulin-resistant women. Obes Surg 2009;19:1564–1573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hardy OT, Perugini RA, Nicoloro SM, et al. Body mass index-independent inflammation in omental adipose tissue associated with insulin resistance in morbid obesity. Surg Obes Relat Dis 2011;7:60–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Elbein SC, Kern PA, Rasouli N, Yao-Borengasser A, Sharma NK, Das SK. Global gene expression profiles of subcutaneous adipose and muscle from glucose-tolerant, insulin-sensitive, and insulin-resistant individuals matched for BMI. Diabetes 2011;60:1019–1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rasouli N, Kern PA, Elbein SC, Sharma NK, Das SK. Improved insulin sensitivity after treatment with PPARγ and PPARα ligands is mediated by genetically modulated transcripts. Pharmacogenet Genomics 2012;22:484–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Greenawalt DM, Dobrin R, Chudin E, et al. A survey of the genetics of stomach, liver, and adipose gene expression from a morbidly obese cohort. Genome Res 2011;21:1008–1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hughes TR, Mao M, Jones AR, et al. Expression profiling using microarrays fabricated by an ink-jet oligonucleotide synthesizer. Nat Biotechnol 2001;19:342–347 [DOI] [PubMed] [Google Scholar]
- 30.Sears DD, Hsiao G, Hsiao A, et al. Mechanisms of human insulin resistance and thiazolidinedione-mediated insulin sensitization. Proc Natl Acad Sci USA 2009;106:18745–18750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 1985;28:412–419 [DOI] [PubMed] [Google Scholar]
- 32.Alssema M, Dekker JM, Nijpels G, Stehouwer CD, Bouter LM, Heine RJ, Hoorn Study Proinsulin concentration is an independent predictor of all-cause and cardiovascular mortality: an 11-year follow-up of the Hoorn Study. Diabetes Care 2005;28:860–865 [DOI] [PubMed] [Google Scholar]
- 33.Bhatia V, Viswanathan P. Insulin resistance and PPAR insulin sensitizers. Curr Opin Investig Drugs 2006;7:891–897 [PubMed] [Google Scholar]
- 34.Vozarova B, Weyer C, Lindsay RS, Pratley RE, Bogardus C, Tataranni PA. High white blood cell count is associated with a worsening of insulin sensitivity and predicts the development of type 2 diabetes. Diabetes 2002;51:455–461 [DOI] [PubMed] [Google Scholar]
- 35.Anderson EK, Gutierrez DA, Hasty AH. Adipose tissue recruitment of leukocytes. Curr Opin Lipidol 2010;21:172–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annu Rev Immunol 2011;29:415–445 [DOI] [PubMed] [Google Scholar]
- 37.Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest 2007;117:175–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu J, Divoux A, Sun J, et al. Genetic deficiency and pharmacological stabilization of mast cells reduce diet-induced obesity and diabetes in mice. Nat Med 2009;15:940–945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Feuerer M, Herrero L, Cipolletta D, et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med 2009;15:930–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Colca JR, McDonald WG, Waldon DJ, et al. Identification of a novel mitochondrial protein (“mitoNEET”) cross-linked specifically by a thiazolidinedione photoprobe. Am J Physiol Endocrinol Metab 2004;286:E252–E260 [DOI] [PubMed] [Google Scholar]
- 41.Mori MA, Liu M, Bezy O, et al. A systems biology approach identifies inflammatory abnormalities between mouse strains prior to development of metabolic disease. Diabetes 2010;59:2960–2971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Solomon DH, Massarotti E, Garg R, Liu J, Canning C, Schneeweiss S. Association between disease-modifying antirheumatic drugs and diabetes risk in patients with rheumatoid arthritis and psoriasis. JAMA 2011;305:2525–2531 [DOI] [PubMed] [Google Scholar]
- 43.Fleischman A, Shoelson SE, Bernier R, Goldfine AB. Salsalate improves glycemia and inflammatory parameters in obese young adults. Diabetes Care 2008;31:289–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Alvehus M, Burén J, Sjöström M, Goedecke J, Olsson T. The human visceral fat depot has a unique inflammatory profile. Obesity (Silver Spring) 2010;18:879–883 [DOI] [PubMed] [Google Scholar]
- 45.Arner P, Bernard S, Salehpour M, et al. Dynamics of human adipose lipid turnover in health and metabolic disease. Nature 2011;478:110–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hoffstedt J, Arner E, Wahrenberg H, et al. Regional impact of adipose tissue morphology on the metabolic profile in morbid obesity. Diabetologia 2010;53:2496–2503 [DOI] [PubMed] [Google Scholar]
- 47.Arner P, Arner E, Hammarstedt A, Smith U. Genetic predisposition for type 2 diabetes, but not for overweight/obesity, is associated with a restricted adipogenesis. PLoS One 2011;6:e18284. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.