

Abstract
Amino, bromo, nitro, and naphthalene functionalized UiO-66 metal–organic frameworks have been synthesized through reticular chemistry. UiO-66–NH2 is shown to be suitable for postsynthetic modification with a variety of anhydrides to generate new, functionalized frameworks.
Metal–organic frameworks (MOFs), which are comprised of metal ions or metal clusters connected by organic ligands,1–3 have been increasingly studied in the past decade for a variety of applications. Through the careful choice of organic ligands and metal precursors, a vast number of topologies can be rationally designed and synthesized.1,4,5 Indeed, Yaghi and co-workers have previously demonstrated the isoreticular synthesis of topologically identical frameworks with a series of related organic ligands.6 Due to their high porosity, these hybrid materials have been extensively studied for gas storage,7–12 separation,13,14 catalysis,15 and drug delivery16,17 applications. However, a major limitation of MOFs, especially those based on zinc-carboxylate secondary building units (SBUs), is their low chemical stability to atmospheric moisture and protic media.18
Several recent studies have shown that specialized functionalities can be introduced into porous MOFs through a postsynthetic modification (PSM) approach.19 However, access to novel functionalities through PSM and their subsequent exploitation in new MOF applications are still limited by the inherent chemical stability of the parent framework. Because of this limitation, utilization of PSM in more chemically robust systems has been increasingly investigated.20 Lillerud et al. synthesized the first example of a zirconium(IV) dicarboxylate porous material (UiO-66). Owing to the highly oxophilic nature of zirconium(IV), the Zr6-cluster SBU formed in these MOFs makes these materials very resistant towards various solvents and elevated pressures.21 Recently, Cr(CO)3 was successfully grafted to the arene moieties of UiO-66 through thermal decomposition in a postsynthetic fashion.22 Subsequent photolysis of the Cr(CO)3 modified material with UV light in an N2 atmosphere led to photoinduced substitution of one of the CO ligands with an N2 molecule. However, prolonged exposure to UV light (>30 min) led to decomposition of the arene–Cr(CO)3 species. In an effort to expand the number of chemically robust materials available for covalent PSM, we sought to incorporate a variety of organic functionalities into the UiO-66 framework through a combination of reticular chemistry (Scheme 1) and PSM (Scheme 2). The synthesis of –NH2, –Br, –NO2, and naphthalene-substituted UiO-66 analogues is presented. With respect to PSM, we described the synthesis and modification of an amine functionalized UiO-66 analogue. Overall, the UiO-66 framework is found to be a robust motif that is highly amenable to isoreticular functionalization and modification.
Scheme 1.
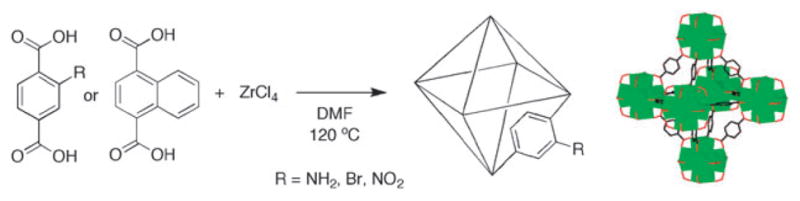
Synthesis of isoreticular UiO-66 functionalized analogues UiO-66–NH2, UiO-66–Br, UiO-66–NO2 (R = NH2, Br, or NO2), and UiO-66-1,4–Naph. The UiO-66 framework with its Zr6O6 cuboctahedron SBU (green) is schematically represented as an octahedron.
Scheme 2.

Postsynthetic modification of UiO-66–NH2 with various anhydrides.
The syntheses of –NH2, –Br, –NO2, and naphthalene functionalized UiO-66 analogues were performed using a slightly modified procedure of Lillerud et al.21 To obtain these materials, equal molar amounts of ZrCl4 and terephthalic acid precursors (0.35 mmol) were combined in DMF (~0.09 M) and heated.‡ Under these conditions all of the functionalized terephthalic acid precursors led to materials possessing powder X-ray diffraction (PXRD) patterns that were remarkably consistent with that reported for the UiO-66 topology (Fig. 1).21,23 The thermal stability of the UiO-66 analogues varied by functionality. While the bromide and naphthalene analogues UiO-66–Br and UiO-66-1,4–Naph show thermal stability comparable to the parent UiO-66 material, the amino and nitro functionalized analogues UiO-66–NH2 and UiO-66–NO2 showed significant weight losses (25% to 30%) at ~400 °C, presumably due to ligand decomposition (Fig. 2).
Fig. 1.

PXRD patterns of UiO-66 derivative materials.
Fig. 2.

Thermogravimetric analysis of UiO-66 (black), UiO-66–NH2 (red), UiO-66–Br (brown), UiO-66–NO2 (green), UiO-66-1,4–Naph (purple), UiO-66–AM1 (orange), UiO-66–AM4 (navy blue), UiO-66–AM7 (cyan), and UiO-66–AMMal (gray).
The covalent postsynthetic modification of UiO-66–NH2 was explored using a series of alkyl anhydrides to produce amide modified frameworks designated UiO-66–AM (Scheme 2). In a typical reaction UiO-66–NH2 (~60 mg, 0.2 mmol-NH2) was suspended in 2 mL of CHCl3, treated with one equivalent of anhydride, and heated at 55 °C for 24 h, followed by extensive rinsing with methanol to extract by-products from the porous solids.‡ As described in our earlier studies, modification was confirmed by nuclear magnetic resonance (NMR) spectroscopy.20,24
Samples of PSM treated UiO-66–NH2 materials were digested with HF and [D6]DMSO for examination by 1H NMR spectroscopy. Digestion of unmodified UiO-66–NH2 predominantly displayed resonances associated with 2-amino-1,4-benzene dicarboxylic acid, but some minor impurities were also observed in the aromatic region. These impurities appear to be associated with the amino-functionalized benzene-dicarboxylate (BDC) ligand, as they were not observed upon digestion of UiO-66, UiO-66–Br, UiO-66–NO2, or UiO-66–Naph (Fig. S1, ESI†). Nonetheless, the 1H NMR spectra of modified samples showed a distinct downfield shift of the aromatic proton resonances associated with the BDC ligand (Fig. 3). The percent conversion of the amine groups in the framework to amides was determined by comparing the relative integrated areas of the aromatic resonances (corresponding to the C-3 position of the BDC ring) between the modified and unmodified BDC ligands. The results are summarized in Table 1. As previously described for other MOF systems, conversion to the amide products decreased with increasing size (chain length) of the anhydride with yields of 88%, 49%, and 25% for acetic (–AM1), valeric (–AM4), and octanoic anhydride (–AM7), respectively. Additionally, reaction with the maleic anhydride produced UiO-66–AMMal with 25% conversion. The latter modification introduces a free carboxylate group into the framework, which could not be readily obtained by direct solvothermal synthesis and has been shown to generate acidic materials with catalytic properties.25,26
Fig. 3.

1HNMR spectra of digested UiO-66 modified samples. Black circles indicate unmodified NH2–BDC and red squares are from modified BDC ligands.
Table 1.
Conversion of UiO-66–NH2 to UiO-66–AM derivatives using different anhydrides
| UiO-66– | Anhydride | % Conversion | STDa |
|---|---|---|---|
| AM1 | Acetic | 88 | ±6 |
| AM4 | Valeric | 49 | ±7 |
| AM7 | Octanoic | 25 | ±6 |
| AMMal | Maleic | 25 | ±5 |
Percent conversion values are averaged from at least three independent experiments.
Additional evidence for modification was obtained via infrared spectroscopy (IR) on UiO-66–AM1, UiO-66–AM4, UiO-66–AM7, and UiO-66–AMMal, which displayed a weak, but reproducible stretch around 1700 cm−1 associated with the carbonyl moieties of the amide functionality (Fig. S2 and S3, ESI†). The presence of the amide substituents was also confirmed by electrospray ionization mass spectrometry (ESI-MS) of digested modified samples (Fig. S4–S7, ESI†). Consistent with the UiO-66–NH2 precursor framework, the amide-functionalized frameworks UiO-66–AM1, UiO-66–AM4, UiO-66–AM7, and UiO-66–AMMal show somewhat decreased thermal stabilities (Fig. 2) relative to UiO-66.
The effects of functionalization on the porosity of the UiO-66 systems were investigated by measuring the Brunauer–Emmett–Teller (BET) surface areas with N2 adsorption at 77 K. The results are summarized in Table 2. As shown by the surface measurements, the amino moieties in UiO-66–NH2 do not show much effect on the porosity of the UiO-66 system; however, increasingly larger functional groups lead to diminished porosities (e.g. –Br, –NO2, –Naph). Similarly, longer aliphatic chains introduced through PSM progressively reduced the inherent porosity of UiO-66–AM compounds.
Table 2.
BET surface areas of UiO-66 functionalized systems (m2 g−1)
| BET surface areaa | Langmuir surface areaa | |
|---|---|---|
| UiO-66 | 1110 | 1311 |
| UiO-66–NH2 | 1112 | 1313 |
| UiO-66–Br | 851 | 1004 |
| UiO-66–NO2 | 756 | 893 |
| UiO-66-1,4–Naph | 615 | 732 |
| UiO-66–AM1 | 818 | 965 |
| UiO-66–AM4 | 717 | 847 |
| UiO-66–AM7 | 646 | 762 |
| UiO-66–AMMal | 814 | 962 |
The results are the average from two independent experiments.
In summary, we have synthesized and characterized –NH2, –Br, –NO2, and naphthalene functionalized UiO-66 analogues in an isoreticular manner. In comparison to UiO-66, the thermal stability of the amino and nitro functionalized UiO-66–NH2 and UiO-66–NO2 is somewhat decreased. The postsynthetic modification of UiO-66–NH2 was accomplished with aliphatic and cyclic anhydrides and verified through a combination of 1H NMR, IR, and ESI-MS analysis. The chemical stability of the UiO-66 framework and facile incorporation of functionalized or extended species, through reticular synthesis or postsynthetic modification, show that this class of porous solid can serve as a tunable, microporous scaffold for novel applications in separations, catalysis, and biotechnology.
Acknowledgments
We thank Dr Y. Su (U.C.S.D.) for performing the mass spectrometry experiments. This work was supported by U.C.S.D., the NSF (new MOF synthesis CHE-0952370; instrumentation grants CHE-9709183, CHE-0116662 and CHE-0741968), and the Department of Energy (DE-FG02-08ER46519, MOF modification for gas sorption, PXRD instrumentation). S.J.G. was supported by a supplement to NCI grant 3R01 CA095298-07S1.
Footnotes
Electronic supplementary information (ESI) available: Synthetic details, characterization of all compounds, and details of cleavage assays. Scheme S1, Fig. S1–S10. See DOI: 10.1039/c0cc02990d
Synthesis of isoreticular UiO-66 frameworks. In a typical reaction, the functionalized terephthalic acids (0.35 mmol) along with ZrCl4 (0.35 mmol) and DMF (4 mL) were placed in a Teflon lined autoclave and heated at 120 °C for 24 h. The microcrystalline powders were then isolated by centrifugation and heated at 100 °C for 1–2 h. Residual DMF and terephthalic acid precursors were removed from microcrystalline UiO-66 (~60 mg) by methanol rinsing (3 × 15 mL), soaking for 3 d in methanol, and subsequent heating at 150 °C for 5 h.
PSM on UiO-66-NH2 framework. Activated UiO-66–NH2 (~60 mg, 0.2 mmol-NH2) was treated with a 2 mL solution of CHCl3 containing the anhydride (0.2 mmol) and heated at 55 °C for 24 h. After the reaction was complete the sample was rinsed and intermittently centrifuged with CHCl3 (3 × 15 mL) and soaked for 3 d. The CHCl3 rinsed sample was subsequently rinsed with methanol (3 × 15 mL) soaked for 3 d and dried at 150 °C for up to 2 h.
Notes and references
- 1.Yaghi OM, O’Keeffe M, Ockwig NW, Chae HK, Eddaoudi M, Kim J. Nature. 2003;423:705–714. doi: 10.1038/nature01650. [DOI] [PubMed] [Google Scholar]
- 2.Kitagawa S, Kitaura R, Noro S. Angew Chem, Int Ed. 2004;43:2334–2375. doi: 10.1002/anie.200300610. [DOI] [PubMed] [Google Scholar]
- 3.Ferey G. Chem Soc Rev. 2008;37:191–214. doi: 10.1039/b618320b. [DOI] [PubMed] [Google Scholar]
- 4.Eddaoudi M, Moler DB, Li H, Chen B, Reineke TM, O’Keeffe M, Yaghi OM. Acc Chem Res. 2001;34:319–330. doi: 10.1021/ar000034b. [DOI] [PubMed] [Google Scholar]
- 5.Ockwig NW, Delgado-Friedrichs O, O’Keeffe M, Yaghi OM. Acc Chem Res. 2005;38:176–182. doi: 10.1021/ar020022l. [DOI] [PubMed] [Google Scholar]
- 6.Eddaoudi M, Kim J, Rosi N, Vodak D, Wachter J, O’Keeffe M, Yaghi OM. Science. 2002;295:469–472. doi: 10.1126/science.1067208. [DOI] [PubMed] [Google Scholar]
- 7.Rowsell JLC, Yaghi OM. Angew Chem, Int Ed. 2005;44:4670–4679. doi: 10.1002/anie.200462786. [DOI] [PubMed] [Google Scholar]
- 8.Latroche M, Surble S, Serre C, Mellot-Draznieks C, Llewellyn PL, Lee JH, Chang JS, Jhung SH, Ferey G. Angew Chem, Int Ed. 2006;45:8227–8231. doi: 10.1002/anie.200600105. [DOI] [PubMed] [Google Scholar]
- 9.Dinca M, Dailly A, Liu Y, Brown CM, Neumann DA, Long JR. J Am Chem Soc. 2006;128:16876–16883. doi: 10.1021/ja0656853. [DOI] [PubMed] [Google Scholar]
- 10.Britt D, Tranchemontagne D, Yaghi OM. Proc Natl Acad Sci U S A. 2008;105:11623–11627. doi: 10.1073/pnas.0804900105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Furukawa H, Yaghi OM. J Am Chem Soc. 2009;131:8875–8883. doi: 10.1021/ja9015765. [DOI] [PubMed] [Google Scholar]
- 12.Ma S, Zhou HC. Chem Commun. 2010;46:44–53. doi: 10.1039/b916295j. [DOI] [PubMed] [Google Scholar]
- 13.Nuzhdin AL, Dybtsev DN, Bryliakov KP, Talsi EP, Fedin VP. J Am Chem Soc. 2007;129:12958–12959. doi: 10.1021/ja076276p. [DOI] [PubMed] [Google Scholar]
- 14.Ahmad R, Wong-Foy AG, Matzger AJ. Langmuir. 2009;25:11977–11979. doi: 10.1021/la902276a. [DOI] [PubMed] [Google Scholar]
- 15.Lee J, Farha OK, Roberts J, Scheidt KA, Nguyen ST, Hupp JT. Chem Soc Rev. 2009;38:1450–1459. doi: 10.1039/b807080f. [DOI] [PubMed] [Google Scholar]
- 16.McKinlay AC, Morris RE, Horcajada P, Ferey G, Gref R, Couvreur P, Serre C. Angew Chem, Int Ed. 2010;49:6260–6266. doi: 10.1002/anie.201000048. [DOI] [PubMed] [Google Scholar]
- 17.Taylor-Pashow KM, Della Rocca J, Xie Z, Tran S, Lin W. J Am Chem Soc. 2009;131:14261–14263. doi: 10.1021/ja906198y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaye SS, Dailly A, Yaghi OM, Long JR. J Am Chem Soc. 2007;129:14176–14177. doi: 10.1021/ja076877g. [DOI] [PubMed] [Google Scholar]
- 19.Wang Z, Cohen SM. Chem Soc Rev. 2009;38:1315–1329. doi: 10.1039/b802258p. [DOI] [PubMed] [Google Scholar]
- 20.Volkringer C, Cohen SM. Angew Chem, Int Ed. 2010;49:4644–4648. doi: 10.1002/anie.201001527. [DOI] [PubMed] [Google Scholar]
- 21.Cavka JH, Jakobsen S, Olsbye U, Guillou N, Lamberti C, Bordiga S, Lillerud KP. J Am Chem Soc. 2008;130:13850–13851. doi: 10.1021/ja8057953. [DOI] [PubMed] [Google Scholar]
- 22.Chavan S, Vitillo JG, Uddin MJ, Bonino F, Lamberti C, Groppo E, Lillerud KP, Bordiga S. Chem Mater. 2010;22:4602–4611. [Google Scholar]
- 23.Guillerm V, Gross S, Serre C, Devic T, Bauer M, Ferey G. Chem Commun. 2010;46:767–769. doi: 10.1039/b914919h. [DOI] [PubMed] [Google Scholar]
- 24.Wang Z, Tanabe KK, Cohen SM. Inorg Chem. 2009;48:296–306. doi: 10.1021/ic801837t. [DOI] [PubMed] [Google Scholar]
- 25.Garibay SJ, Wang Z, Tanabe KK, Cohen SM. Inorg Chem. 2009;48:7341–7349. doi: 10.1021/ic900796n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garibay SJ, Wang Z, Cohen SM. Inorg Chem. 2010;49:8086–8091. doi: 10.1021/ic1011549. [DOI] [PMC free article] [PubMed] [Google Scholar]