

Abstract
Previously we showed that transgenic mice expressing human HLA-DR3 gene are susceptible to PLP91-110 induced experimental autoimmune encephalomyelitis (EAE) and can serve as an animal model of multiple sclerosis (MS). HLA-DR3 mice with EAE showed increased number of CD8 T cells indicating their important role in disease pathogenesis. The role of CD8 T cells in MS, an inflammatory demyelinating disease of CNS, has been enigmatic as it has been assigned both regulatory and pathogenic roles. Therefore, to evaluate the role of CD8 T cells, we generated CD8 deficient HLA-DR3 transgenic mice (DR3.CD8-/-). Immunization with PLP91-110 led to more severe EAE in DR3.CD8-/- mice compared to HLA-DR3 mice indicating a regulatory role for CD8 T cells. Interestingly, DR3.CD8-/- mice with EAE showed decreased CNS pathology compared to DR3 mice thus suggesting a pathogenic role for CD8 T cells. We show that these two subsets of CD8 T cells can be differentiated based on the surface expression of CD122 (IL-2 Rβ chain). CD8 T cells expressing CD122 (CD8+CD122+) play a regulatory role while CD8+CD122- T cells act as a pathogenic subset. CD122 expressing CD8 T cells are the regulatory subset of CD8 T cells and regulate the encephalitogenic CD4 T cells either through direct modulation of antigen presenting cells or through the release of immuno-regulatory cytokines such as IL-10, IFNγ and TGFβ. We also showed that adoptive transfer of CD8CD122-T cells caused increased spinal cord demyelination indicating that these are pathogenic subset of CD8 T cells. Our study suggests that CD8+ T cells play both regulatory as well as pathogenic role in disease pathogenesis of EAE. A better understanding of these subsets could aid in designing novel therapy for MS patients.
Keywords: Experimental autoimmune encephalomyelitis, Multiple sclerosis. HLA class II transgenic mice, demyelination, cytokine, CD8 regulatory T cells
1.0 Introduction
Multiple sclerosis (MS) is a chronic demyelinating disease of the CNS of unknown etiology [1-3]. Collective evidence suggests that disease may result from T cell driven aberrant immune responses to a number of myelin antigens. Autoreactive T cells circulate in the periphery, and upon activation, cross the blood-brain barrier (BBB) to initiate a local inflammatory response or directly target cells inside the CNS. As a result, demyelination occurs, followed by damage to axons possibly due to the reduced capacity of oligodendrocytes to repair myelin sheaths. The demyelination process is the consequence of an inflammatory response by infiltrating CD4, CD8 T cells and macrophages in the white matter.
Previously we have shown that transgenic mice expressing MS associated HLA-DR3 gene can be used as an animal model to study role of HLA class II genes in immuno-pathogenesis of MS [4-6]. The disease was dependent on CD4 T cells as no EAE was seen in MHC class II -/- (Aβ -/-) mice. However, the disease was characterized by increase in the frequency of CD8 T cells number in both periphery as well as CNS indicating an important role of CD8 T cells in disease process. Most studies in MS and EAE have focused on the role of CD4 T cells in the pathogenesis of disease, with the underlying assumption that, like classical EAE, MS is also predominantly mediated and regulated by CD4 T cells. However, several factors point towards an important role of CD8 T cells in MS [7-10]. First, CD8 T cell clones specific for myelin antigens have been isolated from the periphery of MS patients and normal donors [7, 9]. Second, MHC class I genes are in linkage disequilibrium with the MHC class II genes associated with susceptibility to MS [11]. Third, CD8 T cells are the most common subset of T cells found in brain of MS patients [12] and are statistically associated with axonal injury. Interestingly, the MHC class I restricted CD8 T 3 cells outnumber CD4 T cells almost ten fold in MS lesion [12, 13] and are present both in the perivascular infiltrate as well as in brain parenchyma.
MS pathology is characterized by demyelinated plaques with glial scar formation in CNS [14] and CD8 T cells have the potential to kill microglia, astrocytes, oligodendrocytes and neurons [15, 16]. Myelin specific CD8 T cells have been shown to induce inflammation and demyelination in animal model of MS [17, 18]. Sun et al showed that MOG35-55 specific CD8 T cells induced severe and more destructive disease in B6 mice [18]. Similarly, MBP79-87 specific CD8 T cells clone induced CNS autoimmunity [17].
However, all CD8 T cells in MS patients are not of the effector phenotype as recent studies have shown that CD8 regulatory T cells are present in MS patients and play an important role in regulating disease [19-23]. Initial studies on the role of CD8 T cells in EAE, indicated that CD8 T cells might play a regulatory role [24, 25]. Glatiramer Acetate (GA), a FDA approved drug to treat MS, has been shown to modulate immune responses by inducing regulatory/suppressor CD8 T cells [22].
Thus, there is uncertainty regarding role of CD8 T cells in EAE/MS as it had been assigned either a regulatory or a pathogenic role. To the best of our knowledge, there are no reports so far showing regulatory as well as pathogenic role of CD8 T cells in a single model of EAE. Therefore, we undertook this study to understand if CD8 T cells can play both regulatory as well as pathogenic role in PLP91-110 induced EAE in HLA-DR3 transgenic (Tg) mice. We observed that HLA-DR3 Tg mice lacking CD8 T cells (DR3.CD8-/-) showed increased disease incidence as well as higher average clinical scores compared to CD8 sufficient DR3 Tg mice indicating a regulatory role of CD8 T cells. In contrast DR3.CD8-/- Tg mice showed less CNS pathology compared to DR3 Tg mice indicating that CD8 T cells are also important for causing CNS pathology. Based on above results, we believe that like CD4 T cells, CD8 T cells play a dual role in disease pathogenesis of EAE, with one subset playing a regulatory role whereas the other subset plays a pathologic role in CNS. We have characterized the regulatory subset as CD8+CD122+ T cells, and were FoxP3lo, FR4 lo, GITR lo, CTLA-4hi and CD103hi. We also show that CD8+CD122- subset was responsible for inducing CNS pathology.
2.0 Material and methods
2.1 Transgenic (Tg) mice
The HLA-DR3 (DRB1*0301), Tg mice were produced, as previously described [5]. To generate DR3.β2m-/-.Aβknockout mice, DR3 Tg mice, were mated with β2m deficient mice (gift of Dr. Beverly Koller, University of North Carolina, Chapel Hill, NC) [26]. Similarly, DR3 Tg mice were mated with CD8 deficient mice on C57BL/6 background (a kind gift from Dr. Tak Mak, University Health Network, University of Toronto) to produce DR3.CD8-/-.Aβ lines. These mice were intercrossed for several generations. All mice were bred and maintained in the pathogen free Immunogenetics Mouse Colony of Mayo Clinic according to National Institutes of Health and institutional guidelines.
2.2 Flow cytometry
Expression of HLA-DR, β2m and CD8 molecules on peripheral blood lymphocytes (PBLs) were analyzed by flow cytometry using monoclonal antibodies (mAbs) L227 (HLA-DR), s19.8.503 (mouse β2m) and Ly 2-53-7.6 (CD8). Surface expression of CD4 (GK1.5), CD8 (53.6.72), CD25 (PC61), CD28 (37.51), CD11c+ DCs (HL3), monocytes/macrophages (M1/70), CD62L (MEL14) NK cells (PK136), CD80 (1G10), CD86 (2D10), CD103 (M290), CD152/CTLA-4 (UC 10), GITR (DTA1), and PD1 (J43) were analyzed using fluorescent conjugated mAb from BD Biosciences. Intracellular levels of FoxP3 were performed using with APC–anti-Foxp3 (FJK-16s) from eBioscience.
2.3 T cell proliferation assay and disease induction
Twenty-amino acid-long synthetic peptide PLP91-110 (YTTGAVRQIFGDYKTTICGK) was synthesized at the peptide core facility of Mayo Clinic, Rochester, MN. Mice were immunized subcutaneously with PLP91-110 (100 μg) in CFA. Mice were sacrificed 10 days after immunization, draining lymph nodes removed and challenged in vitro [27]. For disease induction 12-14 weeks old Tg mice were immunized subcutaneously in both flanks with 100 mg of PLP91-110 emulsified in CFA containing Mycobacterium tuberculosis H37Ra (400 μg/mice). Pertussis toxin (Sigma Chemicals, St. louis, Mo, USA; 100ng) was injected i.v. at day 0 and 48 h post immunization. Mice were observed daily for clinical symptoms as described previously [28].
2.4 Cytokines
Splenocytes were collected 3 weeks post immunization and stimulated with PLP91-110 peptide. Supernatants were collected from T-cell culture 48 hrs after peptide stimulation. The concentration of cytokines (IFN-γ, IL-2, IL-4, IL-6, IL-10, IL-12 and TNF-α) in the supernatant was measured by sandwich ELISA using pairs of relevant anti-cytokine monoclonal antibodies according to manufacturer's protocol (Pharmingen, San Deigo, California, USA).
2.5 Harvesting and morphology of the CNS
At time of sacrifice mice were perfused with Trump's fixative via intracardiac puncture. Spinal cords were dissected and cut into one mm blocks. Every third block was embedded in glycol methacrylate and stained with a modified erichrome stain with a cresyl violet counter-stain. The remaining spinal cord blocks were embedded in paraffin for immunoperoxidase staining. Detailed morphologic analysis was performed on 10-15 coronal spinal cord sections from each animal as described previously [29]. Each quadrant from every third spinal cord block from each animal was analyzed for the presence or absence of gray matter disease, meningeal inflammation and demyelination without knowledge of genotype or experimental group. The score was expressed as the percentage of spinal cord quadrants examine with pathologic abnormality. A maximum score of 100 indicated that there was particular pathologic abnormality in every quadrant of all spinal cord section of that particular mouse. For Brain pathology, following perfusion with Trump's fixative, two coronal cuts were made in the intact brain at the time of removal from the skull (one section through the optic chiasm and a second section through the infundibulum). This allowed for systematic analysis of the pathology of the cortex, corpus callosum, hippocampus, brainstem, striatum, and cerebellum. The tissue was embedded in paraffin. The resulting slides were then stained with hematoxylin and eosin. Each area of brain was graded on a 4-point scale as described previously [29]. The image was taken (10X or 40X magnification) using an Olympus Provis A×70 microscope (Leeds Precision Instruments, Inc., Minneapolis, MN, USA) fitted with a DP70 digital camera.
2.6 Adoptive transfer of CD4+ and CD8 T cells in PLP91-110 immunized DR3.CD8-/-.Aβ-/- mice
To study effect of CD4 or CD8 T cells in regulation of EAE in DR3.CD8-/-.Aβ-/- mice, EAE was induced in DR3.CD8-/-.Aβ-/- mice by administration of PLP91-110 (as mentioned above). Purified CD4, or CD8 T cells were adoptively transferred 5 days postimmunization and animals were monitored for EAE. For isolation of CD4 or CD8 T cells, splenocytes from naïve DR3.Aβ-/- mice were first incubated at 37°C in RPMI 1640 culture media (BioWhittaker), to allow adherent monocytes/DCs to attach to the plastic. Non-adherent cells were collected after 1 hr of incubation, stained either with FITC-anti-CD4 or FITC-CD8 (BD Biosciences). And purified by cell sorting using a FACS IV (BD Biosciences, San Jose, California, USA) and purity was always ≥95%.
2.7 Isolation and purification of different subset of CD8+ T cells
CD8 T cells were isolated from splenocytes of naïve DR3.Aβ-/- Tg mice using BD IMag™ anti-mouse CD8 particles according to manufacturer's protocol (BD Biosciences, San Diego, CA, USA). Purified CD8+ were labeled with FITC-CD8 and PE-CD122 (BD Biosciences) and CD8+CD122+ or CD8+CD122- populations were isolated by cell sorting using a FACS IV (BD Biosciences, San Jose, California, USA) and purity was always ≥95%. CD8+CD25+/CD8+CD25- or CD8+CD28+/CD8+CD28- cell population were purified similarly using CD25 or CD28 specific antibodies.
2.8 In vitro regulatory Assay and Transwell studies
To study the regulatory function of various subset of CD8 T cells, CD8+CD122+/CD8+CD122-, CD8+CD25+/CD8+CD25- T cells were cultured with PLP91-110 specific CD4 T cells in various effector to regulatory T cells ratio. Antigen specific CD4 T cells were isolated from draining lymph node of PLP91-110 immunized HLA-DR3 Tg mice and plated at 1 ×105cells/well in presence or absence of 20μg/ml of PLP91-110. Purified CD8+CD122+ or CD8+CD122- T cells were added at 2.5, 5 or 10 × 104 cells/well either in direct contact or separated from effector CD4 T cells in Corning HTS transwell 96 well plates. Two set of experiments were run in parallel, with one set used for T cells proliferation measurement, while the other set was used for collection of supernatant for cytokine analysis. Similar experiments were setup with CD8+CD25+/CD8+CD25-or CD8+CD28+/CD8+CD28- T cells. PLP91-110 specific CD4 T cells were purified using BD IMag™ anti-mouse CD4 particles according to manufacturer's protocol (BD Biosciences, San Deigo, California, USA).
2.9 Modulation of antigen presenting cells by regulatory CD8 T cells
To study effect of regulatory CD8 T cells on antigen presenting cells, CD8+CD122+ or CD8+CD122- were cultured with DR3-bone marrow-dendritic cells (BM-DCs) either directly or separated by Maxicell 0.4-μM tissue culture inserts (Corning) in 24 well plates. After 48 hrs, cells were gently washed to remove CD8 T cells (direct contact assay) and loosely adherent dendritic cells were collected. These CD8 T cells conditioned BM-DCs were used either as antigen presenting cells to PLP91-110 specific CD4 T cells in a standard T cell recall response assay or for analysis of cell surface expression of various co-stimulatory molecule such as MHC class II, B7.1, and B7.2. To obtain BM-DCs, bone marrow cells were isolated from DR3.Aβ° mice and cultured in presence of IL-4 and GM-CSF as described previously [30].
2.10 Adoptive transfer of CD8+CD122+ TR/S or CD+CD122- TEff cells and CD8 T cells in PLP91-110 immunized DR3.CD8-/-.Aβ-/- mice
To study effect of CD8+CD122+ TR/S or CD+CD122- TEffin regulation of EAE in DR3.CD8-/-.Aβ;/- mice, EAE was induced in DR3.CD8-/-.Aβ-/- mice by administration of PLP91-110 (as mentioned above). CD8+CD122+ TR/S or CD+CD122- TEff, isolated from naïve DR3.Aβ-/- mice as described before and were adoptively transferred 5 days post-immunization into DR3.CD8-/-.Aβ;/- mice. These animals were monitored for clinical sign of disease and scored using standard EAE scoring method.
2.11 Mixed glial culture and cytotoxicity assay
Mixed glial cells were prepared from 2 days old DR3.Aβ-/- pups as described previously [31]. On the basis of glial fibrillary acidic protein (GFAP) staining, astrocytes constituted >75% of mixed glial population. The splenocytes were harvested from PLP91-110 immunized DR3.Aβ-/- and DR3.CD8-/-.Aβ-/- mice and various concentrations of splenocytes (0.5 to 10 millions/well) were added on mixed glial cells in 24 wells plate (for cytotoxicity assay) and in 8 well slide chamber (for GFAP imuunocytochemistry). After 24h of incubation, chamber slides were stained using florescence conjugated antibody against GFAP (Clone G-A-5, Sigma, USA) to examine the status of activated astrocytes under experimental condition. Cell cytotoxicity of mixed glial cells was examined by MTT assay in 24 well plates as described previously [31].
3.0 Results
3.1 PLP91-110 induced severe disease in DR3 Tg mice lacking CD8 T cells
We previously showed that PLP91-110 induced EAE in DR3.Aβ-/- Tg mice [5]. The disease was mediated by PLP91-110 specific CD4 T cells as no disease was seen in DR3.CD4-/- mice (lacking CD4 T cells). To investigate the role of CD8 T cells in PLP91-110 induced EAE in DR3.Aβ-/- Tg mice, we generated DR3 mice lacking the β2m molecule (DR3.β2m-/-). These mice had very few (<1%) CD8 T cells as they lack a functional class-I molecule (data not shown). Immunization of DR3.β2m-/- mice with PLP91-110 led to severe disease in 100% of mice compared to 65% in DR3 Tg mice. The clinical disease in DR3.β2m-/- mice were characterized by an early onset (9±1 Vs 13±1.5 days, p<0.05) and higher average clinical score compared to DR3 mice (Table IA and B). EAE was very severe in DR3.β2m-/- mice and had to be sacrificed at the end of 3 weeks. Thus, data from DR3.β2m-/- mice indicated that CD8 T cells may play a regulatory role in PLP91-110 induced EAE in DR3.Aβ-/- Tg mice. Since DR3.β2m-/- mice also lack NKT cells, it is possible that either CD8 T cells or NKT cells or both might be playing a critical role in regulating PLP specific encephalitogenic CD4 T cells. Therefore, to delineate specific roles of CD8 T cells in PLP91-110 induced EAE in DR3 Tg mice; we generated DR3.CD8-/- mice by crossing DR3 lines to CD8-/-mice (Fig. 1A). These mice developed normally, have <2% CD8 cells (Fig. 1B), showed no gross abnormalities and class II expression as similar to parental DR3 line (Fig. 1C). Similar to DR3.b2m-/- Tg mice, DR3.CD8-/- mice also develop very severe clinical disease with all the mice showing symptoms as early as 9 days post-immunization and were paralytic by day 14 (Table IB and Fig. 1D). The experimental group had to be sacrificed by 25 days post-immunization as they were almost moribund. At the same point DR3 mice showed only hind limb paralysis with disease incidence of around 65% and none of the mice became moribund or needed to be sacrificed even at the end of the study (day 40 post-immunization). These set of experiments clearly indicate that CD8 T cells may play a regulatory role in our EAE model.
Table I.
PLP91-110 induced EAE in HLA Tg micea
| Mouse strain | Disease incidence (%) | Mean onset of disease ± SD (Days) | Number of mice with maximum severity score | ||||||
|---|---|---|---|---|---|---|---|---|---|
| A) | 0 | 1 | 2 | 3 | 4 | 5 | |||
| B10 | 0/15 (0) | - | 15 | - | - | - | - | - | |
| Aβ-/- | 0/15 (0) | - | 15 | - | - | - | - | - | |
| DR3.Aβ-/- | 10/15 (67%) | 13±1.5 | 5 | - | 1 | 8 | - | 1 | |
| B) | |||||||||
| DR3.β2m-/-.Aβ/- | 20/20 (100%) | 9.5±1.3 | 0 | - | - | 8 | 2 | 10 | |
| DR3.CD8-/-.Aβ/- | 19/20 (95%) | 10.5±1.2 | 1 | - | - | 9 | 2 | 7 | |
| DR3.CD4-/-.Aβ/- | 0/15 (0) | - | 15 | - | - | - | - | - | |
Mice were immunized with 100μg of PLP peptide/400μg Mtb in CFA and Ptx was administered at 0 and 48 h post immunization. Mice were scored daily for disease as mentioned in material and methods. The data is from three experiments combined.
p<0.01 Anova Ranks for Group A and B
Figure 1. CD8 deficient HLA-DR3 (DR3.CD8-/-.AβTg mice develop severe EAE (PLP91-110induced) compared to CD8 sufficient HLA-DR3 (DR3.Aβ-/-) Tg mice.
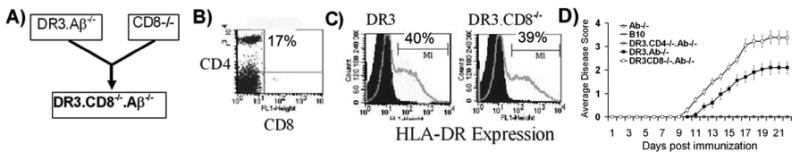
(A) DR3.CD8-/-.Aβ-/- mice were generated by mating DR3 Tg mice with CD8 deficient mice on C57BL/6 background. (B) These mice develop normally and lack CD8 T cells. (C) DR3.CD8-/-.Aβ-/-express HLA-DR molecule and levels of surface HLA-DR molecule were comparable to DR expression observed in HLA- DR3.Aβ/- Tg mice. (D) DR3.CD8-/-.Aβ-/- (n= 10) mice show earlier disease onset, higher disease incidence and increased disease severity compared to DR3.Aβ-/- mice (n=8), while no disease was observed in DR3.CD4-/-.Aβ-/- (n= 10), B10 (n= 10) or MHC class II -/- (Aβ-/-) mice (n= 10). Five to ten mice per group were immunized with 100μg of PLP91-110 and scored daily for disease (as stated in material and methods) and the daily mean disease score for each group is plotted. Error bars represent the standard error of mean. To analyze surface expression, splenocytes were isolated from DR3.Aβ-/- or DR3.CD8-/-.Aβ-/- Tg mice and analyzed for cell surface expression of CD4 (GK1.5), CD8 (53.6) and HLA-DR (L227 clone) by flow cytometry. Numbers in scatter plot and histograms indicate the percentage of positive cells. Data represent one of three experiments performed at different time points.
3.2 DR3.CD8-/- Tg mice with EAE showed less severe inflammation and demyelination
Since DR3.CD8-/- mice developed very severe paralytic disease, we expected increased CNS pathology in DR3.CD8-/- mice compared to CD8 sufficient DR3 mice. However, analysis of spinal cord sections from DR3.CD8-/- mice with EAE showed less demyelination compared to CD8 sufficient DR3 Tg mice (8%±2.6 Vs 20%±3.1, p<0.01) (Fig. 2A). Inflammation in the spinal cord was similar in both CD8 deficient and CD8 sufficient DR3 Tg mice (26%±7.8 Vs. 17%±4.1, p=ns) (Fig. 2B). The brain sections from CD8 sufficient DR3 Tg mice showed more inflammation and demyelination compared to those from CD8 deficient DR3 Tg mice (Fig. 2C). Brain pathology in DR3 Tg mice was characterized by inflammation in meningeal, brain stem, corpus callosum as well as cerebellum region (Fig. 2D). Similar pathology was observed in DR3.b2m-/- mice with EAE, which showed less CNS pathology compared to CD8 sufficient DR3 Tg mice (data not shown). In contrast to clinical disease phenotype which points towards a regulatory role of CD8 T cells, histopathology data indicates that CD8 T cells are required for inducing pathology in CNS.
Figure 2. CD8 deficient HLA-DR3 (DR3.CD8-/-.Aβ-/-) Tg mice with EAE develop less inflammation and demyelination in CNS compared to CD8 sufficient HLA-DR3 (DR3.Aβ-/-) Tg mice.
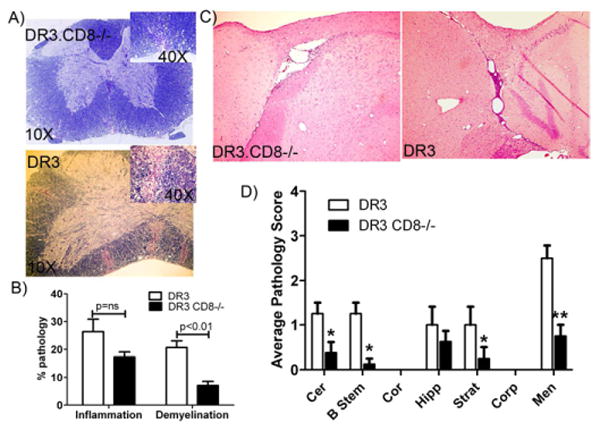
Representative photomicrograph of inflammatory lesions in spinal cord (A) and brain (C of DR3.CD8-/-.Aβ;° and DR3.AβTg mice with EAE. A) The photographs (10×) of spinal cord sections show increased inflammation and demyelination in CD8 sufficient DR3.Aβ-/- mice compared to CD8 deficient DR3CD8-/-Aβ-/- mice. Inset picture show a magnified image (40×) showing demyelinating regions. B) Quantitative analysis of spinal cord pathology also showed th DR3.Aβ-/- mice (n=5) with EAE have a higher pathology score (percent of spinal cord quadrants showing inflammation and demyelination for each mouse (mean ± SD) compared to DR3.CD8-/-.A&β-/- mice (n=5). C) Brain pathology was characterized with widespread inflammation and demyelination in DR3.Aβ-/- compared to mild meningeal inflammation in DR3.CD8-/-.Aβ-/- mice with EAE. Inset picture show a magnified image (40×) showing demyelinating regions. D) Quantitative analysis of brain pathology also showed that DR3.Aβ-/- (n=5) mice with EAE have a higher pathology score (mean ± SD) compared to DR3.CD8-/-.Aβ-/- mice (n=5). These figures are representative of one of three experiments performed at different time point. *p<0.05, **p<0.005 as compared to DR3.CD8-/-.Aβ-/- mice. The image was taken (10X magnification) using an Olympus Provis A×70 microscope (Leeds Precision Instruments, Inc., Minneapolis, MN, USA) fitted with a DP70 digital camera.
3.3 Adoptive transfer of CD8 T cells exhibited dual effect in DR3.CD8-/- mice
Since we observed both regulatory as well as pathogenic role of CD8 T cells in our EAE model, we hypothesized that CD8 T cells play a dual role in pathogenesis of EAE. To examine this possibility, we performed adoptive transfer of CD8 T cells in DR3.CD8-/- Tg mice with a rationale that CD8 T cells have two different subsets, an effector subset and a regulatory subset. We isolated CD8 T cells from naïve DR3 Tg mice and adaptively transferred them in CD8 deficient DR3 (DR3.CD8-/-) Tg mice immunized with PLP91-110. As shown in Table II, transfer of CD8 T cells showed differential effect depending on the number of cells the animal received. Transfer of 1 × 106 CD8 T cells in to DR3.CD8-/- Tg mice had no effect on disease severity or incidence, however showed more demyelination in the spinal cord compared to DR3.CD8-/- receiving no cells (Fig. 3 A). These mice also showed increased inflammation and demyelination in the brain (Fig. 3 B). As a control, we also adoptively transferred CD4 T cells from naïve DR3 Tg mice to DR3.CD8-/- Tg mice and observed no difference in disease incidence or CNS pathology (Table II, Fig 3A and B). Transfer of higher number of CD8 T cells (10 × 106 cells) led to decrease in disease incidence with only 60% of mice developing EAE indicating presence of a regulatory CD8 subset (Table II). The mice that developed EAE showed less severe clinical disease compared to CD8 sufficient DR3 mice or DR3.CD8-/- mice receiving 1×106 CD8 T cells or 10×106 CD4 T cells. However DR3.CD8-/- mice with EAE, showed increased inflammation and demyelination in spinal cord. Adoptive transfer data indicates that regulatory subset might constitute a small subset of CD8 T cells, whereas the majority of CD8 T cells may act as effector cells.
Table II. Modulation of PLP91-110 induced EAE in DR3.CD8-/- Tg mice by adoptive transfer of CD8 and CD4 subset of T cells.
| Mouse strain | Adoptive transfer of cells | Disease incidence (%) | Number of mice with maximum severity score | |||||
|---|---|---|---|---|---|---|---|---|
|
|
||||||||
| 0 | 1 | 2 | 3 | 4 | 5 | |||
| DR3.CD8-/-.Aβ/- | PBS | 5/5 (100%) | - | - | - | 3 | 1 | 1 |
| DR3.CD8-/-.Aβ-/- | CD8 T cells (1×106 cells) | 5/5 (100%) | - | - | - | 4 | - | 1 |
| DR3.CD8-/-.Aβ-/- | CD8 T cells (10×106 cells) | 3/5 (
 %) %) |
2 | - | 2 | 1 | - | - |
| DR3.CD8-/-.Aβ-/- | CD4 T cells (10×106 cells) | 5/5 (100%) | - | - | - | 3 | 1 | 1 |
Anova on RANKS p=0.019
Figure. 3. Adoptive transfer of CD8 T cells but not CD4 T cells leads to increased CNS pathology in CD8 deficient DR3.CD8-/-.Aβ-/-Tg mice.
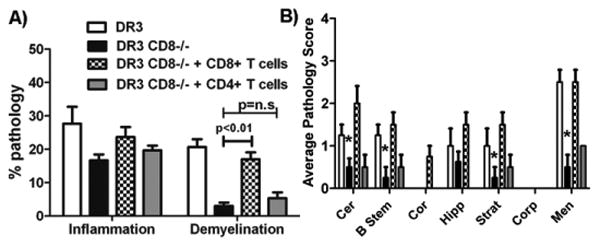
A) Quantitative analysis of spinal cord pathology showed that DR3.CD8-/-.Aβ-/- (n=5) mice receiving CD8 T cells have a higher pathology score compared to DR3.CD8-/-.Aβ-/- (n=5) mice receiving CD4 T cells or PBS only. B) Similarly quantitative analysis of brain pathology showed that DR3.CD8-/-.Aβ-/- mice (n=5) receiving CD8 T cells have a higher pathology score compared to compared to DR3.CD8-/-.Aβ-/-(n=5) mice receiving CD4 T cells or PBS only. * p<0.01 compared to demyelination in DR3 mice with disease. EAE was induced in CD8 deficient DR3.CD8-/-.Aβ-/- Tg mice (N=20) described in methods and randomly divided into 3 groups receiving CD4, CD8 or PBS only. CD4 and CD8 T cells were isolated from spleen of naïve DR3 Tg mice and 10 × 106 cells were adoptively transferred (i.v.) in to immunized DR3.CD8-/-.Aβ-/-(n=5) mice on day 5 post- immunization. Mice were monitored for disease as described in methods and sacrificed at day 25 post-immunization for CNS histopathology.
3.4 Regulatory subset of CD8 T cells express CD122 and can suppress CD4 T cell proliferation
Our findings suggest the presence of different CD8 T cell population comprising both a regulatory as well as an effector subset. There are number of markers which have been associated with regulatory/suppressor CD8 T cells such as presence of CD122 or CD25 or absence of CD28 molecule [32-37]. We analyzed expression of CD25, CD28 and CD122 on CD8 T cells in our HLA transgenic mice and observed higher expression of CD122 on CD8 T cells compared to CD4 T cells in splenocytes of naïve DR3.Aβ/- Tg mice (Fig. 4A). In contrast CD25 expression was more abundant in the CD4 T cell subset. Therefore, we isolated different subsets of CD8 T cells such as CD8+CD25+, CD8+CD28- and CD8+CD122+ T cell subsets using FACS sorting and analyzed their function using an in-vitro approach. PLP91-110 specific and HLA-DR3 restricted CD4 T cells were used as effector cells and cultured with medium alone or in the presence of various regulatory subsets of CD8 T cells at different regulatory to effector cell ratios. Irradiated splenocytes from HLA-DR3 mice, pulsed with PLP91-110 were used as APCs. We did not observe any suppressive effect on proliferation of antigen specific CD4 T cells, when CD8+CD25+ or CD8+CD28- were used as the CD8 T cell subset (Fig. 4B). However, CD8+CD122+ T cells significantly suppressed proliferation of PLP91-110 specific CD4 T cells (Fig. 4B) in a dose dependent manner as maximum suppression was observed at 1:1 (CD8 to CD4) ratio followed by 1:2 and 1:4 doses. No suppression in CD4 T cell proliferation was seen when CD8+CD122- T cells were added in culture (Fig. 4C). Thus CD8+CD122+ subset of CD8 T cells is the regulatory/suppressor T cells (CD8 TR/S cells) in our EAE model.
Figure. 4. CD8 T cells from naïve DR3 Tg mice, express higher level of CD122 (IL-2β chain) compared to CD4 T cells and suppress proliferation of PLP91-110specific CD4 T cells.
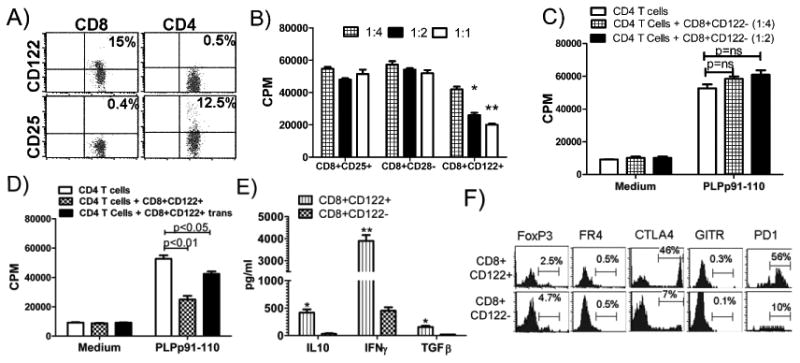
A) Splenocytes were isolated from naïve DR3.Aβ;° mice, stained for surface expression of indicated molecule using fluorescence conjugated antibodies and analyzed using flow cytometry. Numbers in scatter plots indicate the percentage of positive cells. Data represent one of three experiments performed at different time points. B) CD122+ subset of CD8 T cells suppressed proliferation of PLP91-110 specific CD4 T cells in a dose dependent manner, while CD25+ and CD28- subset showed no effect on CD4 T cell proliferation. CD122+ CD25+ and CD28- subsets of CD8 T cells were sorted using FACS Vanage Cell sorter and cultured in different ratio with PLP91-110 specific CD4 T cells (5×105 cells) together with DCs pulsed with antigen. *p<0.05, **p<0.01 as compared to CD4 T cells alone; C) CD122- subset of CD8 T cells showed no effect on PLP91-110 specific CD4 T cell proliferation. The proliferative response was assessed by pulsing the cultures with [3H]thymidine for the last 16 h. The data are presented as the mean CPM ± SD and are average of three independent experiments. NS: not significant compared to CD4 T cells alone in presence of antigen pulsed DCs. D) CD8+CD122+ T cells suppress proliferation of PLP91-110 specific CD4 T cells through both cell to cell contact as well as soluble mediators. CD8+CD122+ T cells (3×105 cells) were cultured with PLP91-110 specific CD4 T cells (3×105 cells) either together or separated by a transwell membrane. Antigen pulsed DCs were used as antigen presenting cells. E) CD8+CD122+ T cells produced high level of IFNγ, IL-10 and TGFβ compared to CD8+CD122- subset. CD122+ and CD122- subset of CD8 T cells were sorted using FACS Vanage Cell sorter and stimulated with plate bound anti-CD3 and anti-CD28. Cell free supernatants were collected after 48 hrs and analyzed using standard ELISA. The data presented are average of three independent experiments. *** p<0.001; **p<0.01, *p<0.05; compared to cytokine secreted by CD8+CD122- cells. F) CD8+CD122+ T cells express higher level of inhibitory molecules CTLA-4 and PD1 compared to CD8+CD122- subset. Splenocytes were isolated from naïve DR3.Aβ-/- mice and stained for surface expression of indicated molecule using fluorescence conjugated antibodies and analyzed using flow cytometry. The analysis was performed with CellQuest software. The data are representative of three independent experiments.
3.5 Suppression is mediated through both direct cell to cell contacts as well as through soluble cytokine mediators
CD8 TR/S cells can show their suppressive effect either by direct cell to cell contact or through chemical mediators such as cytokines. To determine the mechanism of suppression by CD8+CD122+ T cells, we isolated CD4 T cells from DR3.CD8-/- Tg mice immunized with PLP91-110 and cultured these cells either directly with CD8+CD122+ T cells or in transwell plate separated by a membrane. Maximum suppression was seen when both CD4 T cells as well as regulatory CD8 T cells were in direct contact (Fig. 4D). When they were separated by a membrane, T cell proliferation was inhibited by only 40% compared to ≥75% (direct contact) indicating that CD8+CD122+ T cells suppress antigen specific CD4 T cells by both cell to cell contact as well as through potential cytokine mediators. We also analyzed levels of different cytokines in culture and observed that CD8+CD122+ T cells secreted higher levels of IFNγ, IL-10 and TGFβ compared to CD8+CD122- T cells (Fig. 4E). Expression of certain surface markers such as FoxP3, CTLA4, B7H1, CD103, Fas and PD1 have been associated with suppressive function of T cells, therefore we analyzed expression of these molecules on CD8+CD122+ Tregs and CD8+CD122- T effector cells. CD8+CD122+ T cells expressed higher levels of CTLA4, CD103 and PD1 compared to CD8+CD122- T cells (Fig. 4E). CD8+CD122+ Tregs did not express FoxP3, GITR and FR4, molecules associated with CD4 regulatory T cells.
3.6 CD8+CD122+ TR/S cells suppress CD4 T cells proliferation through modulation of APC
In order to dissect the mechanism of CD8 TR/S induced cell-contact dependent suppression of antigen specific CD4 T cells; we analyzed the antigen presentation function of DCs. We hypothesized that CD8+CD122+ T cells may suppress antigen specific CD4 T cells proliferation through modulation of DCs, the professional antigen presenting cells. To address this, bone marrow derived DCs (BM-DCs) were isolated from DR3 mice and cultured in absence or presence of either CD8+CD122+ or CD8+CD122- T cells. After 24 hr incubation, non-adherent cells (CD8 T cells) were removed and conditioned DCs were used as APCs to stimulate DR3 restricted PLP91-110 specific CD4 T cells. DCs cultured with CD8+CD122+ T cells showed decreased antigen presentation compared to those cultured with CD8+CD122- T cells or medium control (Fig. 5A). These results indicated that CD8+CD122+ T cells suppressed antigen specific T cell proliferation by modulating antigen presentation function of DCs. Dendritic cells cultured with CD8+CD122+ T cells showed decreased levels of activation marker such as B7.1, B7.2 and HLA-DR compared to normal DCs cultured with medium (Fig. 5 B).
Figure. 5. CD8+CD122+ TR/Scells suppress proliferation of PLP91-110specific CD4 T cells by modulating antigen presenting dendritic cells.
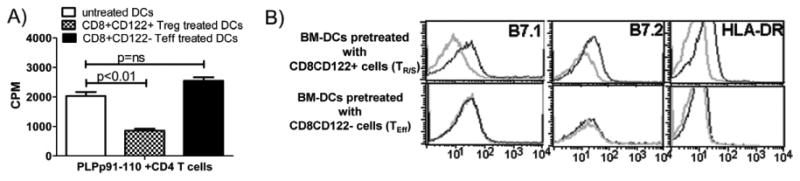
A) Bone marrow derived dendritic cells (BM-DCs), pre-cultured with CD8+CD122+ TR/S cells showed decreased antigen presentation capacity compared BM-DCs cultured with CD8+CD122- T cells or medium only. BM-DCs from long bone of DR3.Aβ-/- mice were cultured with CD8+CD122+, CD8+CD122- T cells or medium for 24 hrs. These pre-conditioned BM-DCs were pulsed with antigen and used as APCs to PLP91-110 specific CD4 T cells. The proliferative response was assessed by pulsing the cultures with [3H]thymidine for the last 16 h. The data are presented as the mean CPM ± SD and are average of three independent experiments. *** p<0.001; **p<0.01, *p<0.05; NS: not significant compared to CD4 T cells alone in presence of antigen pulsed DCs. B) BM-DCs pre-cultured with CD8+CD122+ T cells showed decreased expression of costimulatory molecules (HLA-DR, B7.1 and B7.2) compared BM-DCs cultured with CD8+CD122- T cells. The pre-conditioned DCs were prepared as before and analyzed for surface expression of co-stimulatory molecule by flow cytometry. The data are representative of three independent experiments.
3.7 CD8 sufficient T cells cause increased cytotoxicity in mixed glial culture compared to CD8 deficient cells
To further examine if CD8 cells have ability to kill glial cells, we prepared mixed glial cultures of 1-3 days old pups from CD8 sufficient or CD8 deficient DR3 Tg mice. Splenocytes from DR3 or DR3CD8-/- mice immunized with PLP91-110 were isolated at the peak of disease (16 days post-immunization). We isolated total splenocytes instead of only CD8 T cells as DR3CD8-/- mice lack CD8 T cells. These CD8 sufficient or deficient splenocytes were added to mixed glial cultures at 0.5× 106 or 2 × 106 or 10 × 106 cells per well. After 24 hr of incubation, cytotoxicity in these two populations was assayed by directly staining for astrocytes or by analyzing cell viability using standard MTT assay. CD8 sufficient splenocytes population induced more killing compared to CD8 deficient splenocytes population at all tested cell concentrations (Fig 6A and B). This response was dose dependent since maximum killing was observed only at the highest cell concentration. Similarly using MTT assay we observed that CD8 sufficient splenocytes caused more astrocytes killing than CD8 deficient splenocytes. To confirm a direct role of effector CD8 T cells in glial killing, we analyzed effect of CD8+ T cells isolated from CD8 sufficient DR3 Tg mice on mixed glial culture and observed that these cells induced killing similar to CD8 sufficient splenocytes (data not shown).
Figure. 6. CD8 sufficient splenocytes induce increased cytotoxicity in mixed glial culture compared to CD8 deficient cells.
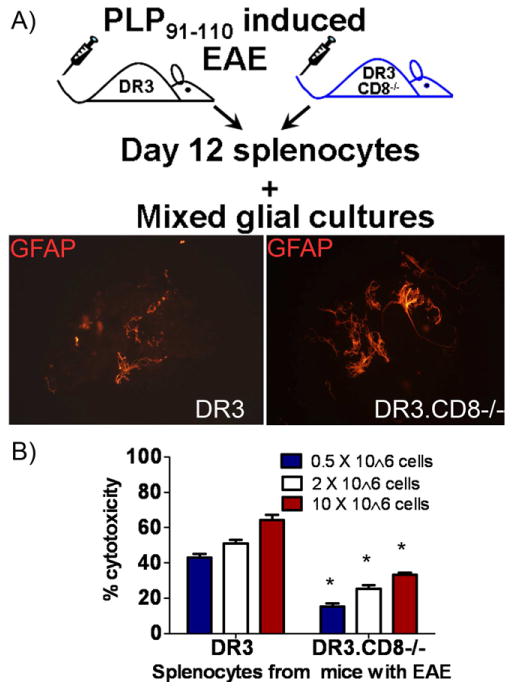
DR3.Aβ-/- and DR3.CD8-/-.Aβ-/- mice were immunized with PLP as described before. (A) twelve days post-immunization, total splenocytes were harvested and various concentrations of splenocytes (0.5 to 10 millions/well) were added on mixed glial cells as described in methods. After 24h of incubation, slides were stained for GFAP to examine the status of activated astrocytes under experimental condition; The image was taken (40X magnification) using an Olympus Provis A×70 microscope (Leeds Precision Instruments, Inc., Minneapolis, MN, USA) fitted with a DP70 digital camera. (B) Cell cytotoxicity of mixed glial cells was examined by MTT assay in 24 well plates as described in methods. The data represents three separate experiments done in triplicates. *p<0.05 compared to DR3.Aβ-/- and treatment.
3.8 Adoptive transfer of CD8CD122+ TR/S cells suppress EAE, while CD8CD122- TEff cells caused increased CNS pathology
To examine if CD8+CD122+ T cells can modulate ongoing EAE, we isolated CD8+CD122+ TR/S and CD8+CD122- T cell subsets from naïve HLA-DR3 Tg mice. These CD8 T cells subsets were adoptively transferred into HLA- DR3.CD8-/- Tg mice immunized with PLP91-110 peptide. Mice received 2 injection of CD8+CD122+ TR/S or CD8+CD122- TEff cell subsets or PBS at day -1 and day 5 post-immunization. Mice were monitored for weight loss and clinical signs of EAE. Mice receiving PBS or CD8+CD122- TEff cells showed no major change in disease incidence or severity (Table III), however, group receiving regulatory CD8 T cell subset showed decreased incidence (50% Vs 100%) and less severe disease (mean clinical score 1±1.4 Vs 3.6±0.9 Vs. 3.75±0.96, p<0.05) compared to PBS control or CD8+CD122- T cells injected group. Analysis of CNS pathology showed that mice receiving CD8+CD122- TEff cells had increased spinal cord demyelination compared to the PBS group (37.9±5.4 Vs 8.3±4.5, p<0.01) (Fig. 7B, and 7C). Thus our results indicate that CD8+CD122+ TR/S cells can suppress EAE whereas CD8+CD122- TEff cells can induce CNS pathology.
Table III.
Modulation of PLP91-110 induced EAE in DR3.CD8-/- Tg mice by adoptive transfer of CD8+CD122+ and CD8+CD122- subset of T cells
| Mouse strain | Adoptive transfer of cells | Disease Incidence (%) | Number of mice with maximum severity score | |||||
|---|---|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | 4 | 5 | |||
DR3.CD
 -/-.Aβ-/- -/-.Aβ-/-
|
PBS | 5/5 (100%) | - | - | - | 3 | 1 | 1 |
| DR3.CD8-/-.Aβ-/- | CD8+CD122- T cells 2×106 cells | 4/4 (100%) | - | - | - | 2 | 1 | 1 |
| DR3.CD
-/-.Aβ-/-
|
CD8+CD122+ T cells 0.5×106 cells | 2/4 (50%) | 2 | - | 1 | 1 | - | - |
Anova on RANKS p=0.046
Figure.7. Adoptive transfer of CD8+CD122- TEffcells lead to increased spinal cord demyelination.
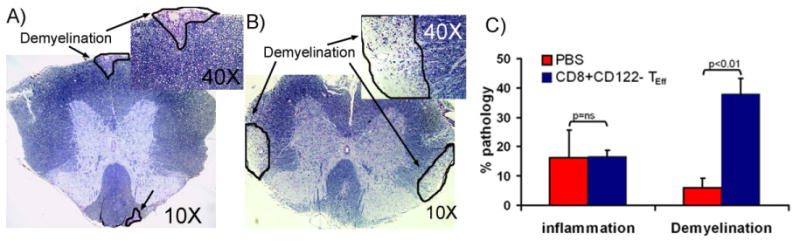
Representative photomicrograph (10×) of flammatory lesions in spinal cord of DR3.CD8-/-.Aβ/- receiving CD8+CD122+ TR/S cells (A) or CD8+CD122- TEff cells (B). The photographs of spinal cord sections (10×) show increased demyelination in mice receiving CD8+CD122- TEff cells compared to those receiving CD8+CD122+ TR/S cells. Inset picture show a magnified image (40×)showing demyelinating regions. Black arrow points the area with demyelination. The image was taken (10X or 40X magnification) using an Olympus Provis A×70 microscope (Leeds Precision Instruments, Inc., Minneapolis, MN, USA) fitted with a DP70 digital camera. (C) Quantitative analysis of spinal cord pathology also showed that DR3.CD8-/-.Aβ-/-(n=3) mice receiving CD8+CD122- TEff cells had increased demyelination compared to DR3.CD8-/-.Aβ-/- (n=3) mice receiving PBS. CD122+ or CD122- subset of CD8 T cells were isolated from spleen of naïve DR3 Tg mice and adoptively transferred (i.v.) in to immunized DR3.CD8-/-.Aβ-/- Tg (n=3) mice on day 5 post-immunization. The mice in control group just received PBS. Mice were monitored for disease and sacrificed at day 25 post-immunization for CNS histopathology.
4.0 Discussion
The role of CD8 T-cells in MS/EAE has been enigmatic as it has been assigned either a regulatory or pathogenic role in disease [17, 23, 38, 39]. Here we report for the first time a dual role of CD8 T cells in a single animal model. Based on our data, we show that CD8 T cells act both as pathogenic/effector as well as regulatory cells in immuno-pathogenesis of PLP91-110 induced EAE in HLA-DR3 Tg mice. We show that CD8+CD122+ T cells are the regulatory subset of CD8 T cells which suppress CD4 T cells by inhibiting antigen presentation pathway by both direct cell to cell contact as well as by secreting suppressive cytokine such as IL-10 and TGFβ. In contract CD8+CD122- T cells are the pathogenic subset.
There is a direct correlation between number of CD8 T cells and extent of axonal damage. Initial studies investigating the role of CD8 T cells in EAE showed that CD8 T cells may have a regulatory function as CD8-/- mice or depletion of CD8 T cells resulted in increased disease susceptibility and severity [25, 40]. In a study comparing development of EAE in CD4-/-and CD8-/- DBA/l mice, Abdul-majid et. al. [41] reported that CD8-/- mice develop less inflammation and demyelination compared to CD8 sufficient mice [41]. However, some studies have also shown a pathogenic role of CD8 T cells in animal models [17, 18, 38]. Immunostaining of postmortem brain sections from MS patients have shown that CD4 T cells are more restricted to perivascular cuffs, whereas CD8 T cells were observed in the parenchyma of lesions highlighting an important role of CD8 T cells in neuronal injury and demyelination in MS [12]. Although CD8 T cells have been assigned both regulatory as well as pathogenic role, ours is the first model showing both phenotype of CD8 T cells in a single animal model. These results are not surprising as CD4 T cells also have dual role in immuno-pathogenesis of EAE and other diseases [42, 43]. On one hand antigen specific auto-reactive CD4 T cells are pathogenic, while CD4+CD25+ FoxP3+ T cells have been shown to be regulatory in nature. Our adoptive transfer experiment further confirmed that CD8 T cells are both regulatory as well as pathogenic cells. We observed only the pathogenic characteristic of CD8 T cells when small numbers of CD8 T cells were transferred, while regulatory effect was observed only on adoptive transfer of large number of CD8 T cells. This differential dose effect can be explained by the fact that regulatory CD8 T cells population constitute only a small fraction of total CD8 T cells and a minimal number of cells might be required for its suppressive effect. Similar effect has been reported previously in other animal models examining the role of IL-2 in CD4+CD25+ Tregs, where a protective effect was observed only on adoptive transfer of large number of splenocytes or thymocytes [44].
In recent years, there have been increased interests in understanding the role of regulatory subset of CD8 T cells [45]. A number of markers have been described for regulatory CD8 T cell subsets. Some groups have shown that CD8 regulatory T cells express Qa-1 antigen [24], while others have shown that CD8+CD28- T cell subset is a regulatory subset which suppresses antigen specific immune response [33]. FoxP3 and CD25 expressing regulatory CD8 T cells have also been reported from humans that can suppress Th1 cells [34]. Recently another subset of CD8 regulatory T cells expressing CD122, a IL-2Rβ chain that have been reported [36, 46-48]. In our experimental model we observed that only CD122 expressing CD8 T cells showed the regulatory/suppressive phenotype. CD8+CD122+ T cells have been shown to be naturally occurring regulatory cells and their absence led to lympho-proliferative disorder [36]. The adoptive transfer of CD8+CD122+ T cell subset in neonates prevented development of abnormal T cells confirming the regulatory role of this subset in maintaining immune homeostasis.
Regulatory T cells can modulate immune response either by secreting soluble mediators (e.g. IL-10, TGFβ) or through cell to cell contact. Therefore, to analyze the mechanism of suppression by CD8+CD122+ regulatory subset in our model, we performed Transwell plate experiments. We observed that CD8+CD122+ regulatory T cells suppressed CD4 T cells response through both soluble mediators as well as by cell to cell direct contact. Maximum suppression was observed when cells were cultured in direct contact indicating that suppressive action of CD8CD122+ T cells might be through cell-contact. This suppressive action might be mediated by modulating antigen presenting cells (DCs), which in turn leads to decreased antigen presentation and T cell response. We also observed that BM-DCs cultured with CD8CD122 regulatory T cell subset showed down regulation of costimulatory molecules (such as MHC class II, B7 and B7.1) associated with antigen presentation.
We also observed that CD8+CD122+ TR/S subset produced increased levels of IFNγ, IL-10 and TGFβ compared to CD8+CD122- subset. Although the role of IL-10 and TGFβ as immunoregulatory cytokine is well established, IFNγ is considered a pro-inflammatory cytokine. However, we and others have shown that IFNγ can also act as an immuno regulatory cytokine [28, 49]. We have shown that a high level of IFNγ is responsible for protective effect in our DR3DQ6 double Tg mice. Lee et al [50] showed that CD8+CD122+ regulatory T cells produce high levels of IFNγ, however the authors did not elaborate on possible immuno-regulatory role of IFNγ in CD8+CD122+ mediated suppression. Additionally, mice lacking IFNγ or IFN-R develop severe and fatal EAE on immunization with myelin antigen indicating an important role of IFNγ in regulating autoreactive T cells and subsequent immune response [51].
We further confirmed regulatory role of CD8+CD122+ T cells by adoptive transfer of CD8+CD122+ TR/S and CD8+CD122- T cell subset into DR3.CD8-/- mice. Adoptive transfer of only CD8+CD122+ TR/S subset suppressed EAE, confirming our hypothesis that CD8+CD122+ T cells play regulatory/suppressive role in our EAE model. Lately it was reported that tolerized DCs expressing B-7H1 in CNS recruit CD8+CD122+ regulatory T cells in CNS and decrease EAE severity [52]. Thus we have identified a regulatory subset of CD8 T cells and have also shown that this regulatory subset modulates EAE by suppressing encephalitogenic T cell proliferation by modulating DCs function as well as through secretion of immunoregulatory cytokines.
We next characterized effector function of CD8 T cells by investigating its ability to kill glial cells in mixed glial cultures. The mixed glial culture cytotoxic assay confirmed our hypothesis as we saw increased astrocytes killing by splenocytes with CD8 T cells. Our adoptive transfer experiment confirmed that CD8CD122- subset is effector subset responsible for causing CNS demyelination as mice receiving these subset showed increased CNS demyelination compared to the mice receiving CD8+CD122+ TR/S subset. Thus our data indicate that CD8 T cells specially CD122- subset of CD8 T cells act as effector T cells and can induce CNS pathology in our EAE model and a similar situation might be true for human MS too.
To the best of our knowledge, this is the first study to show that CD8 T cells play both regulatory as well as effector/pathogenic role in EAE induced by encephalitogenic CD4 T cells. Since recent studies had reported an important role of CD8 T cells in immuno-pathogenesis of MS, a better understanding of these subsets could aid in designing novel therapy for MS patients.
Research Highlights.
CD4+ T cells are required for development of EAE in HLA-DR3 transgenic mice.
CD8+ T cells play both regulatory and pathogenic role in PLP91-110 induced EAE.
CD8+CD122+ T cells are regulatory subset of CD8 T cells.
CD8+CD122+ cells regulate immune response by modulation of antigen presentation.
CD8+CD122- T cells are pathogenic subset of CD8 T cells and induce CNS pathology.
Acknowledgments
We thank Julie Hanson and her staff for breeding and maintaining the various HLA class II transgenic mice used for this study. We also thank Lauri Zoecklein, Louiza Papke and Mable Peirce for excellent technical assistance.
Footnotes
Conflict of Interest: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Libbey JE, Tsunoda I, Fujinami RS. Studies in the modulation of experimental autoimmune encephalomyelitis. J Neuroimmune Pharmacol. 5:168–75. doi: 10.1007/s11481-010-9215-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sospedra M, Martin R. Immunology of multiple sclerosis. Annual review of immunology. 2005;23:683–747. doi: 10.1146/annurev.immunol.23.021704.115707. [DOI] [PubMed] [Google Scholar]
- 3.Tsunoda I, Fujinami RS. Two models for multiple sclerosis: experimental allergic encephalomyelitis and Theiler's murine encephalomyelitis virus. Journal of neuropathology and experimental neurology. 1996;55:673–86. doi: 10.1097/00005072-199606000-00001. [DOI] [PubMed] [Google Scholar]
- 4.Luckey D, Bastakoty D, Mangalam AK. Role of HLA class II genes in susceptibility and resistance to multiple sclerosis: studies using HLA transgenic mice. Journal of autoimmunity. 37:122–8. doi: 10.1016/j.jaut.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mangalam AK, Khare M, Krco C, Rodriguez M, David C. Identification of T cell epitopes on human proteolipid protein and induction of experimental autoimmune encephalomyelitis in HLA class II-transgenic mice. European journal of immunology. 2004;34:280–90. doi: 10.1002/eji.200324597. [DOI] [PubMed] [Google Scholar]
- 6.Mangalam AK, Rajagopalan G, Taneja V, David CS. HLA class II transgenic mice mimic human inflammatory diseases. Advances in immunology. 2008;97:65–147. doi: 10.1016/S0065-2776(08)00002-3. [DOI] [PubMed] [Google Scholar]
- 7.Tsuchida T, Parker KC, Turner RV, McFarland HF, Coligan JE, Biddison WE. Autoreactive CD8+ T-cell responses to human myelin protein-derived peptides. Proc Natl Acad Sci U S A. 1994;91:10859–63. doi: 10.1073/pnas.91.23.10859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jurewicz A, Biddison WE, Antel JP. MHC class I-restricted lysis of human oligodendrocytes by myelin basic protein peptide-specific CD8 T lymphocytes. J Immunol. 1998;160:3056–9. [PubMed] [Google Scholar]
- 9.Biddison WE, Taub DD, Cruikshank WW, Center DM, Connor EW, Honma K. Chemokine and matrix metalloproteinase secretion by myelin proteolipid protein-specific CD8+ T cells: potential roles in inflammation. J Immunol. 1997;158:3046–53. [PubMed] [Google Scholar]
- 10.Lucchinetti CF, Rodriguez M. The controversy surrounding the pathogenesis of the multiple sclerosis lesion. Mayo Clinic proceedings. 1997;72:665–78. doi: 10.1016/S0025-6196(11)63576-3. [DOI] [PubMed] [Google Scholar]
- 11.Bugawan TL, Klitz W, Blair A, Erlich HA. High-resolution HLA class I typing in the CEPH families: analysis of linkage disequilibrium among HLA loci. Tissue Antigens. 2000;56:392–404. doi: 10.1034/j.1399-0039.2000.560502.x. [DOI] [PubMed] [Google Scholar]
- 12.Babbe H, Roers A, Waisman A, Lassmann H, Goebels N, Hohlfeld R, et al. Clonal expansions of CD8(+) T cells dominate the T cell infiltrate in active multiple sclerosis lesions as shown by micromanipulation and single cell polymerase chain reaction. J Exp Med. 2000;192:393–404. doi: 10.1084/jem.192.3.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gay FW, Drye TJ, Dick GW, Esiri MM. The application of multifactorial cluster analysis in the staging of plaques in early multiple sclerosis. Identification and characterization of the primary demyelinating lesion. Brain. 1997;120(Pt 8):1461–83. doi: 10.1093/brain/120.8.1461. [DOI] [PubMed] [Google Scholar]
- 14.Lassmann H. Neuropathology in multiple sclerosis: new concepts. Mult Scler. 1998;4:93–8. doi: 10.1177/135245859800400301. [DOI] [PubMed] [Google Scholar]
- 15.Shresta S, Pham CT, Thomas DA, Graubert TA, Ley TJ. How do cytotoxic lymphocytes kill their targets? Curr Opin Immunol. 1998;10:581–7. doi: 10.1016/s0952-7915(98)80227-6. [DOI] [PubMed] [Google Scholar]
- 16.Trapani JA, Davis J, Sutton VR, Smyth MJ. Proapoptotic functions of cytotoxic lymphocyte granule constituents in vitro and in vivo. Curr Opin Immunol. 2000;12:323–9. doi: 10.1016/s0952-7915(00)00094-7. [DOI] [PubMed] [Google Scholar]
- 17.Huseby ES, Liggitt D, Brabb T, Schnabel B, Ohlen C, Goverman J. A pathogenic role for myelin-specific CD8(+) T cells in a model for multiple sclerosis. J Exp Med. 2001;194:69–76. doi: 10.1084/jem.194.5.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun D, Whitaker JN, Huang Z, Liu D, Coleclough C, Wekerle H, et al. Myelin antigen-specific CD8+ T cells are encephalitogenic and produce severe disease in C57BL/6 mice. J Immunol. 2001;166:579–87. doi: 10.4049/jimmunol.166.12.7579. [DOI] [PubMed] [Google Scholar]
- 19.Aristimuno C, de Andres C, Bartolome M, de las Heras V, Martinez-Gines ML, Arroyo R, et al. Clinical immunology. Vol. 134. Orlando, Fla: IFNbeta-1a therapy for multiple sclerosis expands regulatory CD8+ T cells and decreases memory CD8+ subset: a longitudinal 1-year study; pp. 48–57. [DOI] [PubMed] [Google Scholar]
- 20.Aristimuno C, Navarro J, de Andres C, Martinez-Gines L, Gimenez-Roldan S, Fernandez-Cruz E, et al. Expansion of regulatory CD8+ T-lymphocytes and fall of activated CD8+ T-lymphocytes after i.v. methyl-prednisolone for multiple sclerosis relapse. J Neuroimmunol. 2008;204:31–5. doi: 10.1016/j.jneuroim.2008.08.009. [DOI] [PubMed] [Google Scholar]
- 21.Fletcher JM, Lalor SJ, Sweeney CM, Tubridy N, Mills KH. T cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Clinical and experimental immunology. 162:11. doi: 10.1111/j.1365-2249.2010.04143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tennakoon DK, Mehta RS, Ortega SB, Bhoj V, Racke MK, Karandikar NJ. Therapeutic induction of regulatory, cytotoxic CD8+ T cells in multiple sclerosis. J Immunol. 2006;176:119–29. doi: 10.4049/jimmunol.176.11.7119. [DOI] [PubMed] [Google Scholar]
- 23.Zozulya AL, Wiendl H. The role of CD8 suppressors versus destructors in autoimmune central nervous system inflammation. Human immunology. 2008;69:97–804. doi: 10.1016/j.humimm.2008.07.014. [DOI] [PubMed] [Google Scholar]
- 24.Jiang H, Ware R, Stall A, Flaherty L, Chess L, Pernis B. Murine CD8+ T cells that specifically delete autologous CD4+ T cells expressing V beta 8 TCR: a role of the Qa-1 molecule. Immunity. 1995;2:85–94. doi: 10.1016/s1074-7613(95)80079-4. [DOI] [PubMed] [Google Scholar]
- 25.Jiang H, Zhang SI, Pernis B. Science. Vol. 256. New York, NY: 1992. Role of CD8+ T cells in murine experimental allergic encephalomyelitis; pp. 213–5. [DOI] [PubMed] [Google Scholar]
- 26.Koller BH, Marrack P, Kappler JW, Smithies O. Science. Vol. 248. New York, NY: 1990. Normal development of mice deficient in beta 2M, MHC class I proteins, and CD8+ T cells; pp. 227–30. [DOI] [PubMed] [Google Scholar]
- 27.Das P, Drescher KM, Geluk A, Bradley DS, Rodriguez M, David CS. Complementation between specific HLA-DR and HLA-DQ genes in transgenic mice determines susceptibility to experimental autoimmune encephalomyelitis. Hum Immunol. 2000;61:79–89. doi: 10.1016/s0198-8859(99)00135-4. [DOI] [PubMed] [Google Scholar]
- 28.Mangalam A, Luckey D, Basal E, Behrens M, Rodriguez M, David C. HLA-DQ6 (DQB1*0601)-restricted T cells protect against experimental autoimmune encephalomyelitis in HLA-DR3.DQ6 double-transgenic mice by generating anti-inflammatory IFN-gamma. J Immunol. 2008;180:747–56. doi: 10.4049/jimmunol.180.11.7747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pavelko KD, Howe CL, Drescher KM, Gamez JD, Johnson AJ, Wei T, et al. Interleukin-6 protects anterior horn neurons from lethal virus-induced injury. J Neurosci. 2003;23:81–92. doi: 10.1523/JNEUROSCI.23-02-00481.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Inaba K, Inaba M, Romani N, Aya H, Deguchi M, Ikehara S, et al. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med. 1992;176:693–702. doi: 10.1084/jem.176.6.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Giri S, Nath N, Smith B, Viollet B, Singh AK, Singh I. 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside inhibits proinflammatory response in glial cells: a possible role of AMP-activated protein kinase. J Neurosci. 2004;24:79–87. doi: 10.1523/JNEUROSCI.4288-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen ML, Yan BS, Kozoriz D, Weiner HL. Novel CD8+ Treg suppress EAE by TGF-beta- and IFN-gamma-dependent mechanisms. European journal of immunology. 2009;39:423–35. doi: 10.1002/eji.200939441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cortesini R, LeMaoult J, Ciubotariu R, Cortesini NS. CD8+CD28- T suppressor cells and the induction of antigen-specific, antigen-presenting cell-mediated suppression of Th reactivity. Immunological reviews. 2001;182:01–6. doi: 10.1034/j.1600-065x.2001.1820116.x. [DOI] [PubMed] [Google Scholar]
- 34.Cosmi L, Liotta F, Lazzeri E, Francalanci M, Angeli R, Mazzinghi B, et al. Human CD8+CD25+ thymocytes share phenotypic and functional features with CD4+CD25+ regulatory thymocytes. Blood. 2003;102:107–14. doi: 10.1182/blood-2003-04-1320. [DOI] [PubMed] [Google Scholar]
- 35.Najafian N, Chitnis T, Salama AD, Zhu B, Benou C, Yuan X, et al. Regulatory functions of CD8+CD28- T cells in an autoimmune disease model. J Clin Invest. 2003;112:037–48. doi: 10.1172/JCI17935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rifa'i M, Kawamoto Y, Nakashima I, Suzuki H. Essential roles of CD8+CD122+ regulatory T cells in the maintenance of T cell homeostasis. J Exp Med. 2004;200:123–34. doi: 10.1084/jem.20040395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rifa'i M, Shi Z, Zhang SY, Lee YH, Shiku H, Isobe K, et al. CD8+CD122+ regulatory T cells recognize activated T cells via conventional MHC class I-alphabetaTCR interaction and become IL-10-producing active regulatory cells. International immunology. 2008;20:7–47. doi: 10.1093/intimm/dxn052. [DOI] [PubMed] [Google Scholar]
- 38.Goverman J, Perchellet A, Huseby ES. The role of CD8(+) T cells in multiple sclerosis and its animal models. Current drug targets. 2005;4:39–45. doi: 10.2174/1568010053586264. [DOI] [PubMed] [Google Scholar]
- 39.Whitacre C. Spotlight on CD8 T cells in MS. Blood. 2004;103:3999. [Google Scholar]
- 40.Koh DR, Fung-Leung WP, Ho A, Gray D, Acha-Orbea H, Mak TW. Science. Vol. 256. New York, NY: 1992. Less mortality but more relapses in experimental allergic encephalomyelitis in CD8-/- mice; pp. 210–3. [DOI] [PubMed] [Google Scholar]
- 41.Abdul-Majid KB, Wefer J, Stadelmann C, Stefferl A, Lassmann H, Olsson T, et al. Comparing the pathogenesis of experimental autoimmune encephalomyelitis in CD4-/- and CD8-/- DBA/1 mice defines qualitative roles of different T cell subsets. J Neuroimmunol. 2003;141:0–9. doi: 10.1016/s0165-5728(03)00210-8. [DOI] [PubMed] [Google Scholar]
- 42.Dittel BN. CD4 T cells: Balancing the coming and going of autoimmune-mediated inflammation in the CNS. Brain, behavior, and immunity. 2008;22:421–30. doi: 10.1016/j.bbi.2007.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Goverman JM. Immune tolerance in multiple sclerosis. Immunological reviews. 241:228–40. doi: 10.1111/j.1600-065X.2011.01016.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Klebb G, Autenrieth IB, Haber H, Gillert E, Sadlack B, Smith KA, et al. Interleukin-2 is indispensable for development of immunological self-tolerance. Clinical immunology and immunopathology. 1996;81:282–6. doi: 10.1006/clin.1996.0190. [DOI] [PubMed] [Google Scholar]
- 45.Filaci G, Fenoglio D, Indiveri F. CD8(+) T regulatory/suppressor cells and their relationships with autoreactivity and autoimmunity. Autoimmunity. 44:51–7. doi: 10.3109/08916931003782171. [DOI] [PubMed] [Google Scholar]
- 46.Shi Z, Okuno Y, Rifa'i M, Endharti AT, Akane K, Isobe K, et al. Human CD8+CXCR3+ T cells have the same function as murine CD8+CD122+ Treg. European journal of immunology. 2009;39:2106–19. doi: 10.1002/eji.200939314. [DOI] [PubMed] [Google Scholar]
- 47.Shi Z, Rifa'i M, Lee YH, Shiku H, Isobe K, Suzuki H. Importance of CD80/CD86-CD28 interactions in the recognition of target cells by CD8+CD122+ regulatory T cells. Immunology. 2008;124:121–8. doi: 10.1111/j.1365-2567.2007.02747.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Endharti AT, Rifa IM, Shi Z, Fukuoka Y, Nakahara Y, Kawamoto Y, et al. Cutting edge: CD8+CD122+ regulatory T cells produce IL-10 to suppress IFN-gamma production and proliferation of CD8+ T cells. J Immunol. 2005;175:7093–7. doi: 10.4049/jimmunol.175.11.7093. [DOI] [PubMed] [Google Scholar]
- 49.Willenborg DO, Fordham SA, Staykova MA, Ramshaw IA, Cowden WB. IFN-gamma is ritical to the control of murine autoimmune encephalomyelitis and regulates both in the periphery and in the target tissue: a possible role for nitric oxide. J Immunol. 1999;163:5278–86. [PubMed] [Google Scholar]
- 50.Lee YH, Ishida Y, Rifa'i M, Shi Z, Isobe K, Suzuki H. Essential role of CD8+CD122+ regulatory T cells in the recovery from experimental autoimmune encephalomyelitis. J Immunol. 2008;180:825–32. doi: 10.4049/jimmunol.180.2.825. [DOI] [PubMed] [Google Scholar]
- 51.Willenborg DO, Fordham S, Bernard CC, Cowden WB, Ramshaw IA. IFN-gamma plays a critical down-regulatory role in the induction and effector phase of myelin oligodendrocyte glycoprotein-induced autoimmune encephalomyelitis. J Immunol. 1996;157:3223–7. [PubMed] [Google Scholar]
- 52.Zozulya AL, Ortler S, Fabry Z, Sandor M, Wiendl H. The level of B7 homologue 1 expression on brain DC is decisive for CD8 Treg cell recruitment into the CNS during EAE. European journal of immunology. 2009;39:1536–43. doi: 10.1002/eji.200839165. [DOI] [PMC free article] [PubMed] [Google Scholar]