

Abstract
Introduction
As part of our program to develop estrogen receptor (ER) targeted imaging and therapeutic agents we chose to evaluate 11β-substituted estradiol analogs as a representative scaffold. Previous synthetic studies provided an entry into this class of compounds and other work indicated that 11β-(substituted aryl) estradiol analogs were potent antagonists of the ER. Little information existed about the specific structural features involved in the transition from agonism to antagonism for the 11β-aryl estradiol analogs or their potential as scaffolds for drug conjugation.
Methods
We prepared and characterized a series of 11β-(4-Substituted phenyl) estradiol analogs using modifications of existing synthetic methods. The new compounds, as well as standard steroidal agonists and antagonists, were evaluated as competitive ligands for the ERβ-LBD. Functional assays used the induction of alkaline phosphatase in Ishikawa cells to determine potency of the compounds as ER agonists or antagonists.
Results
The synthetic strategy successfully generated a series of compounds in which the 4-substituent was sequentially modified from hydroxyl to methoxy to azidoethoxy/N,N-dimethylaminoethoxy and eventually to a prototypical 1,4-naphthoquinone-containing moiety. The new compounds all retained high relative binding affinity (RBA) for the ERα-LBD, ranging from 13–83% that of estradiol. No subtype selectivity was observed. More importantly, the transition from agonist to antagonist activity occurs at the 4-methoxy stage where the compound is a mixed antagonist. More notably, antagonism appeared to be more dependent upon the size of the 11β-substituent than upon the nature of the terminal group
Conclusions
We have developed a synthetic strategy that provides facile access to potent 11β-(4-substituted phenyl) estradiol analogs. The resultant compounds retain high affinity for the ERα-LBD and, more importantly, demonstrate potent antagonist activity in cells. Large functionalities distal to the 11β-phenyl ring had little additional effect on either affinity or efficacy, suggesting the incorporation of diverse imaging or biologically active groups can be attached without significantly compromising the ER-binding capacity. Future studies are in progress to exploit the 11β-aryl estradiol analogs as potential drug delivery systems and imaging agents.
Keywords: Steroidal anti-estrogens, Estrogen receptor, Synthesis, Agonist, Antagonist, Conformational analysis
1. Introduction
The estrogen receptor (ER) is a member of the nuclear receptor (NR) superfamily of transcription factors, a group of proteins that mediate a wide variety of physiological and developmental processes. 1,2 Because inappropriate or over-expression of ER is associated with a number of endocrine disorders, such as breast, endometrial and ovarian cancer, and osteoporosis, chemical modulation of the ER-regulated pathways is a critical clinical objective. 3–5 A number of reviews have described the structure of the ER, including its subtypes, and the general mechanism by which binding of the endogenous ligand initiates the events leading to transcription (agonist responses).6–11 The process leading to ER antagonism, although less well defined, has been described with increasing detail.12–14 While many individual steps are involved in the overall process, the initial binding of the ligand to the (apo)receptor to generate a stable complex constitutes the key step that defines the subsequent events. Because virtually all subsequent biological responses, including antagonist responses, are influenced by the conformation of the receptor–ligand complex, studies that enhance our understanding of this initial interaction and its therapeutic implications are valuable.
Our research has primarily focused on the systematic preparation of modified derivatives of estradiol that evaluate the influence of structural properties on receptor affinity, selectivity and efficacy. Although most of our efforts focused on the 17α-position, through the use of the phenyl vinyl moiety,15–22 we also conducted studies evaluating the effect of small 11β-substituents on estrogenic function. 23–28 In particular, we observed, as have others,29–35 the potency- enhancing influence of small substituents, such as methoxy, ethyl and vinyl, at the 11β position of estradiol. Notably, all of these small 11β-alkyl/alkoxy substituted estradiol derivatives were ER agonists.
In our current research program we were interested in developing new drug/imaging-estradiol conjugates for the treatment or diagnosis of diseases that over-express ER, such as hormone responsive breast cancer (Fig. 1). Recent reviews reported that the most common sites of conjugation were the 3,6,16α, 17α and 17β-positions of estradiol, however, in almost all examples, introduction of the therapeutic or imaging groups led to significant loss of ER-binding affinity and therefore target cell selectivity.36,37 Of equal concern to us was the observation that the conjugates were based on agonist structures which would tend to lead to cell proliferation rather than cytotoxicity. The only structural variations that addressed the issues of affinity and efficacy were the 7α-substituted estradiol-conjugates. Upon binding to ER, the steroid scaffold in these analogs apparently rotates around the 3–17 axis and the 7α-group occupies the 11β-binding pocket of the ER. Those few studies that used this approach reported retention of significant ER binding affinity, antagonist properties, and modest cytotoxic activity in breast cancer cell lines.38,39 Although 11β-substituted estradiols have been described as potent ER antagonists, no studies have been reported to date in which the 11β-position was successfully exploited for drug conjugation. Therefore this study was initiated with the aims of developing the chemistry needed to efficiently introduce 11β-substituents, to demonstrate that these 11β-substituted estradiol analogs would retain high ER binding affinity and generate antagonist responses, and that the 11β-substituent could be ultimately modified with terminal groups similar to those found in therapeutic drugs or imaging moieties.
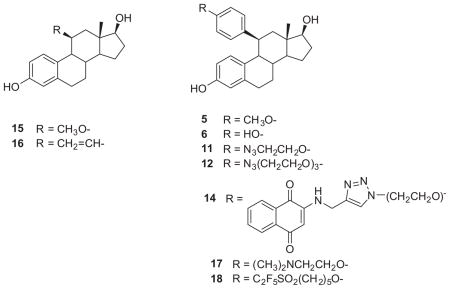
Figure 1.

Drug-estradiol conjugates for targeted drug delivery and primary sites of conjugation.
The strategy in this study addresses each of these aims. As part of earlier studies directed toward high affinity estrogens, we had established synthetic methods for the facile introduction of substituents at the 11β-position using Cu(I)-catalyzed Grignard reactions. 23,27 A significant body of work published by the pharmaceutical industry (Roussel-UCLAF and more recently Aventis) 40–46 demonstrated that estradiol, when substituted with appropriate 11β-aryl groups, were potent anti-estrogens. Borgna et al. subsequently used the 11β-aryl estradiol scaffold as the basis for developing electrophilic agents for alkylating the estrogen receptor.47–49 In those studies, simple 11β-aryl (phenyl/thienyl) estradiol analogs were agonists but that introduction of tamoxifen- like side chains (RU-39411) or substituted alkyoxy phenyl side chains (RU-58668) yielded antagonism.35,40,50–52 Hochberg et al. also reported that within a homologous series of 11β-alkyl estradiol analogs, the transition from agonism to antagonism occurred within a 1–2methylene group transition.53,54 These observations suggested that small structural modifications produced significant alterations in the conformational preferences for helix-12 of the ERα-LBD. Notably, no studies specifically addressed the transition from agonist to antagonist conformations with aryl groups within the 11β-binding pocket or the extent to which binding affinity was affected. Although previous studies suggested that terminal groups beyond the amine of tamoxifen and raloxifene-like antagonists occupy solvent accessible space, none had looked at the influence of the alkoxy group or the heteroatom on the interactions with complementary receptor residues. In this study we describe the synthesis a series of 11β-(4-substituted phenyl) estradiol analogs, their evaluation as probes (affinity and efficacy) for the ER-LBD, and their assessment as scaffolds for diagnostic/therapeutic drugestradiol conjugates.
2. Experimental
2.1. General information
All reagents and solvents were purchased from Aldrich or Fisher Scientific. THF and toluene were distilled from sodium/benzophenone. Reactions were monitored by TLC, performed on 0.2 mm silica gel plastic backed sheets containing F-254 indicator. Visualization on TLC was achieved using UV light, iodine vapor and/or phosphomolybdic acid reagent. Column chromatography was performed with 32–63 μm silica gel packing. Melting points were determined using an Electrotherm capillary melting point apparatus and are uncorrected. 1H NMR spectra were recorded with a Varian Mercury 300 MHz, a Varian 500 MHz or a Bruker 700 MHz spectrometer. DEPT and 13C experiments were performed on a Varian Mercury instrument at 75 MHz. NMR spectra chemical shifts are reported in parts per million downfield from TMS and referenced either to TMS, or internal standard for chloroform-d1, acetone-d6, methanol-d4, and THF-d8 solvent peak. Coupling constants are reported in hertz. High-resolution mass spectra were obtained by electron impact (EI) or fast atom bombardment (FAB) on MStation JMS700 (JEOL) by University of Massachusetts Amherst, Mass Spectrometry Center using sodium iodide as an internal standard. NMR spectra for the intermediates and final compounds are provided in the Supplementary data.
2.2. 3,3,17,17-Diethylenedioxy-5,10-α-epoxy-estr-9(11)-ene 3 and 3,3,17,17-Diethylenedioxy-5,10-β-epoxy-estr-9(11)-ene (2)
Estra-5(10),9(11)-diene 3,17 diethylene ketal 127 (1 g, 2.79 mmol), hexafluoroacetone trihydrate (0.04 mL, 0.279 mmol), pyridine (0.005 mL), 50% hydrogen peroxide (0.3 mL, 4.74 mmol, ca. 18 M) and dichloromethane (10 mL) were charged into a round bottom flask at room temperature under argon atmosphere. The mixture was stirred for 20 h at room temperature (TLC monitoring: ethyl acetate/hexanes, 3:7). After reductive workup (aqueous sodium thiosulfate solution, 2 g in 50 mL of water), the organic layer was washed with water (25 mL × 2), extracted with dichloromethane (30 mL × 2). The organic layer was dried over magnesium sulfate and concentrated under reduced pressure to give 2 as mixture of isomers (ratio of α:β ≈ 3:1, 1H NMR). The crude mixture could be purified from other components by chromatographic separation on a silica gel column (25 g, ethyl acetate/hexanes, 1:4). The combined fractions containing the individual α/β products were concentrated under reduced pressure.
Yield = 0.806 g, 76% Rf = 0.4 (ethyl acetate/hexanes 5:1). 1H NMR (300 MHz, CDCl3, δ 6.05 (m, 1H, 2α), 5.86 (m, 1H, 2β), 0.88 (s, 3H, 2β), 0.89 (s, 3H, 2α). 13C NMR (75 MHz, CDCl3) δ 191.1, 164.9, 132.2, 1145, 55.8.
2.3. 11β-(4-Methoxyphenyl)estra-4,9-diene-3,17 dione (3)
Copper (I) chloride (0.24 g, 2.41 mol, 0.15 equiv) was added at 20 °C (water bath) to a 1 M solution of 4-methoxyphenyl magnesium bromide in THF (5 mL, anhydrous, 2.0 equiv). A solution of the mixture 2 (α- and β-isomers, 0.8 g, 2.14 mmol, 1.0 equiv) in THF (10 mL, anhydrous) was added dropwise over 30 min at 20 °C. The reaction mixture was then stirred for 1 h at 20 °C. Upon completion (TLC monitoring: ethylacetate:hexanes, 3:7), the mixture was poured into a mixture of aqueous ammonium chloride (8 mL, 15 equiv) and methylene chloride (8 mL) at 10–15 °C. The organic layer was separated, washed with water (20 mL × 2), concentrated under reduced pressure to ~5 mL, and diluted with methylene chloride (5 mL). Aqueous hydrochloric acid (6 equiv, 0.47 g in 2.6 mL of water) was added at 0–5 °C. The mixture was stirred for 2 h at 0–5 °C and then diluted with water (20 mL). The pH of the mixture was <1 (pH paper). The organic phase was washed with water (20 mL × 2), neutralized with aqueous 10% sodium bicarbonate solution to pH 8–9, washed with water (30 mL × 3), dried over magnesium sulfate, and concentrated to dryness under vacuum. This crude product (0.66 g) was purified by column chromatography (silica gel, 25 g; ethyl acetate/hexanes, 3:7). The fractions containing the product were combined and concentrated under reduced pressure to compound 3.
Yield = 0.23 g, 25%. 1H NMR (300 MHz, CDCl3): δ 6.72 and 7.01 (AA′BB′, 4H), 5.78 (s, 1H), 4.35 (d, J = 6.6 Hz, 1H), 3.75 (s, 3H), 0.55 (s, 3H).
2.4. 11β-(4-Methoxyphenyl)estra-1,3,5-trien-3-ol-17-one (4)
To a solution of 3 (0.66 g, 1.75 mmol) in methylene chloride (10 mL), acetic anhydride (0.17 mL, d = 1.08 g/mL, 1.75 mmol, 1 equiv) was added over 5 min. Acetyl bromide (0.32 mL, 4.38 mmol, 2.5 equiv) was added dropwise over 5 min, and at a temperature between 18 and 20 °C. The solution was stirred for 5 h at room temperature (TLC monitoring: ethyl acetate/hexanes, 3:7). The mixture was carefully poured into aqueous sodium bicarbonate (10 mL, 1.34 g, 10 equiv). The mixture was stirred for 18 h at room temperature. The organic layer was separated, washed with 1N sodium hydroxide solution (25 mL × 2), water (30 mL × 2) adjusted to pH 5–6, dried over magnesium sulfate, and concentrated under reduced pressure.
The crude product (0.78 g) was dissolved in a mixture of methanol (10 mL) and methylene chloride (5 mL). A solution of potassium hydroxide (0.147 g, 2.63 mmol, 1.5 equiv) in methanol (10 mL) was added dropwise over 5 min at 0–5 °C. The mixture was stirred at 0–5 °C for 2 h (TLC monitory: ethyl acetate/hexanes, 3:7). The organic layer was separated, washed with water (30 mL × 3) to pH ~6, and brine solution (30 mL), dried over magnesium sulfate, and concentrated under vacuum. The crude product was purified using a silica gel column (25 g, ethyl acetate/hexanes, 2:3). The fractions containing the product were combined and concentrated under reduced pressure to give an oily product 4.
Yield = 0.21 g, 26%. Rf = 0.4 (ethyl acetate/hexanes, 3:7). 1H NMR(300 MHz, CDCl3) δ 6.97 (d, J = 8.7 Hz, 2H), 6.82 (d, J = 8.1 Hz, 1H, C2-H), 6.63 (d, J = 8.7 Hz, 2H), 6.61 (s, 1H, C4-H), 6.42 (dd, J = 8.4 Hz, J = 6.4 Hz, 1H, C1-H), 3.85 (t, J = 4.2 Hz, 1H, C11-Hα),3.69 (s, 3H, OCH3), 0.44 (s, 3H, CH3). 13C NMR (75 MHz, CDCl3, δ 219.9, 156.9, 153.3, 150.7, 137.7, 135.4, 130.7, 130.4, 127.9, 115.1, 113.7, 113.3, 55.2, 52.4, 48.4, 47.7, 40.2, 38.3, 35.6, 35.3, 30.2, 27.5, 21.6, 15.4.
2.5. 11β-(4-Methoxyphenyl)estra-1,3,5(10)-trien-3,17β-diol (5)
To a solution (0–5 °C) of compound 4 (55 mg, 0.147 mmol, 1 equiv) in methanol (10 mL), sodium borohydride (6 mg, 0.153 mmol, 1.05 equiv) was added portion wise. The mixture was stirred at 0–5 °C for 1.5 h (TLC monitoring: ethyl acetate/hexanes, 1:1). The excess borohydride was consumed by addition of acetone (5 mL). The suspension was stirred at 0–5 °C for 0.5 h, and poured onto a mixture of cold water (25 mL) and methylene chloride (25 mL). The organic layer was separated, washed with water (30 mL × 2), brine (30 mL), dried over magnesium sulfate, and concentrated to dryness under reduced pressure. The crude product was dissolved in isopropanol (1 mL) at room temperature. The product crystallized as needles upon standing and was collected by filtration. The crystals were rinsed with small amount of cold water and dried under reduced pressure overnight to give pure 5.
Yield = 36 mg, 65% mp: 209–211 °C. Rf = 0.4 (ethyl acetate/hexanes, 3:7). 1H MNR (300 MHz, CDCl3): δ 6.92 (d, J = 8.7 Hz, 2H), 6.75 (d, J = 8.1 Hz, 1H, C2-H), 6.56 (d, J = 8.7 Hz, 2H), 6.53 (s, 1H, C4-H), 6.35 (dd, J = 8.4 Hz, J = 6.4 Hz, 1H, C1-H), 3.88 (t, J = 4.2 Hz, 1H, C11-Hα) 3.64 (s, 3H, OCH3), 0.44 (s, 3H, CH3).
2.6. 11β-(4-Hydroxyphenyl)-estra-1,3,5(10)-triene-3,17β-diol (6)
To a solution of compound 4 (90 mg, 0.25 mmol) in methylene chloride (5 mL, anhydrous), 1 M boron tribromide (0.3 mL in methylene chloride) was added dropwise over 10 min under argon atmosphere at −78 °C (acetone/dry ice bath). The mixture was stirred at −78 °C for 1 h, and then at room temperature for 2 h. The mixture was poured into a beaker containing cold water and sodium bicarbonate (10 mg), and stirred for 10 min. The mixture was extracted with methylene chloride (30 mL × 3). The combined organic layers were washed with water (30 mL), brine (30 mL), dried over magnesium sulfate, and concentrated under reduced pressure. The crude product was purified by column chromatography (silica gel, 25 g; ethyl acetate/hexanes, 1:1). The fractions containing the product were combined and concentrated under vacuum to give pure intermediate 11β-(4-hydroxyphenyl)-estra-1,3,5(10)-trien-17-one-3-ol.
Yield = 92 mg, 99% Rf = 0.3 (ethyl acetate/hexanes = 1:1). 1H NMR(300 MHz, DCCl3,): δ 6.90 (d, J = 8.1 Hz, 2H), 6.80 (d, J = 8.7 Hz, 1H, C2-H), 6.56 (d, J = 3 Hz,1H, C4-H), 6.53 (d, J = 8.4 Hz, 2H), 6.39 (dd, J = 6 Hz, J = 3 Hz, 1H, C1-H), 4.48 (d, J = 11.1 Hz, 1H, C17-Hα), 3.90 (t, J = 9 Hz, 1H, C11-Hβ), 0.38 (s, 3H).
To a solution (0–5 °C) of the intermediate 11β-(4-hydroxyphenyl)- estra-1,3,5(10)-trien-17-one-3-ol (37 mg, 0.098 mmol) in methanol (10 mL), sodium borohydride (4 mg, 0.103 mmol, 1.05 equiv) was added and the reaction mixture was stirred for 1.5 h (TLC monitoring: ethyl acetate/hexanes, 2:3). The excess hydride was consumed by addition of acetone (5 mL). The suspension was stirred for 0.5 h at 0–5 °C and then poured onto a mixture of cold water (25 mL) and methylene chloride (25 mL). The organic layer was washed with water (25 mL × 2), brine (25 mL), dried over magnesium sulfate, and concentrated under the reduced pressure. The crude product was purified by column chromatography (silica gel, 25 g: ethyl acetate/hexanes, 2:3). The fractions containing the product were combined and concentrated under reduced pressure to give product 6.
Yield = 8 mg, 22%. Rf = 0.2 (ethyl acetate/hexanes, 2:3). 1H NMR(300 MHz, CDCl3): δ 6.90 (d, J = 8.1 Hz, 2H), 6.80 (d, J = 8.7 Hz, 1H, C2-H), 6.56 (d, J = 5.5 Hz, 2H), 6.52 (s, 1H, C4-H), 6.39 (dd, J = 8.7 Hz, J = 6.4 Hz, 1H, C1-H), 4.48 (d, J = 11.1 Hz, 1H), 3.91 (t, J = 8.1 Hz, 1H, C11-Hα), 0.31 (s, 3H, CH3).
2.7. 11β-(4-hydroxyphenyl)-estra-4,9-diene-3,17-dione (7)
Copper (I) chloride (35 mg, 0.35 mmol) was added at room temperature to a ca. 1 M solution of 4-trimethylsilyloxyphenyl magnesium bromide in THF (10 mL) under argon atmosphere. A solution of the mixture of 2α/β (ratio ≈3:1, 1H NMR) (760 mg, 2.03 mmol) in THF (10 mL) was added during ≈30 min at room temperature (exothermic). The mixture was then stirred for 1 h at room temperature (TLC monitoring: ethyl acetate/hexanes = 3:7). When the reaction went to completion, the resulting solution was poured into a biphasic mixture of aqueous ammonium chloride (15 equiv, 6 mL) and methylene chloride (8 mL) at 10–15 °C. The organic layer was separated, washed with water (20 mL × 2), concentrated the total volume to ~5 mL, and diluted with methylene chloride (5 mL). Aqueous hydrochloric acid (6 equiv, 0.47 g in 2.6 mL of water) was added at 0–5 °C. This biphasic mixture was stirred for 2 h at 0–5 °C (pH <1, pH paper) and then diluted with water (20 mL). The organic phase was separated, washed with water (20 mL × 2) and carefully neutralized to pH ≈ 8 (10% sodium bicarbonate, ~1.5 mL, pH ~7–8). The neutralized solution washed with water (30 mL × 2). The combined organic layer was dried over magnesium sulfate. Compound 7 (0.66 g, 90.4%) was isolated from a silica gel flash column chromatography (ethyl acetate/hexanes, 3:7).
Yield = 0.66 g, 90% mp 248 °C, 1H NMR (300 MHz, CDCl3: δ 6.75 and 7.07 (AA′BB′, 4H), 5.81 (s, 1H), 4.35 (d, J = 7.2 Hz, 1H), 4.01 (t, J = 6 Hz, 2H), 2.85 (t, J = 6 Hz, 2H), 2.63 (q, J = 7 Hz, 4H), 0.56 (s, 3H). 13C NMR (75 MHz, CDCl3, δ 219.3, 200.0, 156.6, 154.3, 145.6, 135.8, 130.2, 128.2, 123.5, 115.9, 50.8, 47.9, 39.8, 38.2, 38.0, 36.9, 35.6, 31.1, 26.9, 26.0, 22.1, 14.6.
2.8. 2-[2-(2-Azidoethoxy)ethoxy)ethoxy]-4-methylbenzenesulf onate (8)
To a solution of triethylene glycol (3 g, 0.02 mol) in diethyl ether (40 mL), triethylamine (7.8 g, 0.04 mol) was added at room temperature, followed by addition of p-toluenesulfonyl chloride (7.8 g, 0.04 mol) under argon atmosphere. The mixture was stirred at room temperature for 18 h (TLC monitoring, ethyl acetate/hexanes, 1:4). The organic solvents were removed under reduced pressure. The residue was dissolved in methylene chloride (20 mL), washed with sodium bicarbonate (20 mL, saturated), water (20 mL × 2), brine (30 mL), dried over magnesium sulfate and concentrated under vacuum. Colorless crystals of triethylene glycol ditosylate were obtained from ethyl acetate.
Yield = 5.8 g, 64% mp: 75–77 °C. Rf = 0.5 (ethyl acetate/hexanes, 1:4). 1H NMR (300 MHz, CDCl3): δ 7.78 and 7.34 (AA′BB′, 8H), 4.12 (d, 4H), 3.65 (t, 4H), 3.53 (s, 4H), 2.45 (s, 6H). 13C NMR (75 MHz, CDCl3, δ 145.1, 133.3, 130.1, 128.2, 70.9, 69.4, 69.0, 21.8.
To a solution of the triethylene glycol ditosylate (2 g, 4.4 mmol) in ethanol (50 mL), sodium azide (142 mg, 2.18 mmol) was added at room temperature. The mixture was heated at reflux for 2 h (TLC monitoring: ethyl acetate/hexanes, 1:1). The mixture was allowed to cool to room temperature. The colorless solid that formed upon cooling was collected by filtration and washed with small amount of solution of ethyl acetate/hexanes = 1:1. The filtrate was evaporated to dryness under reduced pressure. The oily residue was dissolved in diethyl either (100 mL), washed with water (20 mL × 2), and brine (20 mL). The organic phase was dried over magnesium sulfate, filtered and evaporated to dryness. Column chromatography on silica gel (25 g; ethyl acetate/hexanes, 2:3), gave a colorless oil X. 51
Yield = 0.26 g, 37%. Rf = 0.5 (ethyl acetate/hexanes, 1:1). 1HNMR (300 MHz, CDCl3): δ 7.78 and 7.34 (AA′BB′, 4H), 4.12 (t, 2H), 3.70 (t, 2H), 3.64 (t, 2H), 3.60 (s, 4H), 3.36 (t, 2H), 2.44 (s, 3H). 13C NMR (75 MHz, CDCl3, δ 145.0, 133.3, 130.0, 128.2, 70.0, 70.8, 70.3, 69.5, 69.0, 50.9, 21.8. IR (thin film) 2105 cm−1.
2.9. 11β-4-(2-Azidoethoxy)phenyl-estra-4,9-diene-3,17-dione (9)
11β-(4-Hydroxyphenyl)estra-4,9-diene-3,17-dione 7 (65.0 mg, 0.179 mmol) was dissolved in acetonitrile (5 mL). Cesium carbonate (234 mg, 0.717 mmol) was added to the solution and the homogeneous mixture was stirred for 15 min after which ethylene glycol ditosylate (100 mg, 0.269 mmol) was added. After 13 h, the reaction mixture was poured into a biphasic mixture of ethyl acetate (20 mL) and water (20 mL). The organic layer was washed with water (2 × 25 mL). All of the aqueous layers were combined; sodium chloride was added and then back extracted with ethyl acetate (30 mL). The organic layer was dried over magnesium sulfate, filtered and the solvent removed under reduced pressure. Flash column chromatography with hexane–ethyl acetate afforded (60 mg, 0.107 mmol, 60% yield) of the product 11β-[4-(2-tosyloxyethoxy)-phenyl]-estra-4,9-diene-3,17-dione. 1H NMR (300 MHz, CDCl3): δ 0.53 (s, 3H), 2.44 (s, 3H), 4.10 (t, 2H), 4.33 (t, 2H) 4.38 (d, J = 6.9, 1H), 5.78 (s, 1H), 6.71 (d, J = 8.4, 2H), 7.04 (d, J = 8.5, 2H), 7.33 (d, J = 8.5, 2H), 7.80 (d, J = 8.3, 2H) 13C NMR (500 MHz, acetone-d6): δ 14.23, 20.92, 21.79, 26.03, 27.09, 30.71, 35.06, 36.88, 38.17, 38.38, 39.74, 47.55, 50.74, 65.80, 69.17, 114.77, 122.95, 128.30, 130.14, 133.56, 137.53, 145.29, 155.92, 156.61, 197.54, 217.44.
Rf (dichloromethane/ethyl acetate = 7:3): 0.60.
Intermediate 11β-[4-(2-tosyloxyethoxy)-phenyl]-estra-4,9-diene-3,17-dione (80 mg, 0.143 mmol) was charged to a reaction tube and dissolved in ethanol (5 mL). Sodium azide (37.1 mg, 0.571 mmol) was added to the solution and the reaction was heated to 80 °C. After 4 h the reaction was poured into ice cold ethyl acetate (20 mL) and washed with water (3 × 20 mL). Sodium chloride was added to the combined aqueous layers and then back extracted with ethyl acetate (20 mL). The organic fractions were combined and dried over magnesium sulfate. The magnesium sulfate was filtered and the solvent removed under reduced pressure. Column chromatography using hexane/ethyl acetate afforded 11-[4-(2-azidoethoxy)-phenyl]-estra-4,9-diene-3,17-dione 9 (60 mg, 0.139 mmol, 97% yield). 1H NMR (300 MHz, CDCl3): δ 0.54 (s, 3H), 3.56 (t, 2H) 4.01 (t, 2H), 4.65 (d, J = 6.9, 1H) 5.77 (s, 1H), 6.82 (d, J = 8.8, 2H), 7.07 (d, J = 8.8, 2H).
13C NMR (300 MHz, acetone-d6): δ 14.26, 21.80, 26.02, 27.07, 30.72, 35.07, 36.89, 38.14, 38.35, 39.74, 47.55, 50.32, 50.77, 67.25, 114.77, 122.96, 128.47, 130.00, 137.44, 145.32, 155.94, 156.79, 197.54, 217.44 IR: 2108.98 cm−1 (N3–).
2.10. 11β-(4-((2-(2-(2-Azidoethoxy)ethoxy)ethoxy)phenyl)-estra-4,9-diene-3,17-dione (10)
11β-(4-Hydroxyphenyl)-estra-4,9-dien-3,17-dione 7 (14 mg, 0.04 mmol), potassium carbonate (44 mg, 0.32 mmol) and acetonitrile (2.4 mL, anhydrous) were added to a round bottom flask at room temperature and stirred for 5 min, then azido-triethylene glycol tosylate 8 (26 mg, 0.08 mmol) was added at room temperature. The mixture was stirred at 80 °C for 10 h (TLC monitoring: ethyl acetate/hexanes, 1:1). The reaction mixture was poured onto a mixture of cold water (10 mL) and methylene chloride (20 mL), stirred for 10 min, extracted with methylene chloride (25 mL × 2), washed with water (20 mL), brine (20 mL), dried over magnesium sulfate, and concentrated under reduced pressure. Without further purification, a crude material was obtained containing mixture of product and starting materials.
1H NMR (300 MHz, CDCl3,): δ 7.08 and 6.84 (AA′BB′, J = 8.7 Hz, J = 9.0 Hz, 4H), 5.80 (s, 1H, C4-H), 4.38 (d, J = 6.6 Hz, 2H, C11α-H), 4.10 (t, J = 4.8 Hz, 2H), 3.86 (t, J = 5.4 Hz, 2H), 3.74 (m, 2H), 3.68 (m, 2H), 3.39 (t, J = 5.1 Hz, 2H), 0.55 (s, 3H, C18-CH3). 13C NMR (75 MHz, CDCl3,): δ 219.0, 199.4, 157.2, 156.1, 145.1, 136.3, 130.3, 128.5, 123.6, 114.9, 71.1, 70.9, 70.3, 67.6, 50.9, 50.9, 47.9, 39.8, 38.2, 38.0, 37.0, 35.6, 31.1, 27.0, 26.1, 22.1, 14.6.
2.11. 11β-(4-(2-azidoethoxy)phenyl)-estra-1,3,5(10)-trien-3,17β-diol (11)
11β-[4-(2-Azidoethoxy)-phenyl)-estra-4,9-dien-3,17-dione 8 (19.0 mg, 0.044 mmol) was charged to a round bottom flask and dissolved in methylene chloride (3 mL). To this solution was added acetic anhydride (4.16 μL, 0.044 mmol) and acetyl bromide (8.2 μL, 0.110 mmol). After 4 h the reaction mixture was poured into a solution of sodium bicarbonate (37.0 mg, 0.44 mmol) in water (15 mL). The product was extracted with ethyl acetate (10 mL) and washed with water (3 × 15 mL). The aqueous washes were combined and back extracted with ethyl acetate (30 mL). The organic fractions were combined and dried over magnesium sulfate, which was removed by filtration and the solvent removed under reduced pressure leaving (20 mg, 0.042 mmol, 96% yield) of 3-acetoxy-11β-(2-azidoethoxyphenyl)-estra-1,3,5(10)-triene-17-one. 1H NMR (300 MHz, acetone-d6): δ 0.41 (s, 3H), 2.17 (s, 3H), 2.99 (m, 1H (11-H)), 3.59 (t, 2H) 4.10 (t, 2H), 6.63 (dd, 1H (2-H)), 6.70 (d, J = 8.8, 2H), 6.87 (d, J = 2.4, 1H (4-H)), 7.01 (d, J = 8.5, 1H (1-H)), 7.09 (d, J = 8.3, 2H) 13C NMR (500 MHz, acetone-d6): δ 15.00, 20.35, 21.27, 27.26, 29.96, 34.89, 35.18, 38.39, 40.46, 47.7, 48.00, 50.32, 52.14, 67.05, 113.87, 119.29, 121.90, 127.58, 130.93, 135.82, 136.31, 137.88, 148.75, 156.00, 168.96, 217.27.
Intermediate 3-acetoxy-11β-(2-azidoethoxyphenyl)-estra- 1,3,5(10)-triene-17-one (29.0 mg, 0.06 mmol) was dissolved in methanol (5 mL). To this solution was added sodium borohydride (3.0 mg, 0.079 mmol). After 1 h 10 N sodium hydroxide (0.024 mL, 0.245 mmol) was added and the reaction continued for 16 h. The reaction was poured into an ice cold biphasic mixture of ethyl acetate (20 mL) and water (20 mL), after which the organic layer was washed with water (2 × 25 mL). The aqueous washes were combined, sodium chloride was added and then back extracted with ethyl acetate (30 mL). The organic layer was dried over magnesium sulfate. After filtration the solvent was adsorbed onto Florisil under reduced pressure. Flash chromatography with hexane; ethyl acetate afforded 11β-[4-(2-azidoethoxy)-phenyl]-estra- 1,3,5(10)-triene-3,17β-diol 11 (26 mg, 0.06 mmol, 98% yield). 1H NMR (300 MHz, CDCl3): δ 0.33 (s, 3H), 3.59 (t, 2H), 3.95 (m, 1H 11-H)), 4.09 (t, 2H), 6.37 (dd, 1H (2-H)), 6.55 (d, J = 2.4, 2H), 6.67 (d, J = 8.8, 2H (4-H)), 6.78 (d, J = 8.8, 1H (1-H)), 7.06 (d, J = 8.6, 1H), 13C NMR (500 MHz, acetone-d6): δ 12.98, 23.21, 28.29, 29.75, 30.12, 35.85, 38.70, 43.96, 45.99, 47.68, 50.34, 52.09, 67.02, 81.77, 113.25, 113.54, 115.27, 127.65, 128.12, 129.72, 130.21, 131.03, 137.25, 137.67, 154.00, 155.67 IR: 2086.03, 3351.50 cm−1 HRMS calcd for C26H31N3O3 m/e 433.2365, found m/e 433.2325.
2.12. 11β-(4-((2-(2-(2-Azidoethoxy)ethoxy)ethoxy)phenyl) estra-1,3,5(10)-trien-3,17β-diol (12)
To a round bottom flask, the mixture containing 10 (24 mg, 0.046 mmol) and methylene chloride (5 mL, anhydrous) were added under argon atmosphere at room temperature. Then acetic anhydride (4 μL, 4.7 mg, d = 1.080 g/mL, 0.046 mmol) was added over ~5 min at room temperature, followed by acetyl bromide (9 μL, 14 mg, d = 1.663 g/mL, 0.115 mmol) for 5 min at room temperature under argon atmosphere. The light yellow mixture was stirred at room temperature for 10 h (TLC monitoring: ethyl acetate/hexanes, 1:1). The mixture was then carefully poured into aqueous sodium carbonate (40 mg, in 5 mL of water, 10 equiv), and stirred for 14 h at room temperature. The organic layer was separated, washed with 1 N sodium hydroxide solution (25 mL × 2), water (25 × 2), dried over magnesium sulfate and concentrated under reduced pressure to give the crude 3-acetoxy estra-trien-17-one intermediate (69 mg).
To a solution (0–5 °C) of crude intermediate (69 mg) in methylene chloride (2 mL), a solution of potassium hydroxide (4 mg) in methanol (5 mL) was added at 0–5 °C. The mixture was stirred at 0–5 °C for 4 h (TLC monitoring: ethyl acetate/hexanes, 1:1). The mixture was diluted with methylene chloride (30 mL), washed with water (20 mL × 2), brine (20 mL), dried over magnesium sulfate, and concentrated under reduced pressure. The crude product was purified by column chromatography on silica gel (10 g, ethyl acetate/hexanes, 2:3). The fractions containing the product were combined and evaporated to dryness, yielding the estra-trien-17- one-3-ol intermediate.
Yield = 15 mg, 63% Rf = 0.4 (ethyl acetate/hexanes, 1:1). 1H NMR (300 MHz, CDCl3) 13C NMR (75 MHz, CDCl3) [Spectra in Supplementary data].
To a 0–5 °C solution of the estra-trien-17-one-3-ol intermediate (15 mg, 0.029 mmol) in methanol (10 mL), sodium borohydride (2 mg, 0.03 mmol) was added as one portion. The mixture was stirred for 1 h at 0–5 °C (TLC monitoring: ethyl acetate/hexanes, 1:1). An additional equivalent of sodium borohydride (2 mg, 0.03 mmol) was added and the reaction mixture was stirred for an additional 1.5 h. The excess borohydride was consumed by addition of acetone (5 mL). The suspension was stirred for 0.5 h at 0–5 °C, and then poured into a mixture of ice-water (10 mL) and methylene chloride (30 mL). The organic layer was extracted with methylene chloride (20 mL × 2), washed with water (25 mL × 2) to pH ≈6–7, brine (30 mL), dried over magnesium sulfate, and concentrated under reduced pressure. After a silica gel column chromatography purification (ethyl acetate/hexanes, 1:1), 11β-(4-((2-(2-(2-azidoethoxy) ethoxy)ethoxy)phenyl) estra-1,3,5(10)-trien-3,17β-diol 12 was isolated.
Yield = 12 mg, 80% Rf = 0.5 (ethyl acetate/hexanes, 1:1). 1H NMR (300 MHz, CDCl3, δ 6.63 (d, J = 4.8 Hz, 2H), 6.48 (d, J = 5.4 Hz, 1H), 6.29 (d, J = 5.1 Hz, 2H), 6.25 (d, J = 1.5 Hz, 1H), 6.08 (dd, J = 5 Hz, J = 1. 5 Hz, 1H), 4.00 (t, J = 2.7 Hz, 2H), 3.92 (t, 1H, H-11α), 3.79 (t, J = 3.3 Hz, 1H), 3.70 (m, 3H), 3.65 (m, 4H), 1.77 (t, J = 3 Hz, 1H), 2.96 (t, J = 9 Hz, 1H), 2.84 (s, 1H), 2.80 (d, J = 3 Hz, 1H), 2.49 (dd, J = 1.2 Hz, J = 7.7 Hz, 1H), 1.00–2.30 (m, 10H), 0.32 (s, 3H, C18-CH3) 13C NMR (75 MHz, CDCl3, δ 155.9, 153.3, 137.9, 136.2, 130.8, 127.9, 115.5, 113.8, 113.5, 82.8, 71.0, 70.9, 70.3, 70.1, 67.3, 52.0, 50.9, 47.6, 45.7, 43.8, 38.5, 35.6, 30.7, 30.4, 28.2, 23.4, 13.1. HRMS calcd for C30H39N3O5 m/e 521.2890, found m/e 521.2840.
2.13. N-Propargyl-2-amino-1,4-naphthoquinone (13)
To a solution of 2-methoxyl-1,4-naphthoquinone (2.28 g, 18 mmol) in methylene chloride (30 mL), propargylamine (1 g, 12 mmol) was added at room temperature under argon atmosphere. The mixture was heated at reflux (~45 °C) and stirred for 18 h (TLC monitoring: methylene chloride, 100% and checked by 1H NMR in CDCl3). The solvents were removed under reduced pressure and the crude product was purified column chromatography on silica gel (70 g, dichloromethane, 100%). The fractions containing the product were combined and concentrated under vacuum to give the pure material 13.56
Yield = 0.24 g, 6%. Rf = 0.3 (methylene chloride, 100%). 1H NMR (300 MHz, CDCl3): δ 8.12 (d, 1H), 8.10 (d, 1H), 7.75 (t, 1H), 7.64 (t, 1H), 5.85 (s, 1H), 4.00 (d, 2H), 2.4 (t, 1H). 13C NMR (75 MHz, CDCl3): δ 183.4, 181.7, 150.7, 147.3, 135.0, 133.5, 132.5, 130.7, 126.6, 126.5, 102.8, 73.6, 32.5.
2.14. 2-(((1-(2(2(2-(4–3,17β-Dihydroxy-estra-1,3,5(10)-trien-11β-yl)phenoxy)ethoxy)ethoxy)ethyl-1H-1,2,3-triazol-4-yl)methyl)amino)naphthalenen-1,4-dione (14)
N-Propargyl-2-amino-1,4-naphthoquinone 13 (12 mg, 0.023 mmol) and compound 12 (7.3 mg, 0.035 mmol) were suspended in a mixture of water and t-butyl alcohol (5 mL, water/tbutyl alcohol = 1:1). A freshly prepared solution of sodium ascorbate (23 mg, 0.115 mmol, 5 mol %) in water (0.5 mL) was added, followed by a freshly prepared solution of copper (II) sulfate penta-hydrate (6 mg, 0.023 mmol, 1 mol %) in water (0.5 mL) at room temperature. The heterogeneous mixture was stirred at the same temperature for 18 h (TLC monitoring: ethyl acetate, 100%). The mixture was diluted with ice-water (15 mL), extracted with methylene chloride (30 mL × 3). Combined organic layers were washed with water (20 ml × 2), brine (30 mL), dried over magnesium sulfate, and concentrated to dryness under reduced pressure. The crude product was purified by column chromatography on silica gel (5 g; ethyl acetate/hexanes, 9:1) to give pure 14.
Yield = 8 mg, 48%. 1H NMR (300 MHz, CDCl3: δ 8.08 (q, 2H), 7.90 (s, 1H), 7.73 (t, J = 6.7 Hz, 1H), 7.63 (t, J = 7.0 Hz, 1H), 6.94 (d, J = 8.7 Hz, 1H), 6.75 (d, J = 8.1 Hz, 1H), 6.61 (d,s, 3H), 6.46 (d, J = 9 Hz, 1H), 6.39 (s, 1H, NH), 5.74 (s, 1H), 4.52 (t, J = 4.2 Hz, 2H), 4.22 (m, 2H), 4.02 (q, 2H), 3.91 (br, 1H), 3.83 (d, J = 4.5 Hz, 2H), 3.78 (t, J = 4.2 Hz, 2H), 3.68–3.60 (br, 4H), 0.32 (s, 3H).
2.15. Biological assays
2.15.1. Competitive binding to ERα-LBD
Binding affinities of the 11β-substituted estradiol derivatives relative to E2 were performed in incubations with the LBD of ERα. in lysates of Escherichia coli in which the LBD of human ERα (M250–V595)] is expressed as described57,58 The assay was performed overnight in phosphate buffered saline + 1 mM EDTA at room temperature. The competition for binding of [3H]E2 to the LBD of the E2-derivatives in comparison to E2, relative binding affinity (RBA) was determined over a range of concentrations from 10−12 to 10−6 M. After incubation, the media is aspirated, the plates are washed 3 times and the receptor bound radioactivity absorbed to the plates are extracted with methanol and counted. The results, as RBAs compared to E2, of all receptor studies shown in Table 1, are from 3 experiments performed in duplicate. RBAs represent the ratio of the EC50 of E2 to that of the steroid analog × 100 using the curve fitting program Prism to determine the EC50. Selected estradiol analogs were evaluated as ligands for the ERβ-LBD using a similar protocol.
Table 1.
Relative binding affinity (RBA) and relative stimulatory activity/Ki of 11β-substituted estadiol analogs at ERα-LBD
| ||||
|---|---|---|---|---|
| Compound | RBAa E2 = 100% | RSAb E2 = 100 | Ki(nM)c | Efficacyd |
| 5 | 12.7 ± 1.5 | Max stim = 35–45% | n.d. | Mixed Agonist-Antagonist |
| 6 | 67 ± 8 | 17.0 ± 8.5 | Agonist | |
| 11 | 39 ± 9 | n.d. | Antagonist | |
| 12 | 26 ± 9 | 2.4 ± 0.6 | Antagonist | |
| 14 | 51 ± 13 | 1.1 ± 0.1 | Antagonist | |
| 15 | 19.0±4.6 | 76 ±15 | Agonist | |
| 16 | 22.5±0.7 | 70 ± 5 | Agonist | |
| 17 RU 39411 | 38 ± 9 | 41.5 ± 9.2 | Antagonist | |
| 18 RU 58668 | 83 ± 13 | 5.3 ± 9.2 | Antagonist | |
Relative binding affinity (RBA) values were determined by a competitive binding assay using [3H] estradiol and ERα-LBD, as described in the Experimental Section. The RBA of estradiol (E2) is defined as 100, and the measured RBA values represent the mean of two or more independent determinations.
The relative stimulatory activity (RSA) values for ER agonists were determined using the induction of alkaline phosphastase (AlkP) in human endometrial adenocarcinoma (Ishikawa) cells as described in the Experimental section. The RSA of estradiol (E2) is defined as 100.
The inhibitory constants Ki of each antagonist compound tested at a range of 10−6 to 10−12 M and measured for the inhibition of the action of 10−9M E2 (EC50 ~0.2 nM). Each compound was analyzed in at least 3 separate experiments performed in duplicate. D Agonists generated full stimulatory response in Ishikawa assay in the absence of estradiol. Antagonists generated no stimulatory response in the absence of estradiol but elicited full block of 0.1 nM estradiol effect with increasing concentration of test compound. n.d. = not determined.
2.15.2. Estrogenic potency in Ishikawa cells
The estrogenic potency of the E2-analogs was determined in an estrogen bioassay, the induction of AlkP in human endometrial adenocarcinoma cells (Ishikawa) grown in 96-well microtiter plates as we have previously described.59 The cells are grown in phenol red free medium with estrogen depleted (charcoal stripped) bovine serum in the presence or absence of varying amounts of the steroids, across a dose range of at least 6 orders of magnitude. After 3 days, the cells are washed, frozen and thawed, and then incubated with 5 mM p-nitrophenyl phosphate, a chromogenic substrate for the AlkP enzyme, at pH 9.8. To ensure linear enzymatic analysis, the plates are monitored kinetically for the production of p-nitrophenol at 405 nm. For antagonists, the effect (Ki) of each compound tested at a range of 10−6 to 10−12 M was measured for the inhibition of the action of 10−9M E2 (EC50 ~0.2 nM). Each compound was analyzed in at least 3 separate experiments performed in duplicate. The Ki and RSA (RSA = ratio of 1/EC50 of the steroid analog to that of E2 × 100) were determined using the curve fitting program Prism.
3. Results and discussion
3.1. Synthesis of estrogen receptor ligands
The synthetic targets in this study were the 11β-(4-substituted phenyl) estradiol analogs in which the 4-substituent varies from hydroxy to methoxy to longer substituted azido-alkoxy derivatives. Because the quinone moiety is a key structural feature in anticancer antibiotics, such as, mitomycin and geldanamycin,60,61, and comparable in size to many imaging groups, introduction of the terminal naphthoquinone would provide preliminary information regarding the ability of the ER to tolerate such substituents.
Our synthetic strategy [Schemes 1 and 2] was based on our previous work and incorporated more recent contributions from the literature. 44–46 In this approach, we began with diketal intermediate 1, available from estrone 3-methyl ether as described in our previous study.27 Epoxidation of 1 gave a mixture of α- and β-epoxides 2 (3:1), isolated in a yield of 76% and used directly in the next step. Cu(I)-catalyzed 1,4-Grignard addition (4-methoxyphenyl magnesium bromide) followed by dehydration, deprotection and chromatographic separation gave the 11β-(4-methoxyphenyl)-estra-4,9-diene-3,17-dione 3, in a 25% yield. Aromatization, deprotection and reduction gave 11β-(4-methoxyphenyl) estradiol 5 (18% for 3 steps).62 O-Demethylation of the intermediate estrone 4 with boron tribromide followed by reduction afforded the corresponding 11β-(4-hydroxyphenyl) analog 6 in 21% yield (2 steps). The synthesis of the remaining estradiol analogs is shown in Scheme 2. Because alkylation of 6 could proceed at either phenolic position, we prepared the 11β-(4-hydroxyphenyl)-estra-4,9-diene-3,17-dione 7 via 1,4-Grignard addition of 4-trimethylsilyloxyphenyl magnesium bromide to 2α/β. The resulting phenol 7 was elaborated using Williamson ether synthesis with ethylene glycol ditosylate to give the crude alkylation product which was converted with sodium azide in ethanol to 9. In a similar fashion, 7 was converted to the corresponding 11β-(4-((2-(2-(2-azidoethoxy) ethoxy)ethoxy)phenyl)-estra-4,9-diene-3,17-dione 10. Aroma tization, reduction, and hydrolysis gave 11β-(2-azidoethoxyphenyl) estradiol analog 1163 and 11β-(4-((2-(2-(2-azidoethoxy)ethoxy) ethoxy)phenyl) estradiol analog 12. In anticipation of future studies in which we would introduce a second bioactive or imaging moiety, we elaborated this intermediate with N-propargyl-2-aminonaphthoquinone 13, using the [3+2] Huisgen cycloaddition (‘click’) reaction, to give the terminally functionalized 11β-substituted estradiol analog 14.64–66
Scheme 1.

Synthesis of 11β-(4-methoxy/hydroxyphenyl)-estradiol analogs 5 and 6 Reagents and conditions. (a) H2O2-50%, CF3COCF3·H2O, pyridine, CH2Cl2 (b) Cu(I)Cl, 4-CH3OC6H4MgBr, THF (c) 6N HCl–CH2Cl2 (d) Ac2O, AcBr, CH2Cl2 (e) KOH, CH3OH (f) NaBH4, CH3OH (g) BBr3, CH2Cl2.
Scheme 2.

Synthesis of 11β-(4-(substituted alkoxy)phenyl)-estradiol analogs 11,12,14. Reagents and conditions. (a) Cu(I)Cl, 4-(CH3)3SiOC6H4MgBr, THF (b) 6N HCl–CH2Cl2 (c) TsOCH2CH2OTs, K2CO3, CH3CN (d) NaN3, EtOH (e) 8, Cs2CO3, CH3CN (f) Ac2O, AcBr, CH2Cl2 (g) NaBH4, CH3OH (h) KOH, CH3OH (i) 13, CuSO4·5 H2O, H2O–t-BuOH.
Although the synthesis of the target compounds involved a multi-step process, it should be noted that the individual steps proceeded in satisfactory, although unoptimized, yields. Among the key features of this synthetic scheme was the observation that separation of the two epoxide isomers, 2α and 2β, while possible, was unnecessary. As noted in the literature,40–42 work up of the subsequent Grignard reaction under acidic conditions generated the more stable 11β-substituted product. Depending upon the ultimate target, introduction of the substituent at the 4-oxyphenyl position could either precede aromatization or follow it. In either case, reproducible introduction of phenyl (aromatic) substituents with the β-orientation was critical for generating the desired biological response and high affinity. This route is a more efficient and versatile method than the alternative approach which involves Grignard addition to an 11-oxo derivative followed by eventual deoxygenation.33,34,67
3.2. Biological evaluation of estradiol analogs
The compounds were initially evaluated as competitive inhibitors of estradiol binding using ERα-LBD, and subsequently as agonists or antagonists using the induction of alkaline phosphatase activity in Ishikawa cells.55–58 11β-Methoxy estradiol 15, 11β-vinyl estradiol 16, two agonists which we had previously prepared, 23,68,69 and two steroidal antagonists, RU 39411 17 and RU 58668 18,70,71 were used for comparison purposes. The results of the binding (RBA) and stimulatory (RSA) assays are shown in Table 1. The binding results indicate that all of the steroidal derivatives possessed high relative binding affinity (RBA) for the ERα-LBD. The binding affinity was essentially independent of the efficacy of the estradiol analogs as agonists with small 11β-substituents, such as 15 and 16, had RBA values comparable to the antagonists with much large substituents, such as 12, 14, or 18.
Binding for the antagonists was also not significantly affected by the nature of the atom at the 2-position of the alkoxy moiety. Whether the substituent was azido (11), amino (17) or oxy (12), the affinities were essentially the same (RBA = 26–39%). This finding is of interest as the dimethylamino group of RU39411, comparable to the dialkylamino groups of tamoxifen and raloxifene, is believed to interact with Asp-351 though ionic or hydrogen bonding. 72 No significant differences in RBA values were observed for these compounds, suggesting that other factors may be more important. Clearly the region of the ligand binding domain that is complementary to the 11β-position of the steroid scaffold is capable of accommodating significant structural diversity at that position. It should be noted that the affinity was also not significantly affected by whether the ligand binds to the agonist or antagonist conformation of the receptor.
We were also interested in determining whether the 11β-aryl estradiol derivatives were ER-substype selective. A few studies suggested that simple 11β-substituted estradiols, such as the 11β-methoxyestradiol, are relatively nonselective ligands for the ERα/ERβ subtypes.54,73, Therefore, we evaluated three 11β-(4-substituted phenyl)estradiol analogs (6,17, 18) as ERβ ligands. The observed ERβ-LBD RBA values were 41%, 17% and 27% respectively, vs 67%, 38%, and 83% for the ERα-LBD, indicating that the analogs retained high affinity for the ERβ-subtype but were relatively nonselective. This is consistent with other studies that relate the lack of subtype selectivity to the presence of a similar receptor topography in the 11-beta pocket, with the major difference being Leu384 (ERα) and Met 336 (ERβ).74,75
Of particular interest to us was the effect of 11β-substitution on efficacy, especially for the 4-substituted phenyl derivatives. The 11β-methoxy and 11β-vinyl estradiols 15 and 16 are well established as potent estrogens, and this effect was observed in the current assay. Previous studies had shown that simple 11β-aryl estradiol analogs also were agonists, but that more highly substituted derivatives expressed anti-estrogenic properties. The specific effects on efficacy of functional groups emanating from a 11β-phenyl( aryl) substituent were not well described as most studies tend to focus on variations of the dialkylaminoalkoxy moiety. Very few studies, even with the triphenylethylenes (TPEs) have examined the structural transition from hydroxyphenyl to methoxyphenyl. In their study of triphenyl acrylonitriles (Fig. 2), Dore, et al., observed that the α,α′,β-tris(4-hydroxyphenyl) compound was a full agonist with high binding affinity (RBA = 166%), whereas the α,β-bis(4-hydroxyphenyl)-α′-(4-methoxyphenyl) analog had reduced binding (RBA = 17%) and ‘partial’ agonist responses.76,77 A larger alkoxy group, such as isopropoxy had even lower RBA and agonist effects. In other studies that examined the benzothiophene class of antiestrogens, that is, structurally related to raloxifene (Fig. 2), one can find similar effects. Griffin and co-workers used hydrogen-deuterium exchange mass spectrometry (HDX-MS) to evaluate the stability of ligand–ER-LBD complexes.78,79 In their work which included raloxifene, the de-alkylated 4-hydroxybenzoyl derivative, and other analogs of raloxifene, they determined that the 4-hydroxybenzoyl compound behaved as an agonist, had high binding affinity (RBA ~25%), and gave an HDX-MS profile similar to that for estradiol. The corresponding 4-methoxybenzoyl analog was not evaluated. What was particularly notable in this study, which evaluated a number of raloxifene analogs and other antiestrogens, was that the antagonist complexes appeared to be more stable than the agonist complexes, based upon the slower exchange rates within the ligand binding core of the receptor.
Figure 2.

Evaluation of methoxy/hydroxyl analogs of estrogens/anti-estrogens.
In our study, we examined the influence of the substituents on phenolic group, as well as the contribution of the more distal components. As the results show, the 11β-(4-hydroxyphenyl) estradiol analog exhibited high affinity (RBA = 67%) but was a slightly less potent agonist (RSA = 17%) than estradiol. Capping the terminal phenolic hydroxyl with a methyl group reduced binding affinity somewhat (RBA = 13%), but the compound now displayed mixed agonist/antagonist activity (See Supplementary data for binding curves). This effect is similar to that observed in the triphenylacrylonitrile and benzothiophene studies. What our series provides, and which was not evaluated in the other studies, is the effect of progressive modification of the alkoxy moiety. Extension of the 4-substituent from a methoxy to a 2-(azidoethoxy)-moiety transformed the ligand into essentially a full antagonist with high affinity (RBA = 39%). The distal azido group is a nonbasic, weak hydrogen bonding group which would not interact strongly with the complementary Asp-351 moiety usually associated with antagonist effects. Substitution of the azido-group by the N,N-dimethylamino group yields the known antagonist RU39411 17 (RBA = 38%), which has much higher affinity than tamoxifen (~1%). 80 This substitution provides essentially no change in either binding affinity or antagonist potency, suggesting that neither nitrogen basicity nor hydrogen bonding are the dominating effects at this site. It has been observed previously that replacement of Asp-351 by neutral amino acids did not significantly affect the binding affinity of antagonists but did influence the subsequent recruitment of coregulatory peptides.81 Weatherman and co-workers, noted in their study of GW7604 derivatives, in which a terminal carboxy group of a nonsteroidal antiestrogen was replaced by an amido or methyl ketone moiety, that affinity but not efficacy was affected.82 Further extension of the 4-substituent, as seen in RU58668 18, or the ω-substituted triethyleneglycol derivatives 12 and 14, in which the beta atom is carbon or oxygen gave equally potent ER ligands with roughly comparable anti-estrogenic properties. Introduction of a terminal 1,4-naphthoquinone moiety to give derivative 14 or the fluoroethylsulfonyl group in 18 produced, slight improvements in ERα-LBD binding and maintenance of full antagonist properties. Therefore the key feature that contributes to the antiestrogenic response appears to be the loss of the phenolic group and addition of at least a two carbon moiety.
The results from the current study demonstrated that the transition from agonist to antagonist properties occurs within a very narrow structural range for this class of compounds. One can consider the initial interaction of the steroidal ligand with the (apo)receptor as proceeding via an interaction with a receptor conformation in which the helix-12 component is loosely bound. This initial complex can then undergo transformations to generate either an agonist conformation or an antagonist conformation, depending upon the nature of the 11β-subsituent. Recruitment of coactivator proteins for the agonist conformation or corepressor proteins for the antagonist conformation then leads to the subsequent biological response. [Fig. 3.] For small functional groups at the 11β-position, represented by 11β-methoxy- or vinyl estradiol 15 or 16, the preferred conformation is clearly the agonist conformation. Folding of helix-12 to give a complex similar to that observed for estradiol is favored and yields a stabilized agonist ligand–receptor complex. As observed in this study, 11β-(4-hydroxyphenyl)estradiol 6 also prefers the agonist conformation, presumably by interactions between the 4-hydroxy moiety and the complementary residues, such as Thr-347, in the receptor binding pocket.[Fig. 4A] Such interactions have been used to explain the binding affinities observed with 1,1-bis(4-hydroxyphenyl)ethylene derivatives, such as the cyclofenil analogs.83,84 Replacement of the terminal hydrogen with a methyl group as in 11β-(4-methoxyphenyl) estradiol 5 apparently disrupts those interactions sufficiently, either by loss of hydrogen bonding or by enhanced steric interactions, such that the alternate complex with the antagonist helix-12 conformation is more competitive.[Fig. 4B] The difference in binding energy may be small as the compound displays a significant estrogenic component (30%), even at high concentrations. In addition, HDX-MS experiments suggest that the antagonist conformations are more stable than agonist conformations which may also influence the observed balance.78,79 Further modification of estradiol analogs with longer/larger substituents on the 4-oxyphenyl group results in a clear preference for the antagonist conformation, similar to that formed with tamoxifen, raloxifene and other nonsteroidal antagonists. The substituents in this study that extend beyond the methoxy-group appear to have little additional effect on the binding affinity.
Figure 3.

Proposed binding of 11β-(4-methoxyphenyl)estradiol analog with ERα-LBD. The initial interaction of the ligand with the receptor is reversible as are the conformational states. The equilibrium is influenced by the contributions of interactions of the terminal methoxy group with complementary residues in the ligand binding pocket (LBP) in the different conformations of the receptor.
Figure 4.
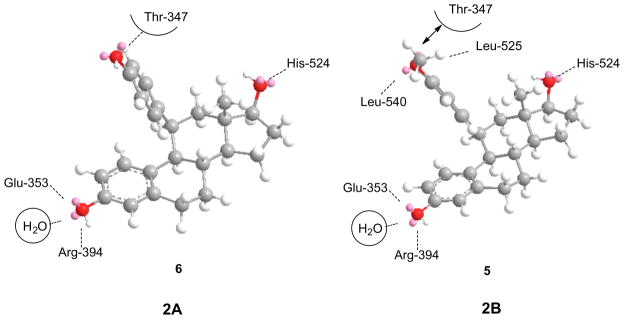
(A, B) Representation of major interactions between the 11β-(4-substituted phenyl)estradiol analogs and the agonist conformation (A) and antagonist conformation (2B) of ERα-LBD. In (A), compound 6 would have strong interactions with the HO-group of Thr-347, in addition to hydrogen bonding with Glu-353 and Arg-394 (3-phenolic – OH) and His-524 (17β-OH), stabilize the agonist conformation. In (B), compound 5 retains the strong hydrogen bonding interactions with Glu-353 and Arg-394 (3-phenolic– OH) and His-524 (17β-OH), but the methoxy-group would have weaker bonding to the HO-of Thr-347. In addition, the steric interactions would disfavor the agonist conformation, while hydrophobic interactions with Leu-525,540 present in helix-12 of the antagonist conformation would be favorable.
One potential consequence of the identification of the agonist-antagonist transition point relates to the assessment of the metabolism of anti-estrogens. With conventional nonsteroidal antiestrogens, such as tamoxifen, primary attention has focused on metabolism of the aromatic ring (4-hydroxytamoxifen) or the terminal amine group (endoxifene).85,86 [Fig. 5] In these cases, metabolism yields pharmacologically active compounds that retain antiestrogenic properties. Further metabolism of the terminal amine to a carboxylic acid, or conjugation with glucoronic acid generates inactive compounds. However, a minor but detectable pathway involves the oxidative cleavage of the dialkylaminoethoxy side chain to give the corresponding phenolic derivative. As we and others have shown, these phenolic compounds (metabolites) are potent estrogenic substances whose levels would need to be suppressed in disorders such as hormone responsive breast cancer. It would be of interest to determine the extent to which these metabolites contribute to the Selective Estrogen Receptor Modulation (SERM) effect.
Figure 5.

Metabolic pathways for tamoxifen. Cleavage of basic side chain yields compound with estrogenic properties.
In considering the design and synthesis of drug-estradiol conjugates for the targeted therapy of ER-expressing breast cancer, most studies of the interaction of anti-estrogens with ERα-LBD have used nonsteroidal agents as the ligands. Crystal structures of complexes between ERα-LBD and tamoxifen or raloxifene have shown that the N,N-dialkylaminoethoxyphenyl moiety extends almost perpendicularly from the nonsteroidal scaffold, into a pocket that has an interaction between Asp-351 and the terminal amino.86–88 In these and other complexes, the position of the terminal amine is essentially at the receptor–solvent interface. For the new compounds that display antagonist properties, one can imagine that the 11β-(4-alkoxyphenyl) group occupies essentially thesame space as the N,N-dialkylaminoethoxyphenyl moiety of the nonsteroidal antagonists. In that scenario, virtually all of the atoms that extend beyond the equivalent of the N,N-dialkylamino group would be external to the receptor surface and therefore extend into the solvent space. Because the interactions between more distal parts of the ligand and the receptor surface are likely to be weaker, receptor affinities should be relatively similar. Although our series does not have sufficient representatives to confirm that hypothesis, the results tend support this proposal. The RBA values for the azidoethoxy and N,N-dialkylaminoethoxy derivatives, as well as the more highly substituted compounds RU58668 (18), 10 and 11, are all roughly equivalent (26–83%), even though the nature of the substituents varies significantly.
The implications of these observations are clear. We have demonstrated that we can easily prepare the 11β-(4-substituted phenyl) estradiol analogs with a variety of terminal substituents. The compounds all retain high binding affinity to ER-LBD and appear to be nonselective for ER-subtypes. For those analogs in which the terminal group is larger than methoxy are potent antiestrogens. Incorporation of a terminal azido-group also permits facile introduction of groups that do not significantly alter either receptor binding or receptor pharmacology. Therefore we should be able to introduce a wide variety of groups- imaging probes or therapeutic moieties at the terminal position. Specific examples of therapeutic agents that could easily undergo this form of ligation are shown below. Amine derivatives of the quinone-containing antibiotics mitomycin C and geldanamycin are well known and preparation of alkyne-modified analogs would constitute appropriate coupling partners.89–91 [Fig. 6]
Figure 6.

Putative alkynylated therapeutic coupling partners for azido-linker substituted steroidal antiestrogen.
4. Conclusions
In this study we have described the successful synthesis of a series of novel 11β-(4-substituted phenyl)estradiol derivatives and their evaluation as ligands for the ERα-LBD. The synthetic pathway provides a facile entry into compounds that can be further elaborated as imaging or therapeutic agents targeting ER. Binding studies demonstrated that all of the new derivatives possess high affinity for ERα-LBD with RBA values in the 23–83% range. Evaluation of several compounds as ERβ-LBD ligands indicated an absence of subtype selectivity. More significantly, the compounds transition from agonists to antagonists within a very narrow structural range, from 4-hydroxy to 4-(2-azidoethoxy) and that affinity are efficacy are not significantly impacted by the nature of the atom at the 2-position of the 4-(ethoxy) group. Most of the new compounds are potent antagonists in which the terminal functional groups have little additional influence on either ER affinity or efficacy. These findings suggest that further elaboration of the terminus with either imaging groups or bioactive moieties should be well tolerated. Studies to explore those applications are in progress and will be described in subsequent publications.
Supplementary Material
Acknowledgments
Support of this research was provided by grants from the National Institutes of Health [PHS 1R01 CA81049 (R.N.H.), PHS 1R01 CA 37799 and R21 MH082252 (R.B.H.)], Department of Energy (DE-SC0001781)[RNH], the Susan G Komen Foundation (BCTR0600659)[RNH] and NSF-IGERT Award # 0504331 (J.A.H.).
Footnotes
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.bmc.2012.04.041.
References and notes
- 1.Evans RM. Science. 1988;240:889. doi: 10.1126/science.3283939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tsai M-J, O’Malley BW. Annu Rev Biochem. 1994;63:451. doi: 10.1146/annurev.bi.63.070194.002315. [DOI] [PubMed] [Google Scholar]
- 3.Gradishar W, Jordan VC. Hematol Oncol Clin North Am. 1999;13:435. doi: 10.1016/s0889-8588(05)70064-2. [DOI] [PubMed] [Google Scholar]
- 4.Clemons M, Goss PN. Eng J Med. 2001;344:276. doi: 10.1056/NEJM200101253440407. [DOI] [PubMed] [Google Scholar]
- 5.Sato M, Grese TA, Dodge JA, Bryant HU, Turner CH. J Med Chem. 1999;42:1. doi: 10.1021/jm980344o. [DOI] [PubMed] [Google Scholar]
- 6.Parker MG. Vitamins and Hormones. 1995;51:267. doi: 10.1016/s0083-6729(08)61041-9. [DOI] [PubMed] [Google Scholar]
- 7.Dickson RB, Stancel GM. J Nat’l Cancer Inst Monograph. 1999;27:135. doi: 10.1093/oxfordjournals.jncimonographs.a024237. [DOI] [PubMed] [Google Scholar]
- 8.Nilsson S, Gustafsson J-Ǻ. Crit Rev Biochem Mol Biol. 2002;37:1. doi: 10.1080/10409230290771438. [DOI] [PubMed] [Google Scholar]
- 9.Hart LL, Davie JR. Biochem Cell Biol. 2002;80:335. doi: 10.1139/o02-038. [DOI] [PubMed] [Google Scholar]
- 10.Krishnan V, Heath H, Bryant HU. Vit Horm. 2001;60:123. doi: 10.1016/s0083-6729(00)60018-3. [DOI] [PubMed] [Google Scholar]
- 11.Biggins JB, Koh JT. Curr Opinion Chem Biol. 2007;11:99. doi: 10.1016/j.cbpa.2006.10.042. [DOI] [PubMed] [Google Scholar]
- 12.Jordan VC. J Med Chem. 2003;46:883. doi: 10.1021/jm020449y. [DOI] [PubMed] [Google Scholar]
- 13.Jordan VC. J Med Chem. 2003;46:1081. doi: 10.1021/jm020450x. [DOI] [PubMed] [Google Scholar]
- 14.McKenna NJ, O’Malley BW. J Steroid Biochem Mol Biol. 2000;74:351. doi: 10.1016/s0960-0760(00)00112-6. [DOI] [PubMed] [Google Scholar]
- 15.Napolitano E, Fiaschi R, Herman LW, Hanson RN. Steroids. 1996;61:384. doi: 10.1016/0039-128x(96)00045-1. [DOI] [PubMed] [Google Scholar]
- 16.Hanson RN, Herman LW, Fiaschi R, Napolitano E. Steroids. 1996;61:718. doi: 10.1016/s0039-128x(96)00201-2. [DOI] [PubMed] [Google Scholar]
- 17.Hanson RN, Lee CY, Friel CJ, Dilis R, Hughes A, DeSombre ER. J Med Chem. 2003;46:2865. doi: 10.1021/jm0205806. [DOI] [PubMed] [Google Scholar]
- 18.Hanson RN, Friel CJ, Dilis R, Hughes A, DeSombre ER. J Med Chem. 2005;48:4300. doi: 10.1021/jm040157s. [DOI] [PubMed] [Google Scholar]
- 19.Hanson RN, Lee CY, Friel C, Hughes A, DeSombre ER. Steroids. 2003;68:143. doi: 10.1016/s0039-128x(02)00165-4. [DOI] [PubMed] [Google Scholar]
- 20.Olmsted SL, Tongcharoensirikul P, McCaskill E, Gandiaga K, Labaree D, Hochberg RB, Hanson RN. Bioorg Med Chem Lett. 2012;22:977. doi: 10.1016/j.bmcl.2011.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hanson RN, Kirss R, McCaskill E, Hua E, Tongcharoensirikul P, Olmsted SL, Labaree D, Hochberg RB. Bioorg Med Chem Lett. 2012;22:1670. doi: 10.1016/j.bmcl.2011.12.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hanson RN, McCaskill E, Tongcharoensirikul P, Dilis R, Labaree D, Hochberg RB. Steroids. 2012;77:471. doi: 10.1016/j.steroids.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hanson RN, Napolitano E, Fiaschi R, Onan KD. J Med Chem. 1990;33:3155. doi: 10.1021/jm00174a010. [DOI] [PubMed] [Google Scholar]
- 24.Hanson RN, Napolitano E, Fiaschi R. Steroids. 1998;63:479. doi: 10.1016/s0039-128x(98)00052-x. [DOI] [PubMed] [Google Scholar]
- 25.Hanson R, Napolitano E, Fiaschi R. J Med Chem. 1998;41:4686. doi: 10.1021/jm9801051. [DOI] [PubMed] [Google Scholar]
- 26.Hanson RN, Dilis R, Tongcharoensirikul P, Hughes A, DeSombre ER. J Med Chem. 2007;50:472. doi: 10.1021/jm060940f. [DOI] [PubMed] [Google Scholar]
- 27.Napolitano E, Fiaschi R, Hanson RN. Gazz Chim Ital. 1990;120:323. [Google Scholar]
- 28.French AN, Napolitano E, VanBrocklin HF, Hanson RN, Welch MJ, Katzenellenbogen JA. Nucl Med Biol. 1993;20:31. doi: 10.1016/0969-8051(93)90134-g. [DOI] [PubMed] [Google Scholar]
- 29.Gantchev TG, Ali H, van Lier JE. J Med Chem. 1994;37:4164. doi: 10.1021/jm00050a013. [DOI] [PubMed] [Google Scholar]
- 30.McElvany KD, Carlson KE, Katzenellenbogen JA, Welch MJ. J Steroid Biochem. 1983;18:635. doi: 10.1016/0022-4731(83)90240-6. [DOI] [PubMed] [Google Scholar]
- 31.Azadian-Boulanger G, Bertin D. Chim Thera. 1973;8:451. [Google Scholar]
- 32.Baran JS, Langford DD, Laos I, Liang CD. Tetrahedron. 1977;33:609. [Google Scholar]
- 33.Napolitano E, Fiaschi R, Carlson KE, Katzenellenbogen JA. J Med Chem. 1995;38:429. doi: 10.1021/jm00003a005. [DOI] [PubMed] [Google Scholar]
- 34.Tedesco R, Fiaschi R, Napolitano E. J Org Chem. 1995;60:5316. [Google Scholar]
- 35.Teutsch G, Belanger A, Philibert D, Tournemine C. Steroids. 1982;39:607. doi: 10.1016/0039-128x(82)90132-5. [DOI] [PubMed] [Google Scholar]
- 36.Biersack B, Schobert R. Curr Med Chem. 2009;16:2324. doi: 10.2174/092986709788453050. [DOI] [PubMed] [Google Scholar]
- 37.Rai S, Paliwal R, Vaidya B, Gupta PN, Mahor S, Khatri K, Goyal AK, Rawat A, Vyas SP. Curr Med Chem. 2007;14:2095. doi: 10.2174/092986707781368432. [DOI] [PubMed] [Google Scholar]
- 38.Mitra K, Marquis JC, Hillier SM, Rye PT, Zayas B, Lee AS, Essigmann JM, Croy RG. J Am Chem Soc. 2002;124:1862. doi: 10.1021/ja017344p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sharma U, Marquis JC, Nicole Dinaut A, Hillier SM, Fedeles B, Rye PT, Essigmann JM, Croy RG. Bioorg Med Chem Lett. 2004;14:3829. doi: 10.1016/j.bmcl.2004.04.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Teutsch G, Belanger Al. Tetrahedron Lett. 1979;22:2051. [Google Scholar]
- 41.Belanger A, Philibert D, Teutsch G. Steroids. 1981;37:361. doi: 10.1016/0039-128x(81)90039-8. [DOI] [PubMed] [Google Scholar]
- 42.Nique F, Van de Velde P, Bremaud J, Hardy M, Philibert D, Teutsch G. J Steroid Biochem Mol Biol. 1994;50:21. doi: 10.1016/0960-0760(94)90168-6. [DOI] [PubMed] [Google Scholar]
- 43.Claussner A, Nedelec L, Nique F, Philibert D, Teutsch G, Van de Velde P. J Steroid Biochem Mol Biol. 1992;41:609. doi: 10.1016/0960-0760(92)90392-v. [DOI] [PubMed] [Google Scholar]
- 44.Larkin JP, Wehrey C, Boffelli P, Lagraulet H, Lemaitre G, Nedelec A, Prat D. Org Proc Res Devel. 2002;6:20. [Google Scholar]
- 45.Prat D, Benedetti F, Girard GF, Bouda LN, Larkin J, Wehrey C, Lenay J. Org Proc Res Devel. 2004;8:219. [Google Scholar]
- 46.Prat D, Benedetti F, NaitBouda L, Girard GF. Tetrahedron Lett. 2004;45:765. [Google Scholar]
- 47.Borgna JL, Scali J. Eur J Biochem. 1991;199:575. doi: 10.1111/j.1432-1033.1991.tb16157.x. [DOI] [PubMed] [Google Scholar]
- 48.Aliau S, Delettre G, Mattras H, El Garrouj D, Nique F, Teutsch G, Borgna J-L. J Med Chem. 2000;43:613. doi: 10.1021/jm990179s. [DOI] [PubMed] [Google Scholar]
- 49.Aliau S, Mattras H, Borgna J-L. J Steroid Biochem Mol Biol. 2006;98:111. doi: 10.1016/j.jsbmb.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 50.Bouhoute A, Leclercq G. Biochem Pharmacol. 1994;47:748. doi: 10.1016/0006-2952(94)90140-6. [DOI] [PubMed] [Google Scholar]
- 51.Poupaert JH, Lambert DM, Vamecq J, Abul-Hajj YJ. Bioorg Med Chem Lett. 1995;5:839. [Google Scholar]
- 52.Jin L, Borras M, Lacroix M, Legros N, Leclercq G. Steroids. 1995;60:512. doi: 10.1016/0039-128x(95)00079-6. [DOI] [PubMed] [Google Scholar]
- 53.Zhang J, Labaree DC, Mor G, Hochberg RB. J Clin Endocrinol Metab. 2004;89:3527. doi: 10.1210/jc.2003-032005. [DOI] [PubMed] [Google Scholar]
- 54.Zhang JX, Labaree DC, Hochberg RB. J Med Chem. 2005;48:1428. doi: 10.1021/jm049352x. [DOI] [PubMed] [Google Scholar]
- 55.Lundt I, Steiner AJ, Stuetz AE, Tarling CA, Ully S. Bioorg Med Chem. 2006;14:1737. doi: 10.1016/j.bmc.2005.10.021. [DOI] [PubMed] [Google Scholar]
- 56.Peterson S, Gauss W, Kiehne H, Juehling L. Zeitschrift Krebsforschung. 1969;72:162. [PubMed] [Google Scholar]
- 57.Labaree DC, Shang J, Harris HA, O’Connor C, Reynolds TY, Hochberg RB. J Med Chem. 1886;2003:46. doi: 10.1021/jm0204340. [DOI] [PubMed] [Google Scholar]
- 58.Green S, Walter P, Kumar V, Krust A, Bonert JM, Argos P, Chambon P. Nature. 1986;320:134. doi: 10.1038/320134a0. [DOI] [PubMed] [Google Scholar]
- 59.Littlefield BA, Gurpide E, Markiewicz L, McKinley B, Hochberg RB. Endocrinology. 1990;127:2757. doi: 10.1210/endo-127-6-2757. [DOI] [PubMed] [Google Scholar]
- 60.Galm U, Hager MH, Van Lanen SG, Ju J, Thorson JS, Shen B. Chem Rev. 2005;105:739. doi: 10.1021/cr030117g. [DOI] [PubMed] [Google Scholar]
- 61.Taldone T, Sun W, Chiosis G. Bioorg Med Chem. 2009;17:2225. doi: 10.1016/j.bmc.2008.10.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Neef G, Sauer G, Wiechert R. Tetrahedron Lett. 1983;24:5205. [Google Scholar]
- 63.Hendricks JA, Gullà SV, Budil DE, Hanson RN. Bioorg Med Chem Lett. 2012;22:1743. doi: 10.1016/j.bmcl.2011.12.091. [DOI] [PubMed] [Google Scholar]
- 64.Kolb HC, Finn MG, Sharpless KB. Angew Chem, Int Ed. 2001;40:2004. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 65.Gil MV, Arevalo MJ, Lopez O. Synthesis. 2007:1589. [Google Scholar]
- 66.Bock VD, Hiemstra H, van Maarseveen JH. Eur J Org Chem. 2006:51. [Google Scholar]
- 67.Agouridas V, Blazejewski J-C, Magnier E, Popkin ME. J Org Chem. 2005;70:8907. doi: 10.1021/jo051424k. [DOI] [PubMed] [Google Scholar]
- 68.Azadian-Boulanger G, Bertin D. Chim Thera. 1973;8:451. [Google Scholar]
- 69.Napolitano E, Fiaschi R, Hanson RN. J Steroid Biochem Mol Biol. 1990;37:295. doi: 10.1016/0960-0760(90)90341-h. [DOI] [PubMed] [Google Scholar]
- 70.Jin L, Borras M, Lacroix M, Legros N, Leclercq G. Steroids. 1995;60:512. doi: 10.1016/0039-128x(95)00079-6. [DOI] [PubMed] [Google Scholar]
- 71.Van de Velde P, Nique F, Bouchoux F, Bremaud J, Hameau MC, Lucas D, Moratille C, Viet S, Philibert D, et al. J Steroid Biochem Mol Biol. 1994;48:187. doi: 10.1016/0960-0760(94)90144-9. [DOI] [PubMed] [Google Scholar]
- 72.Jordan VC. J Med Chem. 2003;46:883. doi: 10.1021/jm020449y. [DOI] [PubMed] [Google Scholar]
- 73.Shughrue PJ, Lubahn DB, Negro-Vilar A, Korach KS, Merchenthaler I. Proc Natl Acad Sci USA. 1997;94:11008. doi: 10.1073/pnas.94.20.11008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Katzenellenbogen JA, Muthyala R, Katzenellenbogen BS. Pure Appl Chem. 2003;75:2397. [Google Scholar]
- 75.Hsieh RW, Rajan SS, Sharma SK, Guo Y, Desombre ER, Mrksich M, Greene GL. J Biol Chem. 2006;281:17909. doi: 10.1074/jbc.M513684200. [DOI] [PubMed] [Google Scholar]
- 76.Bignon E, Pons M, Crastes de Paulet A, Dore JC, Gilbert J, Abecassis J, Miquel JF, Ojasoo T, Raynaud JP. J Med Chem. 1989;32:2092. doi: 10.1021/jm00129a013. [DOI] [PubMed] [Google Scholar]
- 77.Dore JC, Gilbert J, Bignon E, Crastes de Paulet A, Ojasoo T, Pons M, Raynaud JP, Miquel JF. J Med Chem. 1992;35:573. doi: 10.1021/jm00081a021. [DOI] [PubMed] [Google Scholar]
- 78.Dai SY, Chalmers MJ, Bruning J, Bramlett KS, Osbourne HE, Montrose-Rafizadeh C, Barr RJ, Wang Y, Wang M, Burris TP, Dodge JA, Griffin PR. Proc Natl Acad Sci USA. 2008;105:7171. doi: 10.1073/pnas.0710802105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dai SY, Burris TP, Dodge JA, Montrose-Rafizadeh C, Wang W, Pascal PD, Chalmers MJ, Griffin PR. Biochemistry. 2009;48:9668. doi: 10.1021/bi901149t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Foster AB, Jarman M, Leung O-T, McCague R, LeClerq G, Devleeschouwer N. J Med Chem. 1985;28:1491. doi: 10.1021/jm00148a020. [DOI] [PubMed] [Google Scholar]
- 81.Kim JH, Lee MH, Kim BJ, Kim JH, Han SJ, Kim HY, Stallcup MR. J Mol Endocr. 2005;35:449. doi: 10.1677/jme.1.01846. [DOI] [PubMed] [Google Scholar]
- 82.Fan M, Rickert EM, Chen L, Aftab SA, Nephew KP, Weatherman RV. Breast Cancer Res Treat. 2007;103:37. doi: 10.1007/s10549-006-9353-2. [DOI] [PubMed] [Google Scholar]
- 83.Muthyala RS, Sheng S, Carlson KE, Katzenellenbogen BS, Katzenellenbogen JA. J Med Chem. 2003;46:1589. doi: 10.1021/jm0204800. [DOI] [PubMed] [Google Scholar]
- 84.Seo JW, Comninis JS, Chi DY, Kim DW, Carlson KE, Katzenellenbogen JA. J Med Chem. 2006;49:2496. doi: 10.1021/jm0512037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tamoxifen and raloxifene metabolism.
- 86.Shiau AK, Barstad D, Loria PM, Cheng L, Kushner J, Agard DA, Greene GL. Cell. 1998;95:927. doi: 10.1016/s0092-8674(00)81717-1. [DOI] [PubMed] [Google Scholar]
- 87.Brzozowski AM, Pike ACW, Dauter Z, Hubbard RE, Bonn T, Engström O, Ohman L, Greene GL, Gustafsson J-Å, Carlquist M. Nature. 1997;389:753. doi: 10.1038/39645. [DOI] [PubMed] [Google Scholar]
- 88.Renaud J, Bischoff SF, Buhl T, Floersheim P, Fournier B, Geiser M, Halleux C, Kallen J, Keller H, Ramage P. J Med Chem. 2005;48:364. doi: 10.1021/jm040858p. [DOI] [PubMed] [Google Scholar]
- 89.Paz MM, Kumar GS, Glover M, Waring MJ, Tomasz M. J Med Chem. 2004;47:3308. doi: 10.1021/jm049863j. [DOI] [PubMed] [Google Scholar]
- 90.Lee SH, Kohn H. Org Biomol Chem. 2005;3:471. doi: 10.1039/b414806a. [DOI] [PubMed] [Google Scholar]
- 91.Tian ZQ, Liu Y, Zhang D, Wang Z, Dong SD, Carreras CW, Zhou Y, Rastelli G, Santi DV, Myles DC. Bioorg Med Chem. 2004;12:5317. doi: 10.1016/j.bmc.2004.07.053. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.