

SUMMARY
Piwi-interacting (pi) RNAs are germline-expressed small RNAs linked to epigenetic programming. C. elegans piRNAs are thought to be transcribed as independent gene-like loci. To test this idea and to identify potential Transcription Start (TS) sites for piRNA precursors, we developed CapSeq, an efficient enzymatic method for 5′-anchored RNA profiling. Using CapSeq we identify candidate TS sites, defined by 70–90 nt sequence tags, for over 50% of annotated Pol II loci. Surprisingly, however, these CapSeq tags failed to identify the overwhelming majority of piRNA loci. Instead, we show that the likely piRNA precursors are ~26 nt capped-small (cs) RNAs that initiate precisely 2 nt upstream of mature piRNAs, and that piRNA processing or stability requires a U at the csRNA +3 position. Finally, we identify a heretofore-unrecognized class of piRNAs processed from csRNAs that are expressed at promoters genome wide, nearly doubling the number of piRNAs available for genome surveillance.
INTRODUCTION
Argonaute (AGO) proteins associate with small RNAs to form sequence-directed gene-regulatory complexes that are deeply conserved in eukaryotes (Hutvagner and Simard, 2008). Most organisms encode multiple functionally-distinct AGO family members. These AGOs are loaded with a diversity of small RNA cofactors, produced through a similarly diverse repertoire of small-RNA biogenesis mechanisms (Siomi and Siomi, 2009). AGO-associated small RNAs include micro (mi) RNAs and short-interfering (si) RNAs that are processed from double-stranded (ds) RNA precursors by the RNase III-related enzyme Dicer (Bernstein et al., 2001). In some organisms, AGO-associated small-RNA species are produced, independent of Dicer, by RNA-dependent RNA Polymerase (RdRP) (Gu et al., 2009; Pak and Fire, 2007; Sijen et al., 2007).
piRNAs are Dicer-independent small RNAs that interact with AGOs related to Drosophila Piwi (Aravin et al., 2006; Girard et al., 2006; Grivna et al., 2006; Lau et al., 2006; Ruby et al., 2006). Many piRNA species originate from large genomic clusters, and direct Piwi-dependent transposon silencing, heterochromatin modification and germ cell maintenance (Aravin et al., 2007; Batista et al., 2008; Brennecke et al., 2007; Das et al., 2008; Lin, 2007). In flies and mammals, transposon-directed piRNAs typically map to both strands and are produced by a “ping-pong” amplification cycle, whereby sense piRNAs direct Piwi-dependent cleavage of a primary transcript to generate the 5′ ends of antisense piRNAs and vice versa (Aravin et al., 2007; Brennecke et al., 2007; Gunawardane et al., 2007; Houwing et al., 2007). Recent work suggests that Piwi-bound precursor piRNAs are trimmed by a 3′-to-5′ exonuclease and then methylated on the 2′-OH of the 3′ end residue of the mature piRNA (Kawaoka et al., 2011). In mice an abundant class of piRNAs, pachytene piRNAs, originates from large genomic clusters (Aravin et al., 2006). These piRNAs are not generated by the ping-pong cycle (Aravin et al., 2006; Beyret et al., 2012), but instead appear to be processed directly from a single-strand precursor by an unknown mechanism.
The C. elegans piRNAs, known as 21U-RNAs, are an abundant class of germline-expressed small RNAs that interact with the PIWI ortholog PRG-1 (Batista et al., 2008; Das et al., 2008; Ruby et al., 2006). Similar to mammalian pachytene piRNAs, 21U-RNAs are diverse in sequence and the overwhelming majority lack perfectly complementary RNA targets. Unlike mammalian piRNAs, however, 21U-RNAs do not appear to be processed from long RNA precursors. Instead, they derive from individual gene-like loci that are dispersed within two large clusters on chromosome IV (Cecere et al., 2012; Ruby et al., 2006). Within these clusters, more than 15,000 distinct 21U-RNAs are expressed from both strands and reside within introns and intergenic regions, but are rarely found in coding regions (Batista et al., 2008; Ruby et al., 2006). The presence of a conserved 8 nucleotide (nt) motif and A/T-rich region upstream of each 21U-RNA led Ruby et al. (Ruby et al., 2006) to suggest that 21U-RNAs are independently expressed loci. Consistent with this idea, a recent study identified Forkhead-family transcription factors that associate with the 8 nt motif and whose activity was correlated with 21U-RNA expression (Cecere et al., 2012). Using 5′ RACE, this study also amplified 70 nts of a longer transcript that initiated 2 nt upstream of one of two overlapping 21U-RNAs (21ur-3372 and 21ur-14222), suggesting that these 21U-RNAs may be processed from a transcript greater than 70 nts in length.
In this paper we explore the biogenesis of 21U-RNAs genome wide. To do this, we use two complementary 5′-anchored, RNA deep-sequencing approaches called CIP-TAP and CapSeq. Unlike published Cap Analysis of Gene Expression (CAGE)-related methods involving affinity purification (de Hoon and Hayashizaki, 2008), the CapSeq protocol, developed in this study, utilizes an efficient enzymatic approach to dramatically reduce the background of structural RNA reads and to enrich for 70–90 nt sequence tags corresponding to the capped 5′ ends of longer RNAs transcribed by RNA polymerase II (Pol II). We show that CapSeq identifies pre-mRNAs, trans-spliced mRNAs, primary (pri-) miRNAs, and non-coding RNAs, thus defining candidate Transcription Start (TS) sites for over 50% of annotated genes. This information is absent for most current WormBase annotations. Surprisingly, however, we show that CapSeq reads derive from less than 0.5% of annotated 21U-RNA loci.
Instead, using CIP-TAP cloning (Affymetrix/Cold Spring Harbor Laboratory ENCODE Transcriptome Project, 2009), we show that piRNA loci express an endogenous capped-small (cs) RNA species. csRNAs are native 18–40 nt Pol II products thought to arise through early termination or polymerase pausing, and previous studies have shown that csRNAs are expressed, often bi-directionally, at promoter regions in a variety of organisms (Haussecker et al., 2008; Nechaev et al., 2010; Seila et al., 2009; Taft et al., 2009). However, csRNAs have not been described to date in C. elegans. Our CIP-TAP data, which is far from saturation, detects csRNAs at Pol II promoters genome wide, including over 50% of annotated 21U-RNA loci.
Interestingly, we show that csRNA transcription initiates precisely 2 nt upstream of the corresponding mature 21U-RNA, suggesting that csRNAs are processed into piRNAs by removing the cap plus two nucleotides and by trimming the 3′ end. Furthermore, we provide evidence that a U residue at the +3 position of a csRNA is critical for piRNA processing or stability. In summary, we present a fully enzymatic approach, CapSeq, for 5′ anchored RNA profiling, and use this approach to define the first TS site annotations for many Pol II loci in C. elegans. We show that csRNAs identified by CIP-TAP cloning, rather than longer RNAs identified by CapSeq, are the likely piRNA precursors. Our findings also identify csRNAs transcribed at promoters genome wide as 21U-RNA precursors, nearly doubling the repertoire of piRNAs available for genome surveillance.
RESULTS
CapSeq is an efficient method for 5′ anchored profiling of Pol II transcripts
21U-RNAs are thought to be expressed from thousands of independent loci (Ruby et al., 2006), and could thus comprise approximately 40% of the transcription units in the C. elegans genome. However, systematic RNA-seq methods to profile RNA expression (Allen et al., 2011; Lamm et al., 2011) have not identified likely 21U-RNA precursors. Therefore to increase the chance of identifying 21U-RNA precursors, we sought to generate a comprehensive profile of 5′-anchored RNA sequence tags. To do this we developed CapSeq, which employs a straightforward series of enzymatic treatments to enrich the 5′ ends of Pol II transcripts (Figure 1), with as little as 0.5 – 2 μg of total RNA as starting material (see Extended Experimental Procedures).
Figure 1.
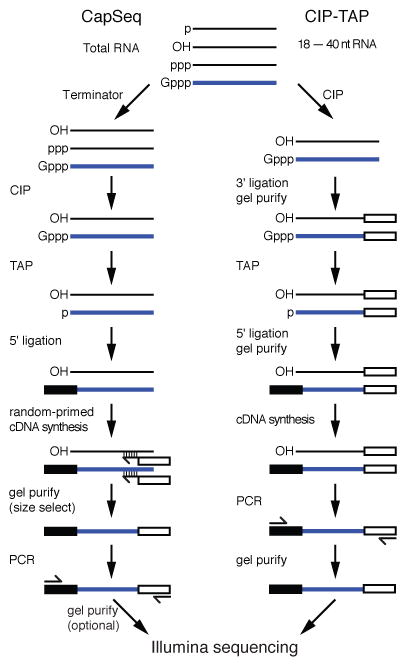
Flowcharts illustrating the CapSeq (left) and CIP-TAP (right) protocols.
Using CapSeq, we generated 5 libraries from 3 different developmental stages (L1, L3, and adult) and obtained ~61 million reads that mapped to the C. elegans genome, including 46 million that mapped to non-structural RNAs. Visual inspection using the genome browser software “Gbrowse” (Stein et al., 2002) revealed that most CapSeq reads are indeed enriched at the 5′ ends of genes transcribed by RNA Pol II (Figure 2A, CapSeq panel). To estimate the fidelity with which CapSeq defines the actual 5′ ends of transcripts, as opposed to internally truncated RNAs, we took advantage of the fact that many worm transcripts contain a capped trans-spliced 5′ leader sequence. Using two different methods, we found that CapSeq enriches approximately 15,000-fold for reads starting at the actual 5′ end (indicated by the 5′-most nt of the spliced leader sequence) over reads starting at any other nucleotide downstream (see Supplemental Discussion).
Figure 2. Comparative analysis of RNA-seq protocols that enrich long- and short-capped RNAs (CapSeq and CIP-TAP respectively) and a protocol (CIP-PNK) that clones uncapped short RNAs such as siRNA, piRNA and miRNA species.
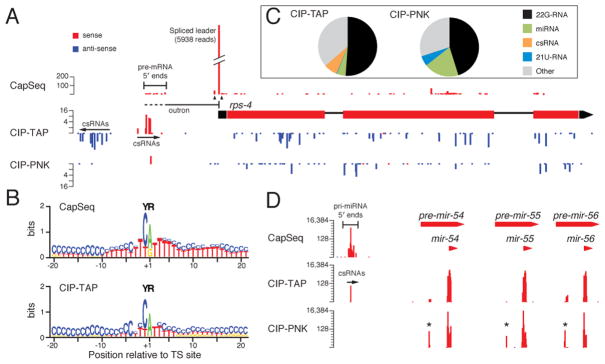
(A) Histograms representing the 5′ ends of mapped reads, as indicated, at a typical protein-coding locus, rps-4. The height of each histogram bar is proportional to the number of reads sharing the same 5′ nt and the scale (log2) is shown. Candidate pre-mRNA 5′ ends and csRNAs are indicated. Trans-splicing at some genes including rps-4 results in removal of the 5′ UTR of the pre-mRNA, called an “outron”, and the addition of a “Spliced leader”. The major trans-splice site for rps-4 is off the scale as indicated by a break in the bar, and the total number of SL-containing reads is indicated. Two minor trans-splice sites flank the major trans-splice site as indicated by triangles below the CapSeq reads. The outron is indicated by a line below the CapSeq reads; dashes indicate the variable 5′ end of the outron. The blue bars beneath the rps-4 coding sequences in the CIP-TAP and CIP-PNK samples correspond to antisense 22G-RNAs.
(B) Schematic representation of the nucleotide composition around candidate TS sites (the +1 position) identified by CapSeq and CIP-TAP reads (here only sense csRNAs). The nucleotide height (in bits) represents the log2 ratio of the frequency observed relative to the expected frequency based on genomic nt composition. The enriched YR motif is indicated.
(C) Pie charts indicating the relative composition of small RNAs recovered in the CIP-TAP and CIP-PNK samples.
(D) Histograms representing the 5′ ends of mapped reads at the mir-54 –56 miRNA cluster. Candidate pri-miRNA 5′ ends and csRNAs are indicated. The asterisks indicate reads corresponding to miRNA star-strands.
Identification of RNA Pol II TS sites and new trans-splice sites
The TS sites for C. elegans genes are poorly mapped, in part because many mRNAs are trans-spliced to a capped ~22 nt spliced leader (SL), which results in removal of the pre-mRNA 5′ end (Blumenthal and Steward, 1997). Our CapSeq data identified 70% of the 15,759 SL trans-splice sites annotated in the WS215 genome release, as well as 5,711 new trans-splice sites (Figure S1A, top panel; Table S1A). Most gene annotations in WormBase simply indicate the 5′ end of the trans-spliced exon, or the position of the AUG codon. The 5′ ends of many non-SL CapSeq reads mapped near, and often upstream of, current 5′ end annotations (Figure 2A and data not shown). We hypothesized that the 5′ ends of these reads could represent TS sites. Consistent with this idea, we noticed that these reads exhibit a strong bias for a 2 nt motif of pyrimidine (Y) purine (R) or “YR”, in which R represents the first nt (+1) in the CapSeq read and Y represents the adjacent 5′ nt (Figure 2B). The YR motif is part of an extended consensus yYRyyy (lower case indicates weaker preference), with a strong preference for A as the R at position +1 and a slight preference for T at flanking positions. The YR motif and extended consensus resemble the initiator element required for RNA Pol II transcription initiation in mammals, plants and flies (de Hoon and Hayashizaki, 2008; Juven-Gershon et al., 2008; Smale and Baltimore, 1989). Using a cutoff of one CapSeq read per 10 million total reads, and a requirement for a YR motif, our CapSeq data predicted approximately 64,000 candidate TS sites genome wide (Table S1B).
In order to pair candidate TS sites with existing annotations, we considered CapSeq reads within a window from 1,000 nt upstream to 100–200 nt downstream of annotated 5′ ends. The 1,000 nt upstream distance was chosen as a conservative upper limit to allow for the possibility of non-annotated 5′ exon sequence or long distances between the TS site and the first splice-acceptor site required for trans-splicing. For most genes, the 3′ limit was arbitrarily set at 200 nt. However, in order to reduce the chance of scoring degradation products as TS sites, this 3′ limit was reduced to 100 nt for very abundantly transcribed genes whose total read counts exceeded 1,000 reads per ten million. Using these criteria, we could assign candidate TS sites to more than 50% of annotations in WS215 (Table S1B), including 52% of annotated protein-coding genes (10,667 genes), 15% of annotated pseudogenes (226), 54% of annotated non-coding RNAs (137), 74% of snoRNAs (102), and 37% of snRNA genes (42). We found that TS sites often appeared to be clustered over regions of several to sometimes more than 50 nt (Figure 2A, CapSeq panel), suggesting that there is an inherent flexibility in transcription initiation mediated by RNA Pol II at these promoters.
We also identified ~20,000 candidate TS sites that did not pair with annotations based on our criteria (Table S1B). These included 12,457 clusters of TS sites that resembled TS-site clusters typical of annotated Pol II genes. The majority of these (84%) were separated from other annotations or from each other by greater than 1 kb. These findings suggest that there are many, as yet, non-annotated Pol II loci in the C. elegans genome and/or that many of the existing annotations are separated from their actual 5′ ends by greater than the arbitrarily set 1 kb limit used for our analysis.
Identification of primary miRNA TS sites
miRNAs are sequentially processed from primary transcripts (pri-miRNAs) synthesized by Pol II. Drosha processes pri-miRNAs into stem-loop precursors (pre-miRNAs) that are exported to the cytoplasm and processed by Dicer into mature miRNAs (Hutvagner and Simard, 2008). In most organisms, the TS sites of pri-miRNAs have not been identified, likely because the original 5′ end is rapidly removed during miRNA maturation. To identify candidate TS sites for miRNA genes, we analyzed CapSeq reads mapping upstream of annotated miRNAs. Because many miRNAs are co-expressed in a single primary transcript (Lau et al., 2001), there are only about 100 unique miRNA loci annotated in the C. elegans genome, encoding ~140 miRNAs. We identified at least 1 candidate TS site for 56 of the 100 annotated miRNA loci corresponding to 74 individual mature miRNAs (Table S1E). As with other Pol II loci, we found that CapSeq reads that mapped upstream of the pre-miRNAs were often clustered within a 50 nt interval (Figure 2D; Table S1E). We found evidence for only a single group of TS sites upstream of each miR cluster, including the mir-54 – 56, mir-35 – 41 and mir-229/64 – 66 clusters (Figure 2D; Table S1E), indicating that each cluster is co-expressed (Lau et al., 2001). Pri-miRNAs were rarely trans-spliced; a total of five SL-containing reads were associated with pri-miRNAs, and all of these were spliced to pri-let-7. The five SL-containing reads mapped ~30 nt upstream of the Drosha-processed pre-let-7 RNA, while 20 non-SL reads mapped approximately 200 nt further upstream. Interestingly, we found that some pri-miRNAs were expressed at levels comparable to the pre-mRNAs of common protein-coding genes (Figure S1E), a finding that differs from previously published RNA-Seq data (Lamm et al., 2011; see Discussion).
Mouse CapSeq
The CapSeq protocol we describe should be useful for 5′-anchored RNA profiling from small quantities of tissue. To confirm that CapSeq can identify TS sites from other species, we performed a pilot study using mouse testis RNA. As shown in Figure 3A, we found that CapSeq reads were strongly biased for the 5′ end of annotated mouse genes. By searching for reads upstream of annotated miRNA loci, we identified candidate TS sites for hundreds of primary mouse miRNA genes (Table S2). We also analyzed reads mapping to mouse piRNA clusters. We found that multiple mouse piRNAs appeared to share a TS site (Figure 3B), consistent with previous studies suggesting that piRNAs may be processed from longer precursor RNAs (Aravin et al., 2006; Girard et al., 2006). Finally, we analyzed the motif surrounding candidate mouse TS sites and observed a clear YR motif within a broader motif of YRNyy, in which R (usually an A) corresponds to the predicted 5′ nt (Figure 3C). Thus our data show that CapSeq is generally useful for identifying Pol II TS sites, and that C. elegans TS sites are similar to mammalian TS sites.
Figure 3. CapSeq analysis of mouse testes RNA.
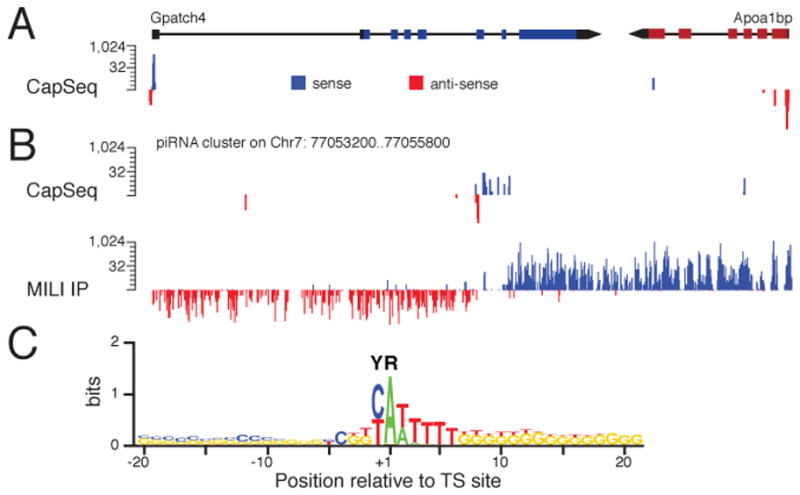
(A and B) Browser views of representative protein-coding and piRNA cluster regions are shown. The histograms (log2 scale) represent the frequency of reads sharing the same 5′ ends from CapSeq or MILI IP (Robine et al., 2009) as indicated. Bidirectional reads were observed around the TS sites of Gpatch4.
(C) Schematic representation of the nucleotide composition around candidate TS sites (YR). The nucleotide height (in bits) represents the log2 ratio of the frequency observed relative to the expected frequency based on genomic nt composition.
See also Table S2.
CIP-TAP cloning identifies promoter associated csRNAs genome wide
Despite the considerable depth of our CapSeq libraries, we were surprised to find that only 217 of over 9000 annotated unique 21U-RNA loci were identified by CapSeq reads. These included a single 88 nt CapSeq read mapping to the locus from which Cecere et al. (2012) identified a 70 nt 5′ RACE product. In contrast, our mouse CapSeq data identified CapSeq reads associated with most of the annotated piRNA loci. These findings could indicate that pre-21U-RNAs are exceptionally unstable or, alternatively, that the actual precursors are shorter than the 70 nt (minimum length) sequence tags amplified by CapSeq. To test the latter idea, we employed CIP-TAP cloning to identify native capped-small (cs) RNAs (Figure 1; Extended Experimental Procedures). Total RNA was size-fractionated to recover 18 – 40 nt RNA species. To select against the recovery of abundant un-capped small RNA species, including 22G-RNAs, miRNAs and mature piRNAs, we treated the sample with CIP to remove 5′ mono- or tri-phosphates, reducing the accessibility of these species to 5′ ligation. The 3′ end of the small RNA was ligated to a linker and gel-purified. Three fourths of the sample was then treated with TAP to decap csRNAs, thus exposing a 5′ monophosphate for 5′ ligation. The remaining one fourth was treated with polynucleotide kinase (PNK) to add a 5′-phosphate onto non-capped small RNA species. The CIP-TAP and CIP-PNK samples were then ligated to a 5′ linker, gel-purified, reverse-transcribed, and PCR-amplified.
Deep sequencing of the CIP-PNK sample revealed abundant 22G-RNAs, miRNAs and piRNAs, but very few small RNAs that mapped to Pol II promoters (Figure 2A). In contrast, the CIP-TAP sample dramatically enriched small RNAs upstream of Pol II loci (Figure 2A). After normalizing to total non-structural reads that match the genome, csRNA reads that map within 1000 nt upstream of WS215 5′ end annotations (including 21U-RNA annotations, see below) were enriched 60-fold in the CIP-TAP sample relative to the CIP-PNK sample (Figure 2C). In contrast, mature miRNAs and 21U-RNA reads were depleted ~4-fold and 17-fold respectively in the CIP-TAP sample. The relative rate at which 22G-RNAs were recovered did not change significantly between the CIP-TAP and CIP-PNK samples. We found that 42% of the sense-oriented reads identified in the CIP-TAP sample corresponded exactly to the 5′ ends of candidate TS sites identified in our CapSeq analysis (Figure 2A; Tables S1B and S1C), supporting the idea that CIP-TAP treatment enriches for C. elegans TS site-associated csRNAs.
csRNAs originate 2 nt upstream of mature 21U-RNAs
Analysis of our CIP-TAP data revealed that csRNAs strongly correlate with 21U-RNAs. There are 9,079 21U-RNA loci that each express a single (non-overlapping) 21U-RNA that matches uniquely in the genome. Among a total of 4.5 million CIP-TAP reads, we identified candidate csRNAs that map to approximately 6,000 of the 9,079 unique 21U-RNA loci. Interestingly, we observed a very strong bias for the 5′ ends of csRNA reads to map 2 nt upstream of the mature 21U-RNA species (~4,600 of 6,000 csRNA/21U-RNA pairs, one tail p = 0, binomial distribution; Figure 4A).
Figure 4. Analysis of annotated 21U-RNA loci.
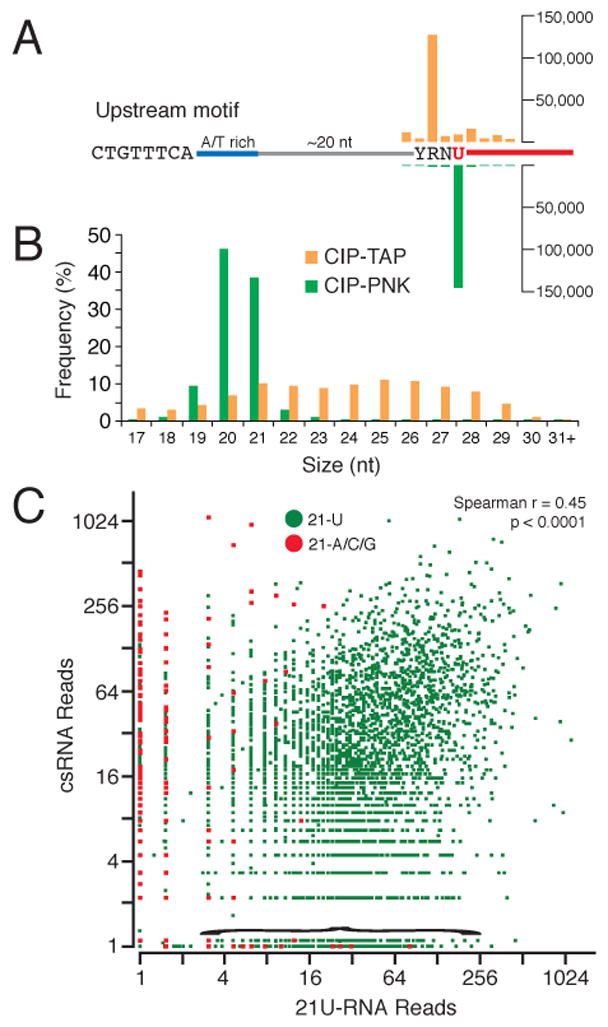
(A) Cumulative analysis of the 5′ ends of unique CIP-TAP (orange) and CIP-PNK (green) sequences with respect to the YRNU motif of a consensus 21U-RNA locus. The scale (linear) is shown. The red segment preceded by U indicates the mature 21U-RNA.
(B) Graph showing the length distribution of CIP-TAP/csRNA reads (orange) and CIP-PNK/21U-RNA reads (green) mapping to 21U-RNA loci.
(C) Graph of csRNA levels plotted against corresponding 21U-RNA levels for annotated 21U locus. The points in red indicate previously annotated 21A/G/C piRNAs. Points near the X-axis (under the bracket) include 22G-RNAs previously mis-annotated as piRNAs.
csRNA reads with 5′ ends that align 2 nt upstream of mature 21U-RNAs (-2 csRNAs) peaked broadly at a length of 25–26 nt (Figure 4B). These -2 csRNAs were enriched approximately 60-fold by CIP-TAP cloning relative to the CIP-PNK cloning method (Gu et al., 2009), which enriches for mature 21U-RNAs. Consistent with a precursor-product relationship, we found that the level of -2 csRNAs recovered by CIP-TAP significantly correlates with the level of mature 21U-RNAs recovered by CIP-PNK (Figure 4C). In contrast, the longer -2 CapSeq reads were not only rarely detected (44 of the 217 loci mentioned above; Figure S2A; Table S3C), but also poorly correlated with corresponding mature 21U-RNA levels, as discussed below. Some loci previously annotated as highly-expressed 21U-RNA loci failed to produce detectable csRNAs (Figure 4C, points just above the x-axis). Visual inspection using Gbrowse revealed that several of these loci are likely to be derived from degraded 22G-RNAs that were mis-annotated as 21U-RNAs (data not shown). Loci with abundant csRNAs that lack corresponding mature 21U-RNAs (Figure 4C, points along the y-axis), define a set of “21U-like” loci discussed below.
The YRNT motif associated with 21U loci represents a transcription start site
As observed for CapSeq reads, CIP-TAP reads genome wide exhibited a strong bias for initiating at a YR motif (Figures 2B and S1A), consistent with the idea that csRNAs are independently initiated Pol II products. As noted above, csRNAs tend to initiate 2 nt upstream of the corresponding mature 21U-RNAs at 21U-RNA loci, which may explain why the majority of 21U-loci exhibit a YRNT motif (Ruby et al., 2006), where R is the first nt of the csRNA and T corresponds to the 5′U of the mature 21U-RNA.
To look for a correlation between the presence of a YR motif and the levels of csRNA and 21U-RNA expression, we considered ~1000 pairs of 21U-RNAs, for which the 5′ ends in each pair are separated by 1 nt. Due to the 1 nt separation, a -2 YR motif can only exist for one member of each paired 21U-RNA. These paired loci, which account for 40% of 21U-RNAs that lack a -2 YR motif, provided an opportunity to examine the expression levels of YR- and non-YR-associated transcripts driven from the same promoter. Consistent with the idea that the YR motif is important for transcription, we found that, regardless of their arrangement (5′ or 3′) at tandem loci, both the csRNAs and corresponding mature 21U-RNAs were 10-fold more abundant for the YR-containing sister than for the non-YR-containing sister (paired t-test, p < 0.0001). Taken together, these findings suggest that the previously defined YRNT motif is a transcription initiation site where R (usually an A) encodes the +1 nt of a pre-21U (csRNA), and T encodes the +3 U, which corresponds to the 5′ end of a mature 21U-RNA (Figure 4A).
A +3 U is required for piRNA production or stability
While analyzing the CIP-TAP data, we noticed that there were many loci within the 21U-RNA clusters on chromosome IV for which abundant csRNAs were detected, but mature 21U-RNAs were not. Altogether, we identified 2,309 csRNA-producing loci that fail to produce mature 21U-RNAs (Table S3A). The csRNA reads obtained from these loci were similar in both size and abundance to those derived from canonical 21U-RNA loci (Figures 5A and S2B). Furthermore, most of these loci (65%; Table S3A) exhibited an adjusted motif score greater than 7, typical of canonical 21U-RNA loci with the upstream 8 nt motif (Ruby et al., 2006). Interestingly, the csRNAs produced at these loci lack a +3U – the majority (~60%) contained a YRNA rather than a canonical YRNT motif (Figure 5B). Approximately 400 previously annotated “21U-RNAs” actually start with a 5′ nt other than U (~3% of annotated 21U-RNAs; Batista et al., 2008; Ruby et al., 2006), and we noted that ~60% of these previously detected 21nt-RNAs exhibit corresponding csRNAs. Further examination of these 21U-like loci revealed that the mature 21nt-RNAs (Figure 4C, red) were an average of 10-fold less abundant relative to their corresponding csRNAs than were 21U-RNAs from canonical 21U-RNA loci (Figure 4C, green). These findings suggest that 21U-like loci express csRNAs at normal levels, but that the mature piRNAs are either inefficiently processed or unstable.
Figure 5. Analysis of 21U-like loci.
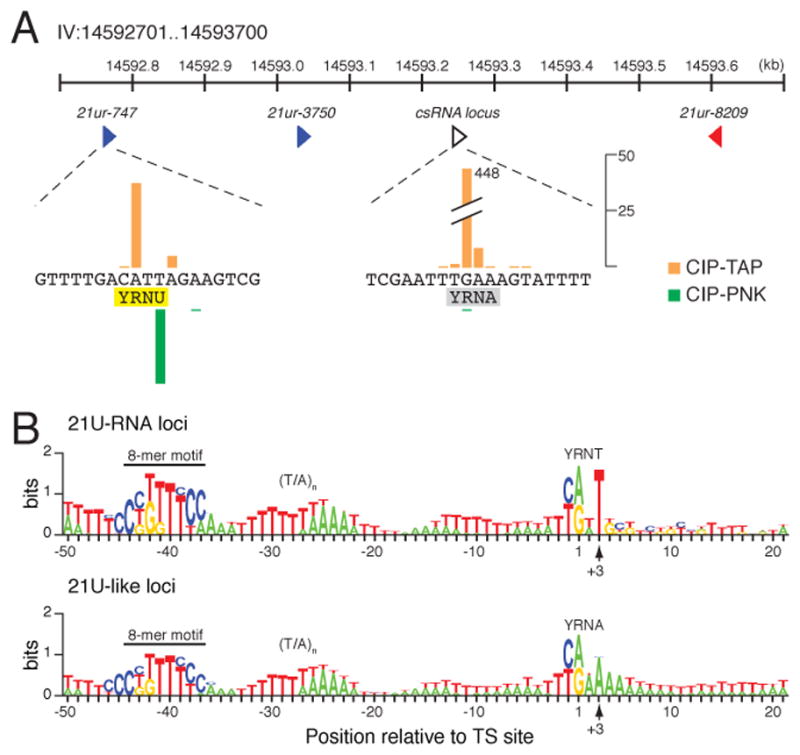
(A) An example of a 21U-like locus with a piRNA cluster on chromosome IV. Both 21ur-747 (blue triangle at left) and a nearby 21U-like or csRNA locus (open triangle) are enlarged to single nucleotide resolution. The bars indicate the number of reads (linear scale provided) sharing the corresponding 5′ nt from CIP-TAP (orange) and CIP-PNK (green), relative to the YRNU motif (yellow) of 21ur-747 and YRNA motif (gray) of the 21U-like locus (open triangle). (B) Schematic representation of the nucleotide composition at canonical 21U-RNA loci (top) and 21U-like loci (bottom). The nucleotide height (in bits) represents the log2 ratio of the frequency observed relative to the expected frequency based on genomic nt composition. The upstream and TS-site (YR) motifs are indicated. The observed 5′ end of mature 21nt-RNAs is indicated by the arrow at the +3 position.
Given the large number of 21U-like loci, we reasoned that polymorphisms in C. elegans wild-isolates might convert the +3 residue of a csRNA to a U at one or more of these loci. Consistent with this possibility, we identified two 21U-RNAs (IV: 17159702–22 and IV: 15903563–83) that were cloned from JU1580 (Felix et al., 2011) and CB4856 (E.Y. and C.M. unpublished data), respectively, that mapped to 21U-like loci in the N2 background. In both cases, independent deep-sequencing data confirmed that the wild-isolates contain SNPs in the corresponding 21U-like loci that change the +3 residue of the csRNA to a U. Taken together, these findings suggest that the YR portion of the YRNT motif is likely sufficient for transcription initiation, while a U at position +3 of the csRNA is important for 21U-RNA processing and/or stability.
Capped-small RNAs produced throughout the genome are processed into 21U-RNAs
The majority of csRNAs produced throughout the genome lack the conserved 8 nt consensus (CTGTTTCA) that was weighted heavily in the annotation of canonical 21U-RNA loci (compare Figures 2B and S1A to Figure 5B). However, as expected by chance, many csRNAs lacking the 8 nt motif, nevertheless exhibit a YRNT motif (Figures 2B and S1A), and thus contain a U at position +3. Therefore, we asked whether this subset of csRNAs, which are associated with TS sites of protein-coding and other Pol II transcripts, might also be processed into mature 21U-RNAs and loaded onto the Piwi Argonaute PRG-1. Indeed, a number of previously annotated 21U-RNAs coincide with csRNAs proximal to protein coding genes on chromosomes other than chromosome IV, where the piRNA clusters reside (data not shown). To investigate this further, we deep sequenced piRNAs enriched by PRG-1 immunoprecipitation (IP). This new IP deep-sequencing data was consistent with previously published and unpublished PRG-1 IP deep-sequencing data (Batista et al., 2008). However, the cloning methods used to generate the previous PRG-1 IP datasets also generated much more noise from degraded mRNAs than did the TAP cloning procedure used here. Altogether, we identified 12,183 new 21U-RNA species (Table S3B), of which ~10,000 exhibit a poor motif score and are associated with TS sites throughout the genome. We refer to this class as “Type-2” 21U-RNAs.
We next asked if Type-2 21U-RNAs exhibit the same 2′-O-methyl modification found on the 3′ ends of canonical 21U-RNAs. Consistent with this idea, examination of a previously published data set (Vasale et al., 2010) revealed that the majority of Type-2 21U-RNAs were resistant to 3′ end oxidization (Figure 6A). Like canonical (or Type-1) 21U-RNAs, Type-2 21U-RNAs were only expressed in the germline, consistent with the germline-specific expression of PRG-1. Soma-specific loci, such as the gut-specific gene vit-1 that produces abundant +3U containing csRNAs did not give rise to 21U-RNAs (data not shown).
Figure 6. Analysis of Type-2 21U-RNA loci.

(A and B) Comparative analysis of reads from RNA-seq protocols (as indicated). The histograms represent the frequency of reads sharing the same 5′ end at the rps-4 locus (A), and at a non-annotated locus on chromosome X (B). The genome coordinates are indicated. A log2 scale is provided for each set of histograms. Mature 21U-RNA reads are enriched in the “oxidized” vs “control” (A) and “PRG-1 IP” vs “Input” (A and B) samples as indicated. Closed triangles indicate the positions of mature 21U-RNAs enriched in the PRG-1 IP. In (B), the red bars correspond to WAGO-dependent 22G-RNAs likely targeting non-annotated transcripts (dashed arrows) of unknown length.
(C) Enlarged regions indicating the precise positions of corresponding CIP-TAP (orange) and PRG-1 IP (green) reads relative to the YRNU motif indicated below the sequence. Note that one of the TS sites for rps-4 is “RR” rather than “YR”. The open triangles above the CIP-TAP bars point to the likely precursor of the mature 21U-RNAs, which are indicated by the closed triangles below the PRG-1 IP bars.
Altogether, our data define more than 12,000 new 21U-RNA species, nearly doubling the total number of piRNAs in C. elegans. Moreover, Type-2 21U-RNAs include several extremely abundant 21U-RNA species. In fact, the single most abundant 21U-RNA is a Type-2 21U-RNA expressed from an X chromosome locus (Figures 6B and 6C). This X-locus is intriguing in that it is one of 6 homologs (all on X) with extensive sequence identity flanking distinct 21U-RNAs (Figures S3A and S3B). The SL1-spliced leader locus and several snRNA loci were also found to produce very abundant 21U-RNA species (Figures S3C and S3D). However, the majority of Type-2 21U-RNA loci produce relatively low levels of mature 21U-RNAs (Table S3B), accounting for approximately 5% of total 21U-RNA levels (see Extended Experimental Procedures).
csRNAs are unlikely to be processed from longer Pol II transcripts
The above findings suggest that -2 csRNAs are very likely the precursors for mature 21U-RNAs, but do not address whether csRNAs might be processed from longer, exceptionally rare, transcripts. To address this possibility, we compared 21U-RNA expression levels to the levels of -2 csRNAs and longer -2 CapSeq RNA reads at Type-1 and Type-2 21U-RNA loci. There are only 44 Type-1 loci with -2 CapSeq reads (Table S3C and Figure 7A). Despite the small sample size, we found a very significant correlation between csRNA and 21U-RNA levels at these loci (Spearman r = 0.69, p < 0.0001; Figure 7A). The correlation coefficient ‘r’ is similar to the one observed previously for all Type-1 21U-RNA loci (Figure 4C). However, we found no correlation between CapSeq reads and 21U-RNA levels (Spearman r =−0.08, p < 0.64; Figure 7A). We also examined the 982 Type-2 loci with -2 csRNA and -2 CapSeq reads, and again found a significant correlation between csRNA and 21U-RNA levels, but no significant correlation between 21U-RNA levels and -2 CapSeq read levels (Figure 7B).
Figure 7. C. elegans piRNAs are processed from capped-small RNAs.

(A and B) Correlation analyses between 21U-RNAs and csRNAs or long-capped RNAs (as indicated) at (A) the 44 Type-1 loci where long-capped RNA reads were obtained by CapSeq, and at (B) 982 Type-2 loci where csRNA and CapSeq reads were both found at +1, relative to the downstream (+3) U of a 21U-RNA. For a perfect correlation, the Spearman rank correlation coefficient (r) = 1 or −1, and for no correlation, r = 0. P-values (p) were calculated using non-parametric correlation model.
(C) Model for the biogenesis of 21U-RNA. Arrows indicate TS sites of Type-1 and Type-2 piRNA loci.
The failure of CapSeq reads to correlate with 21U-RNA expression levels was not due to a lack of depth in the CapSeq data set. There were, in fact, over 40-fold more CapSeq reads than csRNAs reads analyzed in our young adult data sets. Furthermore, as noted above, we frequently observed promoter regions with bidirectional, divergent csRNAs (Figure 2A; Table S1C and S1D). At these bidirectional loci, we found that oppositely oriented -2 csRNAs appeared equally likely to give rise to 21U-RNAs (Figure 6A), whereas longer CapSeq reads were almost exclusively sense oriented (Figure 2A). Moreover, as noted above, even when sense CapSeq reads were present at -2 relative to a Type-2 21U-RNA species, CapSeq reads and mature 21U-RNA levels failed to exhibit a significant correlation. Together these findings suggest that RNA Pol II transcribes csRNAs directly, and that -2 csRNAs (not longer RNA species) are the 21U-RNA precursors.
DISCUSSION
C. elegans piRNA precursors are expressed individually by Pol II as capped-small RNAs
Here we have explored the biogenesis of C. elegans piRNAs (21U-RNAs). To do so, we employed two approaches, CapSeq and CIP-TAP, both of which enrich for the 5′ ends of Pol II transcripts. The CapSeq protocol, designed to select for long-capped RNAs, identified reads mapping to the 5′ ends of thousands of other Pol II genes, but detected reads mapping to only 0.5% of 21U-RNA loci (44 out of 9,079 unique 21U-RNAs). The CIP-TAP protocol, on the other hand, designed to detect capped-small RNAs, identified thousands of candidate 21U-RNA precursor transcripts (more than 50% of 21U loci) that average 26 nt in length and initiate 2 nt upstream of the mature piRNA species. In addition, CIP-TAP identified csRNAs that were associated with many other Pol II promoters, where they were frequently oriented divergently, with the sense csRNA often corresponding to a major TS site for the corresponding longer transcript detected by CapSeq.
Strikingly, germline-expressed csRNAs that contain a U at the +3 position were found to correspond to a previously overlooked class of 21U-RNAs associated with Pol II promoters genome wide. These findings indicate that the U in the YRNU motif is important for 21U-RNA stability, processing, or Piwi Argonaute loading, and that the YR is important for efficient transcription initiation (see Model, Figure 7C). Consistent with this idea, the distance between the conserved upstream 8 nt motif and the putative initiator element (YRNT) is similar to the distance between the TFIIB/TATA and the initiator elements of core TS sites described for other organisms (Juven-Gershon et al., 2008). Based on these findings, we now propose that C. elegans piRNAs be divided into two categories (Figure 7C): Type-1 21U-RNAs, which correspond to the previously defined 21U-RNAs that share an 8 nt upstream motif and are clustered on chromosome IV (Batista et al., 2008; Ruby et al., 2006); and Type-2 21U-RNAs, which need not have an 8 nt motif and are processed from csRNAs derived from the promoters of Pol II genes throughout the genome.
An enzymatic approach for 5′-end anchored transcription profiling
Transcription profiling by deep sequencing has become an increasingly important tool for following gene expression. The CapSeq protocol described here facilitates transcription profiling by using a series of three enzymatic treatments that dramatically enrich for the 5′ ends of Pol II transcripts. Because CapSeq does not require affinity purification to remove structural RNA contaminants, it can be performed on relatively small quantities of RNA. Aside from a single size selection step, the entire procedure is carried out in a PCR tube. Importantly, the CapSeq procedure anchors clones at the 5′ cap of Pol II transcripts, and thus can clone RNAs with or without poly(A) tails. CapSeq provides a quantitative way to profile a diversity of Pol II transcripts, while providing insight on alternative transcription-initiation sites, which may be of potential developmental significance.
Genome-wide identification of Pol II TS sites
The data described here provide the first systematic and comprehensive look at the TS sites of Pol II transcripts in C. elegans. The trans-splicing of SL sequences to the 5′ ends of many mature transcripts confounds the identification of TS sites in C. elegans. Consequently, only a handful of TS sites for C. elegans Pol II transcripts had been identified (Allen et al., 2011; Morton and Blumenthal, 2011). By using CapSeq to clone capped transcripts from several different developmental stages, we have identified candidate TS sites for approximately 50% of the annotated protein-coding genes in C. elegans. In addition, we have identified 5′ ends for Pol II transcripts that are typically under-represented in poly(A)-selected RNA-seq studies, including snRNAs, snoRNAs, SL RNA precursors, and histone mRNAs. In keeping with predictions from previous studies (Allen et al., 2011), we found that an overall 70% of annotated protein-coding genes have trans-spliced forms. Because of the abundance of SL-containing reads, our findings provide a comprehensive measure of alternative spliced-leader usage for most genes, and also provide useful data for refining the prediction of SL splice-acceptor sites.
Sequencing of a mouse testes CapSeq library also revealed a strong enrichment for RNA 5′ ends and for a YR motif at TS sites of mouse Pol II genes. Our CapSeq analysis identified candidate TS sites for many primary miRNA transcripts in both C. elegans and mouse. Altogether, by combining CIP-TAP and CapSeq data, we were able to predict TS sites for 60% of the annotated C. elegans miRNA genes (Table S1E). Surprisingly, we found that expression levels, as inferred from read counts for pri-miRNAs, were comparable to that of many abundant protein-coding genes. In contrast, pri-miRNA transcripts were very rarely detected in data from a previous study that used poly(A) selection RNA-Seq protocols (Lamm et al., 2011), suggesting that either pri-miRNAs lose their poly(A) tail more rapidly than their 5′ cap, or perhaps lack a poly(A) tail entirely. We conclude that CapSeq and CIP-TAP can be used to quantify the activity of a wide variety of Pol II promoters. The approach described here could readily be extended to produce a comprehensive profile of C. elegans or mouse TS sites.
Capped-small RNAs are associated with promoters in C. elegans
Like the longer reads recovered by CapSeq, csRNAs exhibit a consensus Pol II initiator element yYRyyy. Indeed the 5′ ends of csRNAs often coincide with the 5′ ends of CapSeq reads. However, unlike CapSeq reads, csRNAs were frequently bidirectional at promoters, with divergent csRNAs separated by an average of approximately 150 nt. This finding is consistent with the idea that many eukaryotic promoters are intrinsically bidirectional (Seila et al., 2009). In general, for csRNA and CapSeq reads that share a common 5′ end, the abundance of csRNA reads was proportional to the abundance of CapSeq reads, suggesting that csRNAs might be associated with Pol II initiation at active promoters. Despite their correlation with active gene expression, our analysis suggests that csRNAs are relatively low abundance transcripts compared to other small RNAs. Based on our CIP-TAP cloning experiments, we estimate that csRNAs represent less than 1% of the total small RNAs in adult C. elegans.
Capped-small RNAs that flank the TS sites of active promoters have been identified in mammals and Drosophila (Core et al., 2008; Haussecker et al., 2008; Seila et al., 2008; Yamamoto et al., 2007). Our data suggest that csRNAs are most similar to Promoter-Associated Short RNAs (PASRs), which were enriched using a CIP-TAP cloning method and had 5′ ends that frequently coincided with those of capped RNAs identified by CAGE (Affymetrix/Cold Spring Harbor Laboratory ENCODE Transcriptome Project, 2009; Kapranov et al., 2007). Although the biogenesis and function of PASRs remains unknown, it has been speculated that PASRs might reflect Pol II pausing or premature termination, or that they are processed from promoter-associated long-capped RNAs (Affymetrix/Cold Spring Harbor Laboratory ENCODE Transcriptome Project, 2009; Nechaev et al., 2010). Our data are most consistent with the idea that csRNAs are independent transcripts, rather than processing intermediates derived from longer RNAs. If csRNAs were derived from long-capped RNAs, we would have expected a broader size range continuing up to 32 nt (the largest size we could sequence in this experiment). The size of C. elegans csRNAs is similar to the estimated size, approximately 28 nts, of nascent RNA that can be accommodated in the Pol II exit canal (Andrecka et al., 2008; Chen et al., 2009; Proudfoot et al., 2002). This size is also similar to that of csRNAs found associated with promoter-proximal pausing of Pol II, thought to occur at many genes throughout metazoan genomes (Nechaev et al., 2010; Rasmussen and Lis, 1995).
C. elegans piRNAs are processed from capped-small RNAs
Here we have shown that csRNA loci genome wide give rise to 21U-RNAs, and that the relative levels of the -2 csRNA and corresponding mature 21U-RNA are well correlated (Figures 7A and 7B). The only requirement for 21U-RNA production was the presence of a U residue at the +3 position of the csRNA. These findings are consistent with a model in which csRNAs are precursors for 21U-RNA production (Figure 7C). The canonical 21U-RNA loci (Type-1 loci) appear to be specialized to produce csRNAs primarily in one direction. The pattern of RNA expression at these loci was quite distinct from the pattern observed upstream of other Pol II transcripts. Type-1 21U-RNA loci typically produced abundant csRNAs and rarely, if ever, produced longer CapSeq reads (Figure 7C). When multiple csRNAs were produced at 21U-RNA loci, they typically shared the same orientation and their 5′ ends were often separated by less than 5 bp (data not shown). In contrast, other Pol II loci, such as protein-coding genes, produced abundant sense-oriented CapSeq reads, and multiple, relatively low-abundance csRNAs that were often oriented in both directions (Figure 7C). These observations suggest that Type-1 21U-RNA loci somehow focus Pol II initiation and restrict elongation to promote csRNA biogenesis at the expense of longer transcripts. In the future, it will be interesting to learn whether the upstream motif or other features of Type-1 21U-RNA loci govern their tendency to produce csRNAs but not longer Pol II transcripts.
Our findings suggest that a key step in 21U-RNA production is the removal of the cap and two nucleotides (Figure 7C). Although the exact cap structure of csRNAs is not known, we note that removal of a cap-2 structure (m7GpppAmNm) would directly expose the +3 U of a pre-21U for PRG-1 loading. Thus, the piRNA processing machinery could be linked to pathways that decap and turnover csRNAs. However, our findings clearly indicate that a U at positions +2 or +4 cannot substitute for a U at +3, suggesting that PRG-1 does not randomly sample 5′ degradation products, as was proposed for Ago1 and priRNAs in S. pombe (Conte and Mello, 2010; Halic and Moazed, 2010). Additional biochemical and genetic studies are needed to determine the structure and processing of csRNA caps.
The importance of the YR motif for 21U-RNA expression was highlighted by a subset of 21U-RNA loci that produce two mature 21U-RNA species whose 5′ ends are adjacent nucleotides. It is, of course, only possible for one of these 21U-RNAs to be associated with a YR motif, and the YR motif was approximately equally likely to be associated with the 5′ or 3′ 21U-RNA. Interestingly, regardless of relative order, we found that the YR-associated 21U-RNA was an average of ~10 times more abundant than was the non-YR-associated 21U-RNA. In cases where csRNAs were detected for both transcripts (which occurred at 21% of loci), a similar difference in expression level (~10-fold) was also observed between the YR-associated and non-YR-associated csRNAs. These observations support the idea that a YR motif is preferred for strong transcription initiation.
Conclusion
Recent studies have shown that PRG-1 and its piRNA cofactors provide an important first line of defense in a surveillance pathway that distinguishes self from non-self (Ashe et al., 2012; Lee et al., 2012; Shirayama et al., 2012). Importantly, PRG-1/piRNA complexes function in a context that does not require perfect base-pairing, greatly increasing the repertoire of potential target RNAs in C. elegans. The findings described here add to the amazing variety of piRNA biogenesis mechanisms, and identify a new type of piRNA that nearly doubles the number of piRNA species available for genome defense in C. elegans. The finding that small RNAs associated with TS sites are processed and loaded onto an Argonaute also raises an intriguing possibility that Argonaute-small RNA pathways might regulate promoter activity directly.
EXPERIMENTAL PROCEDURES
Worm Strains
The Bristol N2 strain of C. elegans was used in this study and cultured essentially as described (Brenner, 1974).
RNA Cloning and Sequencing
RNA was extracted using TRI Reagent (MRC, Inc.) or phenol. For CapSeq, 0.5 – 2 μg of total RNA was treated with Terminator exonuclease (Epicentre) to degrade rRNAs, calf intestine phosphatase (CIP, NEB) to remove 5′ phosphates, and tobacco acid pyrophosphatase (TAP, Epicentre) to remove 5′ caps. The resulting long-capped RNAs were ligated to a 5′ adapter. First strand cDNA was primed using a pool of random octamers containing a common 5′ sequence corresponding to a 3′ adapter oligo. The first strand cDNA was size selected and then amplified using Illumina adapter oligos.
Small RNA libraries were prepared essentially as described (Gu et al., 2009; Gu et al., 2011). Briefly, for CIP-TAP cloning, 18 – 40 nt RNA was gel purified from 40 μg of total RNA using a 15% PAGE/8M urea gel. The RNA was dephosphorylated with 50 U of CIP in a 100 μl reaction containing 1X NEB Buffer 3 and 0.5 U/μl SUPERase·In™ (Ambion) at 37°C for 1hr, and then extracted with phenol/chloroform, precipitated with isopropanol, ligated to a 3′ linker, and gel-purified. 3/4 of the 3′ ligation product was decapped with 2–4 U TAP in a 10 μl reaction containing 1X TAP buffer and 1 U/μl SUPERase·In™ at 37°C for 1hr (CIP-TAP sample). The remaining 1/4 was phosphorylated with 20 U of PNK (NEB) in a 20 μl reaction containing 1X PNK buffer, 0.5 U/μl SUPERase·In™, and 2 mM ATP at 37°C for 1hr (CIP-PNK control). Each sample was then ligated to a barcoded 5′ linker, gel-purified, reverse-transcribed, and PCR amplified with Solexa linkers.
Additional details are provided in Extended Experimental Procedures. Libraries were sequenced using an Illumina Genome Analyzer II or HiSeq instrument at the UMass Medical School Deep Sequencing Core (Worcester, MA).
Bioinformatics
Sequences were processed and mapped using custom PERL (5.10.1) scripts, Bowtie 0.12.7 (Langmead et al., 2009) and blastn 2.2.25 (Altschul et al., 1990). For C. elegans analysis, reads were mapped to the genome (WormBase release WS215), Repbase 15.10 (Jurka et al., 2005), and miRBase 16 (Kozomara and Griffiths-Jones, 2011). For mouse analysis, reads were aligned to the genome assembly NCBIM37 (Ensembl 67), miRBase 18 and the non-coding RNA database fRNAdb 3.4 (Mituyama et al., 2009). The Generic Genome Browser (GBrowse; Stein et al., 2002) was used to visualize the alignments.
Immunoprecipitation
The PRG-1 IP was performed as described previously (Batista et al., 2008). Small RNAs were extracted from IP and input and cloned using a TAP cloning protocol, as described (Gu et al., 2009).
Supplementary Material
HIGHLIGHTS.
New methodology to enzymatically enrich 5′ ends of Pol II transcripts.
Identification of transcription start sites for most C. elegans Pol II genes.
~26 nt capped-small (cs) RNAs expressed at C. elegans Pol II promoters genome wide.
csRNAs but not longer capped RNAs are the piRNA precursors in C. elegans.
Acknowledgments
We thank members of the Mello lab for useful discussions, Tom Blumenthal for providing spliced leader sequences, Stephen Jones for providing mouse sample, Zhiping Weng for providing information about the mouse database, and Ellen Kittler for deep sequencing. H.-C.L. is supported by a Ruth L. Kirschstein National Research Service Award (GM099372). C.C.M. is a Howard Hughes Medical Institute Investigator and is supported by NIH grant GM058800.
Footnotes
ACCESSION NUMBERS
Illumina data are available from GEO under the series number GSE40053.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Affymetrix/Cold Spring Harbor Laboratory ENCODE Transcriptome Project. Post-transcriptional processing generates a diversity of 5′-modified long and short RNAs. Nature. 2009;457:1028–1032. doi: 10.1038/nature07759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen MA, Hillier LW, Waterston RH, Blumenthal T. A global analysis of C. elegans trans-splicing. Genome Res. 2011;21:255–264. doi: 10.1101/gr.113811.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Andrecka J, Lewis R, Bruckner F, Lehmann E, Cramer P, Michaelis J. Single-molecule tracking of mRNA exiting from RNA polymerase II. Proc Natl Acad Sci U S A. 2008;105:135–140. doi: 10.1073/pnas.0703815105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravin A, Gaidatzis D, Pfeffer S, Lagos-Quintana M, Landgraf P, Iovino N, Morris P, Brownstein MJ, Kuramochi-Miyagawa S, Nakano T, et al. A novel class of small RNAs bind to MILI protein in mouse testes. Nature. 2006;442:203–207. doi: 10.1038/nature04916. [DOI] [PubMed] [Google Scholar]
- Aravin AA, Sachidanandam R, Girard A, Fejes-Toth K, Hannon GJ. Developmentally regulated piRNA clusters implicate MILI in transposon control. Science. 2007;316:744–747. doi: 10.1126/science.1142612. [DOI] [PubMed] [Google Scholar]
- Ashe A, Sapetschnig A, Weick EM, Mitchell J, Bagijn MP, Cording AC, Doebley AL, Goldstein LD, Lehrbach NJ, Le Pen J, et al. piRNAs Can Trigger a Multigenerational Epigenetic Memory in the Germline of C. elegans. Cell. 2012;150:88–99. doi: 10.1016/j.cell.2012.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista PJ, Ruby JG, Claycomb JM, Chiang R, Fahlgren N, Kasschau KD, Chaves DA, Gu W, Vasale JJ, Duan S, et al. PRG-1 and 21U-RNAs interact to form the piRNA complex required for fertility in C. elegans. Mol Cell. 2008;31:67–78. doi: 10.1016/j.molcel.2008.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein E, Caudy AA, Hammond SM, Hannon GJ. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature. 2001;409:363–366. doi: 10.1038/35053110. [DOI] [PubMed] [Google Scholar]
- Beyret E, Liu N, Lin H. piRNA biogenesis during adult spermatogenesis in mice is independent of the ping-pong mechanism. Cell Res. 2012;22:1429–1439. doi: 10.1038/cr.2012.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumenthal T, Steward K. RNA Processing and Gene Structure. In: Riddle DL, Blumenthal T, Meyer BJ, Priess JR, editors. C. elegans II. Cold Spring Harbor Laboratory Press; Cold Spring Harbor: 1997. [PubMed] [Google Scholar]
- Brennecke J, Aravin AA, Stark A, Dus M, Kellis M, Sachidanandam R, Hannon GJ. Discrete small RNA-generating loci as master regulators of transposon activity in Drosophila. Cell. 2007;128:1089–1103. doi: 10.1016/j.cell.2007.01.043. [DOI] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cecere G, Zheng GX, Mansisidor AR, Klymko KE, Grishok A. Promoters Recognized by Forkhead Proteins Exist for Individual 21U-RNAs. Mol Cell. 2012;47:734–745. doi: 10.1016/j.molcel.2012.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CY, Chang CC, Yen CF, Chiu MT, Chang WH. Mapping RNA exit channel on transcribing RNA polymerase II by FRET analysis. Proc Natl Acad Sci U S A. 2009;106:127–132. doi: 10.1073/pnas.0811689106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conte D, Jr, Mello CC. Primal RNAs: The end of the beginning? Cell. 2010;140:452–454. doi: 10.1016/j.cell.2010.02.005. [DOI] [PubMed] [Google Scholar]
- Core LJ, Waterfall JJ, Lis JT. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science. 2008;322:1845–1848. doi: 10.1126/science.1162228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das PP, Bagijn MP, Goldstein LD, Woolford JR, Lehrbach NJ, Sapetschnig A, Buhecha HR, Gilchrist MJ, Howe KL, Stark R, et al. Piwi and piRNAs act upstream of an endogenous siRNA pathway to suppress Tc3 transposon mobility in the Caenorhabditis elegans germline. Molecular cell. 2008;31:79–90. doi: 10.1016/j.molcel.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Hoon M, Hayashizaki Y. Deep cap analysis gene expression (CAGE): genome-wide identification of promoters, quantification of their expression, and network inference. Biotechniques. 2008;44:627–628. 630–632. doi: 10.2144/000112802. [DOI] [PubMed] [Google Scholar]
- Felix MA, Ashe A, Piffaretti J, Wu G, Nuez I, Belicard T, Jiang Y, Zhao G, Franz CJ, Goldstein LD, et al. Natural and experimental infection of Caenorhabditis nematodes by novel viruses related to nodaviruses. PLoS Biol. 2011;9:e1000586. doi: 10.1371/journal.pbio.1000586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girard A, Sachidanandam R, Hannon GJ, Carmell MA. A germline-specific class of small RNAs binds mammalian Piwi proteins. Nature. 2006;442:199–202. doi: 10.1038/nature04917. [DOI] [PubMed] [Google Scholar]
- Grivna ST, Beyret E, Wang Z, Lin H. A novel class of small RNAs in mouse spermatogenic cells. Genes Dev. 2006;20:1709–1714. doi: 10.1101/gad.1434406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu W, Claycomb JM, Batista PJ, Mello CC, Conte D. Cloning Argonaute-associated small RNAs from Caenorhabditis elegans. Methods Mol Biol. 2011;725:251–280. doi: 10.1007/978-1-61779-046-1_17. [DOI] [PubMed] [Google Scholar]
- Gu W, Shirayama M, Conte D, Jr, Vasale J, Batista PJ, Claycomb JM, Moresco JJ, Youngman EM, Keys J, Stoltz MJ, et al. Distinct argonaute-mediated 22G-RNA pathways direct genome surveillance in the C. elegans germline. Molecular cell. 2009;36:231–244. doi: 10.1016/j.molcel.2009.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunawardane LS, Saito K, Nishida KM, Miyoshi K, Kawamura Y, Nagami T, Siomi H, Siomi MC. A slicer-mediated mechanism for repeat-associated siRNA 5′ end formation in Drosophila. Science. 2007;315:1587–1590. doi: 10.1126/science.1140494. [DOI] [PubMed] [Google Scholar]
- Halic M, Moazed D. Dicer-independent primal RNAs trigger RNAi and heterochromatin formation. Cell. 2010;140:504–516. doi: 10.1016/j.cell.2010.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haussecker D, Cao D, Huang Y, Parameswaran P, Fire AZ, Kay MA. Capped small RNAs and MOV10 in human hepatitis delta virus replication. Nature structural & molecular biology. 2008;15:714–721. doi: 10.1038/nsmb.1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houwing S, Kamminga LM, Berezikov E, Cronembold D, Girard A, van den Elst H, Filippov DV, Blaser H, Raz E, Moens CB, et al. A role for Piwi and piRNAs in germ cell maintenance and transposon silencing in Zebrafish. Cell. 2007;129:69–82. doi: 10.1016/j.cell.2007.03.026. [DOI] [PubMed] [Google Scholar]
- Hutvagner G, Simard MJ. Argonaute proteins: key players in RNA silencing. Nat Rev Mol Cell Biol. 2008;9:22–32. doi: 10.1038/nrm2321. [DOI] [PubMed] [Google Scholar]
- Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 2005;110:462–467. doi: 10.1159/000084979. [DOI] [PubMed] [Google Scholar]
- Juven-Gershon T, Hsu JY, Theisen JW, Kadonaga JT. The RNA polymerase II core promoter - the gateway to transcription. Curr Opin Cell Biol. 2008;20:253–259. doi: 10.1016/j.ceb.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapranov P, Cheng J, Dike S, Nix DA, Duttagupta R, Willingham AT, Stadler PF, Hertel J, Hackermuller J, Hofacker IL, et al. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science. 2007;316:1484–1488. doi: 10.1126/science.1138341. [DOI] [PubMed] [Google Scholar]
- Kawaoka S, Izumi N, Katsuma S, Tomari Y. 3′ end formation of PIWI-interacting RNAs in vitro. Mol Cell. 2011;43:1015–1022. doi: 10.1016/j.molcel.2011.07.029. [DOI] [PubMed] [Google Scholar]
- Kozomara A, Griffiths-Jones S. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011;39:D152–157. doi: 10.1093/nar/gkq1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamm AT, Stadler MR, Zhang H, Gent JI, Fire AZ. Multimodal RNA-seq using single-strand, double-strand, and CircLigase-based capture yields a refined and extended description of the C. elegans transcriptome. Genome Res. 2011;21:265–275. doi: 10.1101/gr.108845.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau NC, Lim LP, Weinstein EG, Bartel DP. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science. 2001;294:858–862. doi: 10.1126/science.1065062. [DOI] [PubMed] [Google Scholar]
- Lau NC, Seto AG, Kim J, Kuramochi-Miyagawa S, Nakano T, Bartel DP, Kingston RE. Characterization of the piRNA complex from rat testes. Science. 2006;313:363–367. doi: 10.1126/science.1130164. [DOI] [PubMed] [Google Scholar]
- Lee HC, Gu W, Shirayama M, Youngman E, Conte D, Jr, Mello CC. C. elegans piRNAs Mediate the Genome-wide Surveillance of Germline Transcripts. Cell. 2012;150:78–87. doi: 10.1016/j.cell.2012.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H. piRNAs in the germ line. Science. 2007;316:397. doi: 10.1126/science.1137543. [DOI] [PubMed] [Google Scholar]
- Mituyama T, Yamada K, Hattori E, Okida H, Ono Y, Terai G, Yoshizawa A, Komori T, Asai K. The Functional RNA Database 3.0: databases to support mining and annotation of functional RNAs. Nucleic Acids Res. 2009;37:D89–92. doi: 10.1093/nar/gkn805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton JJ, Blumenthal T. Identification of transcription start sites of trans-spliced genes: uncovering unusual operon arrangements. RNA. 2011;17:327–337. doi: 10.1261/rna.2447111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nechaev S, Fargo DC, dos Santos G, Liu L, Gao Y, Adelman K. Global analysis of short RNAs reveals widespread promoter-proximal stalling and arrest of Pol II in Drosophila. Science. 2010;327:335–338. doi: 10.1126/science.1181421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pak J, Fire A. Distinct populations of primary and secondary effectors during RNAi in C. elegans. Science. 2007;315:241–244. doi: 10.1126/science.1132839. [DOI] [PubMed] [Google Scholar]
- Proudfoot NJ, Furger A, Dye MJ. Integrating mRNA processing with transcription. Cell. 2002;108:501–512. doi: 10.1016/s0092-8674(02)00617-7. [DOI] [PubMed] [Google Scholar]
- Rasmussen EB, Lis JT. Short transcripts of the ternary complex provide insight into RNA polymerase II elongational pausing. J Mol Biol. 1995;252:522–535. doi: 10.1006/jmbi.1995.0517. [DOI] [PubMed] [Google Scholar]
- Robine N, Lau NC, Balla S, Jin Z, Okamura K, Kuramochi-Miyagawa S, Blower MD, Lai EC. A broadly conserved pathway generates 3′UTR-directed primary piRNAs. Curr Biol. 2009;19:2066–2076. doi: 10.1016/j.cub.2009.11.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruby JG, Jan C, Player C, Axtell MJ, Lee W, Nusbaum C, Ge H, Bartel DP. Large-scale sequencing reveals 21U-RNAs and additional microRNAs and endogenous siRNAs in C. elegans. Cell. 2006;127:1193–1207. doi: 10.1016/j.cell.2006.10.040. [DOI] [PubMed] [Google Scholar]
- Seila AC, Calabrese JM, Levine SS, Yeo GW, Rahl PB, Flynn RA, Young RA, Sharp PA. Divergent transcription from active promoters. Science. 2008;322:1849–1851. doi: 10.1126/science.1162253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seila AC, Core LJ, Lis JT, Sharp PA. Divergent transcription: a new feature of active promoters. Cell Cycle. 2009;8:2557–2564. doi: 10.4161/cc.8.16.9305. [DOI] [PubMed] [Google Scholar]
- Shirayama M, Seth M, Lee HC, Gu W, Ishidate T, Conte D, Jr, Mello CC. piRNAs Initiate an Epigenetic Memory of Nonself RNA in the C. elegans Germline. Cell. 2012;150:65–77. doi: 10.1016/j.cell.2012.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sijen T, Steiner FA, Thijssen KL, Plasterk RH. Secondary siRNAs result from unprimed RNA synthesis and form a distinct class. Science. 2007;315:244–247. doi: 10.1126/science.1136699. [DOI] [PubMed] [Google Scholar]
- Siomi H, Siomi MC. On the road to reading the RNA-interference code. Nature. 2009;457:396–404. doi: 10.1038/nature07754. [DOI] [PubMed] [Google Scholar]
- Smale ST, Baltimore D. The “initiator” as a transcription control element. Cell. 1989;57:103–113. doi: 10.1016/0092-8674(89)90176-1. [DOI] [PubMed] [Google Scholar]
- Stein LD, Mungall C, Shu S, Caudy M, Mangone M, Day A, Nickerson E, Stajich JE, Harris TW, Arva A, et al. The generic genome browser: a building block for a model organism system database. Genome Res. 2002;12:1599–1610. doi: 10.1101/gr.403602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto YY, Ichida H, Matsui M, Obokata J, Sakurai T, Satou M, Seki M, Shinozaki K, Abe T. Identification of plant promoter constituents by analysis of local distribution of short sequences. BMC Genomics. 2007;8:67. doi: 10.1186/1471-2164-8-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.