

Abstract
This study was undertaken to test the hypothesis that short-term exposure (4 h) to physiological hyperinsulinemia in normal, healthy subjects without a family history of diabetes would induce a low grade inflammatory response independently of glycemic status. Twelve normal glucose tolerant subjects received a 4-h euglycemic hyperinsulinemic clamp with biopsies of the vastus lateralis muscle. Microarray analysis identified 121 probe sets that were significantly altered in response to physiological hyperinsulinemia while maintaining euglycemia. In normal, healthy human subjects insulin increased the mRNAs of a number of inflammatory genes (CCL2, CXCL2 and THBD) and transcription factors (ATF3, BHLHB2, HES1, KLF10, JUNB, FOS, and FOSB). A number of other genes were upregulated in response to insulin, including RRAD, MT, and SGK. CITED2, a known coactivator of PPARα, was significantly downregulated. SGK and CITED2 are located at chromosome 6q23, where we previously detected strong linkage to fasting plasma insulin concentrations. We independently validated the mRNA expression changes in an additional five subjects and closely paralleled the results observed in the original 12 subjects. A saline infusion in healthy, normal glucose-tolerant subjects without family history of diabetes demonstrated that the genes altered during the euglycemic hyperinsulinemic clamp were due to hyperinsulinemia and were unrelated to the biopsy procedure per se. The results of the present study demonstrate that insulin acutely regulates the levels of mRNAs involved in inflammation and transcription and identifies several candidate genes, including HES1 and BHLHB2, for further investigation.
Keywords: gene expression, muscle, insulin action, euglycemic hyperinsulinemic clamp, inflammation
insulin resistance is a common pathological state associated with obesity, hypertension, atherosclerotic cardiovascular disease, and type 2 diabetes mellitus (11). This cluster of abnormalities, referred to as the insulin resistance or metabolic syndrome, is thought to be related to the underlying insulin resistance (34). Despite the severe impairment in insulin action in insulin resistant states, glucose tolerance remains normal because the pancreatic β-cells are able to augment their insulin secretory capacity to offset the insulin resistance (14, 19). Hyperinsulinemia (increased insulin levels) occurs as a result of the increase in insulin secretion from the β-cells. Over time, the excessive rates of insulin secretion cannot be maintained, leading to the development of glucose intolerance and type 2 diabetes (11, 14).
Despite intensive investigation, the underlying cause of insulin resistance remains unknown. Many cellular defects have been shown to contribute to the pathophysiology of insulin resistance in type 2 diabetes, including impaired insulin signaling (4, 10), reduced insulin-stimulated glucose uptake, decreased hexokinase II expression and activity, diminished glycogen synthase activity, and decreased pyruvate dehydrogenase activity (8, 15, 26, 30, 39, 42). Impaired mitochondrial function has been associated with an increase in toxic lipid metabolites (fatty acyl-CoA, diacylglycerol, ceramide) in skeletal muscle of subjects with prediabetes and type 2 diabetes, and these toxic lipid metabolites have been implicated as a cause of the insulin resistance (1, 20, 23, 27, 29). We (36) previously reported that a physiological increase in plasma FFA concentration in normal healthy subjects resulted in changes in gene expression in skeletal muscle that are consistent with an inflammatory response. In addition, we also demonstrated that changes in the composition of the extracellular matrix are a general characteristic feature of insulin-resistant muscle (7).
Insulin has been shown to be a risk factor for the development of atherosclerotic cardiovascular disease (11). The effects of insulin on arterial tissues include proliferation of smooth muscle cells, enhanced cholesterol synthesis and low-density lipoprotein-receptor activity, increased formation and decreased regression of lipid plaques, stimulation of connective tissue synthesis, and stimulation of growth factors (11). In the present study, we set out to test the hypothesis that short-term exposure (4 h) to physiological hyperinsulinemia in normal, glucose-tolerant, healthy subjects without a family history of diabetes would induce a low-grade inflammatory response, independently of glycemic status, in skeletal muscle. By combining the euglycemic insulin clamp technique (13) with microarray profiling, we tested this hypothesis and, at the same time, identified novel pathways that are regulated by insulin in skeletal muscle.
MATERIALS AND METHODS
Subjects
Healthy, normal glucose-tolerant subjects without a family history of diabetes participated in the study. All subjects were free of any organ system disease as determined by medical history, physical examination, screening chemistries, complete blood cell count, urinalysis, and electrocardiogram. Normal glucose tolerance was established by a 75-g oral glucose tolerance test (OGTT) performed 1 wk prior to the clamp study. Percent body fat was determined by DEXA using a Hologic Delphi A scanner (Hologic, Bedford, MA). Subjects were not taking any medications and demonstrated stable body weight (±3lbs)over the 6 mo prior to the study. None of the subjects participated in any heavy exercise, and they were instructed not to exercise for 3 days before the study. All subjects gave informed written consent prior to their participation in the study. The study protocol was approved by the Institutional Review Board of the University of Texas Health Science Center at San Antonio.
Study design
All subjects consumed a weight-maintaining diet containing 50% carbohydrate, 35% fat, and 15% protein over the 3 days prior to study and were instructed not to participate in any heavy exercise. After a 10-h overnight fast, all subjects reported to the General Clinical Research Center (GCRC) at 0800, and catheters were placed into an antecubital vein (for infusion of all test substances) and retrogradely into a vein on the dorsum of the hand (for blood withdrawal). The hand was placed in a heated box (65°C) to achieve arterialization of the venous blood. Twelve subjects received a 4-h euglycemic hyperinsulinemic (80 mU· m−2·min−1) clamp, which was performed with infusion of [3-3H]glucose to determine rates of glucose appearance and disposal, as previously described (10, 13). Briefly, 120 min prior to the start of the insulin clamp, a primed (25 μCi) continuous (0.25 μCi/min) infusion of [3-3H]glucose (DuPont-NEN, Boston, MA) was started and continued throughout the clamp to determine rates of total body glucose disposal and endogenous (primarily hepatic) glucose production (13). During the 30 min prior to the start of the insulin infusion and throughout the insulin clamp, plasma glucose concentration was determined every 5 min, and a variable glucose infusion was adjusted on the basis of the negative feedback principle to maintain the plasma glucose concentration at each subject's basal value. Plasma insulin and plasma tritiated glucose specific activity were measured every 10–15 min. Under euglycemic hyperinsulinemic conditions, the majority (~85%) of glucose disposal occurs in skeletal muscle (12). Biopsies of the vastus lateralis muscle were taken at −60, 30, and 240 min of the insulin infusion, as previously described (10). A control saline (0.2 ml·m−2·min−1) infusion was performed in three additional subjects, with a vastus lateralis muscle biopsy taken basally and at the end of the saline infusion. After completion of the microarray/RT-PCR determinations (see below), we studied an additional five subjects to validate our original observations. These five subjects received a 3-h euglycemic hyperinsulinemic (80 mU·m−2·min−1) clamp with tritiated glucose, as described above. Vastus lateralis muscle biopsies were performed at $60 min and after 180 min of insulin infusion. Muscle biopsy specimens (~200 mg) were frozen immediately in liquid nitrogen and stored until processed.
Muscle biopsy processing
Muscle biopsies from each subject were homogenized in RNAStat solution (Tel-Test, Friendswood, TX), using a Polytron homogenizer (Brinkmann Instruments Westbury, NY). Total RNA was purified with RNeasy and DNase I treatment (Qiagen, Chatsworth, CA). RNA isolated from the insulin infusion biopsies was used in the microarray and quantitative RT-PCR studies described below. The RNA isolated from the control saline infusion biopsies was used for the quantitative RT-PCR studies.
Microarray analysis: target preparation, hybridization, staining, scanning, and image analysis
RNA was prepared for hybridization to Affymetrix (Santa Clara, CA) HG-U133A arrays according to the manufacturer's instructions. A description of the protocol has been described previously (36). The probe arrays were scanned twice and the stored images aligned and analyzed using the GeneChip Operating System software (GCOS; Affymetrix). GeneChip quality assessments using GCOS demonstrated that the average percent present call across all arrays was 28 ± 1%. Average scaling values and noise levels were 6.8 ± 0.9 and 4.7 ± 0.2, respectively. bioB, bioC, bioD and cre were present on all arrays with the exception of bioB, which was present only 50% of the time, as expected. 3'/5' GAPDH and actin expression ratios were well within the acceptable range: 1.2 ± 0.1 and 0.8 ± 0.1, respectively.
Microarray data expression and analysis
The Affymetrix data acquisition programs in GCOS automatically generate a cell intensity file (CEL) from the stored images that contain a single intensity value for each probe cell on the array. The CEL data files have been deposited in the Gene Expression Omnibus (GEO) database as series GSE9105. The CEL files were imported into the R software package (http://www.r-project.org), and the probe level data were converted to expression measures using the Affy package from Bioconductor. Expression values for each mRNA were obtained by GC-Robust Multi-Array Analysis (GCRMA). CEL files were normalized together, and the expression values obtained were submitted to analysis with linear models of microarray data (LIMMA). To correct for multiple testing, P values were adjusted using the method of Benjamini and Hochberg by setting the false discovery rate (FDR) to 5% and a fold change of >1.5.
Functional classification of the significantly increased or decreased genes was performed using database for annotation, visualization and integrated discovery (DAVID) (http://david.niaid.nih.gov), Onto-Express (http://vortex.cs.wayne.edu/ontoexpress/), and expression analysis systemic explorer (EASE; downloaded from DAVID). DAVID uses clustering algorithms to classify related genes into functionally related groups. EASE discovers categories of genes that are overrepresented in the significant gene list compared with the total set of genes represented on the microarray. Such overrepresented categories represent biological “themes” of a given list of significant genes. The categories are sorted into three major gene ontology (GO) groups: 1) molecular function, 2) biological processes, and 3) cellular components. Onto-Express was used in the present study to translate the list of differentially expressed insulin-regulated genes into chromosome location.
Analysis of published microarray data
We compared the results of our microarray analysis with other previously published datasets by using the Web-based approach described by Parikh et al. (28). Briefly, we obtained normalized data files that were downloaded from either the Diabetes Genome Anatomy Project (DGAP; http://www.diabetesgenome.org) or the GEO database (http://www.ncbi.nlm.nih.gov/geo/). Using the Mann-Whitney U-test, a nonparametric statistical technique to identify mRNA expression changes, we compared the results in the DGAP and GEO databases with our list of mRNAs that were significantly altered following insulin infusion.
Quantitative real-time PCR
Muscle expression of various genes was determined using a one-step quantitative real-time PCR (QRT-PCR) from total RNA (Applied Biosystems 7900HT). The quantity of mRNA for each gene of interest for each sample was normalized to that of 18S ribosomal RNA, using the comparative (2−ΔΔCT) method as described previously (36). Statistical comparisons were done using paired t-tests. Sequences of the primers and probes used for mRNA quantification by QRT-PCR can be supplied upon request.
Substrate and hormone determinations
Plasma glucose concentration was determined by the glucose oxidase method on a Beckman Glucose Analyzer (Beckman Instruments, Fullerton, CA). Plasma insulin was determined by radioimmunoassay (Diagnostic Products, Los Angeles, CA). Plasma tritiated glucose specific activity was determined on Somogyi precipitates of plasma as previously described (5).
Statistical analysis
Data were expressed as the means ± SE. Statistical significance of the difference between means was determined using paired or nonpaired Student's t-tests where appropriate. Correlations were analyzed using Pearson's product moment test. For all analyses, P < 0.05 was considered to be statistically significant. Statistical analysis of microarray data and QRT-PCR data were described above.
RESULTS
Subjects
Twelve healthy subjects without a family history of diabetes (5 females and 7 males) with a mean age (±SE) of 38 ± 4 yr and body mass index of 25.1 ± 0.8 kg/m2 participated in the 4-h euglycemic hyperinsulinemic clamp study. These subjects had a lean body mass and percent body fat of 52.9 ± 4.9 kg and 29.6 ± 4.1%, respectively. The fasting plasma glucose (FPG) and fasting plasma insulin (FPI) concentrations were 5.2 ± 0.2 mmol/l, and 27.8 ± 6.9 pmol/l, respectively. The 2-h plasma glucose concentration during the OGTT was 5.7 ± 0.3 mmol/l and the insulin-stimulated glucose disposal rate (Rd) was 10.3 ± 0.5 mg·kg−1·min−1. Steady-state plasma glucose (SSPG) and insulin (SSPI) concentrations during the insulin clamp were 5.3 ± 0.1 mmol/l and 826.5 ± 48.6 pmol/l, respectively. Basal endogenous glucose production (basal EGP) was 2.0 ± 0.2 mg·kg−1·min−1 and was suppressed to 0.10 ± 0.01 mg·kg−1·min−1 during the clamp. An additional five subjects matched for age, body mass index, FPG, and Rd (45 ± 3 yr, 27.2 ± 1.4 kg/m2, 5.3 ± 0.1 mmol/l, and 7.1 ± 1.1 mg·kg−1·min−1, respectively) participated in a 3-h euglycemic hyperinsulinemic clamp study. Three subjects (age 28 ± 4 yr, body mass index 23.8 ± 2.4 kg/m2, FPG 5.2 ± 0.3 mmol/l, and Rd 8.0 ± 1.4 mg·kg−1·min−1) participated in the control saline infusion.
Analysis of microarray data
Microarray analysis was performed on the muscle biopsies obtained from the 12 subjects who received a 4-h euglycemic hyperinsulinemic clamp (Fig. 1). Using a false discovery rate of <5% and a fold change of >1.5, there were no individual probe sets with significantly increased or decreased expression after 30 min of insulin infusion compared with the basal state. In contrast, 121 probe sets were significantly altered (117 upregulated and 4 downregulated) after 240 min of insulin infusion compared with the basal state [Supplementary Tables 1 and 2, respectively (see online)]. The 117 upregulated probe sets represent in total 93 genes, since most genes are represented more than once due to the presence of more than one probe set on the HG-U133A GeneChip. The expression of 105 probe sets (96 increased and 9 decreased) was altered after 240 min compared with their expression 30 min after the start of the insulin infusion (Supplementary Tables 3 and 4, respectively). The 96 probe sets with significantly increased expression at 240 vs. 30 min of insulin infusion also were significantly altered at 240 min vs. the basal state. We identified five additional probe sets that were decreased at 240 vs. 30 min of the insulin infusion (Supplementary Table 4). Of note, one of these genes was thioredoxin-interacting protein (TXNIP), which has been implicated in insulin-stimulated peripheral glucose uptake in humans (28). The results of the present study indicate that most of the detectable changes occurred 30 min after the start of insulin.
Fig. 1.
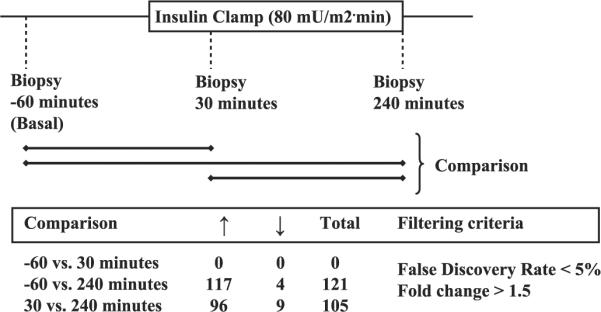
Number of probe sets upregulated or downregulated following insulin infusion.
Functional classification of genes
Gene functional classification clustering analysis was performed in DAVID on the genes that were upregulated (n = 93) by insulin. Four functional groups were identified from the list of upregulated genes: 1) transcription binding, activity, and regulation, 2) protein kinase activity, 3) inflammation and chemokine activity, and 4) metal ion binding (Table 1). Additional information on the clustering of the genes in each group is displayed in Supplementary Figs. 1–4. No functional groups were identified for the four genes downregulated by insulin.
Table 1.
Results of functional classification clustering analysis performed in DAVID (http://david.abcc.ncifcrf.gov) on the genes that were upregulated (n = 93) by insulin
| Gene Functional Classification | Gene Name | Gene Symbol |
|---|---|---|
| Transcription binding, activity and regulation (n = 18) | nuclear receptor subfamily 4, group a, member 3 | NR4A3 |
| kruppel-like factor 10 | KLF10 | |
| v-maf musculoaponeurotic fibrosarcoma oncogene homolog f (avian) | MAFF | |
| v-ets erythroblastosis virus e26 oncogene homolog 2 (avian) | ETS2 | |
| nuclear factor, interleukin 3 regulated | NFIL3 | |
| jun b proto-oncogene | JUNB | |
| interferon, gamma-inducible protein 16 | IFI16 | |
| v-myc myelocytomatosis viral oncogene homolog (avian) | MYC | |
| activating transcription factor 3 | ATF3 | |
| btg family, member 2 | BTG2 | |
| fbj murine osteosarcoma viral oncogene homolog b | FOSB | |
| early growth response 1 | EGR1 | |
| v-fos fbj murine osteosarcoma viral oncogene homolog | FOS | |
| basic helix-loop-helix domain containing, class b, 2 | BHLHB2 | |
| tgfb-induced factor (tale family homeobox) | TGIF | |
| hairy and enhancer of split 1, (drosophila) | HES1 | |
| tsc22 domain family, member 2 | TSC22D2 | |
| ccaat/enhancer-binding protein (c/ebp), delta | CEBPD | |
| Protein kinase activity (n = 4) | pim-1 oncogene | PIM1 |
| snf1-like kinase | SNF1LK | |
| serum/glucocorticoid-regulated kinase | SGK | |
| tribbles homolog 1 | TRIB1 | |
| Inflammation and chemokine activity (n = 4) | chemokine (C-C motif) ligand 2 | CCL2 |
| chemokine (C-C motif) ligand 8 | CCL8 | |
| chemokine (C-X-C motif) ligand 2 | CXCL2 | |
| interleukin 1 receptor, type I | IL1R1 | |
| Metal ion binding (n = 6) | metallothionein 2e | MT2A |
| metallothionein 1f | MT1F | |
| metallothionein 1e | MT1E | |
| metallothionein 1h | MT1H | |
| metallothionein 1m | MT1M | |
| metallothionein 1x | MT1X |
EASE analysis on the genes that were upregulated (n = 93) by insulin yielded 42 significant gene categories, divided among three GO systems: biological process (n = 28), cellular component (n = 1), and molecular function (n = 13). The gene categories identified for each significant GO system are depicted in Supplementary Table 5. Briefly, for the biological process system, the majority of the genes coded for proteins involved in cell growth (50%) and regulation of transcription (31%). In addition, genes in the molecular function system were involved in RNA/DNA binding (74%) and transcription (33%). The only cellular component system identified was the nucleus, where 43% of the genes were localized.
The chromosomal location of the differentially expressed genes following insulin infusion was determined using the program Onto-Express. The location of the insulin-regulated genes was distributed among all chromosomes except Y, 13, and 7. Statistical analysis performed in Onto-Express yielded no chromosome that contained more insulin-regulated genes than expected (Supplementary Fig. 5).
Insulin signaling gene analysis
Because the microarray analysis identified significant increases in some genes involved in the insulin-signaling pathway including RRAD, KRAS, KLF10, and FOS (Supplementary Table 1), we looked at the microarray expression values of other insulin-signaling genes. Following 240 min of insulin infusion, the expression of 24 insulin-signaling genes increased (all P < 0.05, without correcting for multiple testing; Table 2). Many of these mRNA expression changes did not achieve statistical significance when corrected for multiple testing.
Table 2.
Individual insulin-signaling genes with increased expression following 240 min of insulin infusion
| Probe Set ID | Gene Name | Gene Symbol | Basal (mean ± SE) | Insulin (mean ± SE) | P Value |
|---|---|---|---|---|---|
| 201435_s_at | eukaryotic translation initiation factor 4E | EIF4E | 7.7±0.2 | 8.5±0.3 | 0.001 |
| 201436_at | eukaryotic translation initiation factor 4E | EIF4E | 5.2±0.2 | 5.8±0.2 | 0.017 |
| 201437_s_at | eukaryotic translation initiation factor 4E | EIF4E | 6.7±0.2 | 7.9±0.2 | 0.00002 |
| 203719_at | excision repair cross-complementing rodent repair deficiency, 1 | ERCC1 | 6.6±0.1 | 6.7±0.1 | 0.052 |
| 209189_at | v-fos FBJ murine osteosarcoma viral oncogene homolog | FOS | 3.2±0.1 | 10.2±0.8 | 0.00001 |
| 215075_s_at | growth factor receptor-bound protein 2 | GRB2 | 7.4±0.1 | 7.6±0.1 | 0.032 |
| 202934_at | hexokinase II | HK2 | 5.0±0.4 | 6.4±0.4 | 0.001 |
| 213792_s_at | insulin receptor | INSR | 7.0±0.2 | 7.4±0.2 | 0.039 |
| 209184_s_at | insulin receptor substrate 2 | IRS2 | 3.3±0.1 | 4.0±0.3 | 0.010 |
| 209185_s_at | insulin receptor substrate 2 | IRS2 | 8.1±0.3 | 9.0±0.4 | 0.023 |
| 202393_s_at | Kruppel-like factor 10 | KLF10 | 9.3±0.3 | 11.6±0.1 | 0.000001 |
| 204009_s_at | v-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog | KRAS | 6.1±0.4 | 6.9±0.2 | 0.006 |
| 214352_s_at | v-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog | KRAS | 6.2±0.6 | 7.9±0.3 | 0.002 |
| 202068_s_at | Low-density lipoprotein receptor | LDLR | 2.3±0.1 | 3.4±0.4 | 0.013 |
| 212688_at | phosphoinositide-3-kinase, catalytic, beta polypeptide | PIK3CB | 6.2±0.2 | 6.6±0.2 | 0.006 |
| 212240_s_at | phosphoinositide-3-kinase, regulatory subunit 1 (p85 alpha) | PIK3R1 | 7.8±0.2 | 9.0±0.2 | 0.002 |
| 202743_at | phosphoinositide-3-kinase, regulatory subunit 3 (p55, gamma) | PIK3R3 | 3.3±0.2 | 4.7±0.4 | 0.001 |
| 209678_s_at | protein kinase C, iota | PRKCI | 7.4±0.3 | 7.8±0.2 | 0.030 |
| 212610_at | protein tyrosine phosphatase, nonreceptor type 11 | PTPN11 | 10.3±0.2 | 10.5 + 0.1 | 0.040 |
| 212590_at | related RAS viral (r-ras) oncogene homolog 2 | RRAS2 | 3.5±0.2 | 4.0±0.3 | 0.024 |
| 212589_at | Sterol carrier protein 2 | SCP2 | 8.4±0.3 | 9.0±0.2 | 0.001 |
| 210512_s_at | vascular endothelial growth factor | VEGF | 10.2±0.2 | 10.9±0.1 | 0.00008 |
| 211527_x_at | vascular endothelial growth factor | VEGF | 3.8±0.1 | 4.0±0.1 | 0.033 |
| 212171_x_at | vascular endothelial growth factor | VEGF | 8.8±0.1 | 9.1±0.2 | 0.040 |
Significance tested by a Student's paired two-tailed t-test (not corrected for multiple testing); P < 0.05.
Analysis of published datasets
Parikh et al. (28) examined mRNA expression in human muscle biopsies from normal glucose-tolerant subjects before and after a 2-h euglycemic hyperinsulinemic clamp. Analysis of the normalized dataset of Parikh et al. using a Mann-Whitney U-test identified six genes (KCNJ2, SOCS3 BHLHB2, HES1, MT1H, and TXNIP) that were significantly altered and in common with our insulin-regulated genes (Supplementary Table 6). Hansen et al. (21) reported that the expression of three of these genes (HES1, BHLHB2, and TXNIP) was significantly increased by insulin in human myotubes. Rome et al. (37) also reported that HES1 mRNA expression was significantly increased during a 3-h euglycemic hyperinsulinemic clamp. Analysis of the normalized dataset of Patti et al. (29), using a Mann Whitney U-test, identified a significant increase in the mRNA expression of HES1 in type 2 diabetics compared with normal, healthy subjects without a family history of diabetes (Supplementary Table 6). Similarly, BHLHB2 and MT1H mRNAs were both significantly increased in diabetic subjects compared with normal glucose-tolerant subjects when we analyzed the dataset of Mootha et al. (27).
Correlations of microarray data with subject characteristics
To identify associations between insulin-regulated gene expression and insulin-stimulated glucose disposal, we performed correlation analysis using Pearson's product moment test. Insulin-stimulated whole body (primarily represents muscle) glucose disposal was strongly and inversely correlated with ADH1B, CYR61, and BHLHB2 mRNA expression (r = –0.74, –0.65, and –0.57, respectively). We also observed a negative correlation between basal EGP and serum/glucocorticoid-regulated kinase (SGK) mRNA expression (r = −0.74, P < 0.01), while DNAJB1, HBEGF, and TRIB1 mRNA gene expression was strongly correlated with fasting plasma insulin concentration (r = 0.74, 0.65, and 0.58, respectively). These and other correlations are depicted in Supplementary Table 7.
QRT-PCR analysis to confirm microarray data
To validate the microarray data, we quantitated the changes in mRNA levels of 20 genes (18 of which increased and 2 of which decreased) using QRT-PCR in the twelve subjects who received a 4-h euglycemic hyperinsulinemic clamp. These genes displayed wide differences in their response to insulin on the microarray (from −2.3 to 7.7-fold change). The correlation coefficient (r) between the genes quantitated by QRT-PCR and microarray analysis was 0.79 (P < 0.01) (Supplementary Fig. 6). Of note, the QRT-PCR fold changes were larger than the changes observed with microarray analysis. This is consistent with previous studies from our laboratory in which microarray analysis tends to underestimate the changes in gene expression (36). In addition, we independently validated the mRNA expression changes in an additional five subjects who received a 3-h euglycemic hyperinsulinemic clamp. mRNA expression in these 5 subjects, who were studied after the microarray/QRTPCR analyses were completed in the first 12 subjects, closely paralleled that in the original 12 individuals (Table 3). We also observed significant interindividual variation in the basal mRNA levels in these normal, healthy subjects. This natural variation in mRNA expression most likely represents the complex interplay of intrinsic (i.e., age, sex, ethnicity, and genes), physiological, and environmental factors. This observation is consistent with other studies that have identified significant interindividual variation in basal mRNA expression in healthy subjects (18, 33, 43).
Table 3.
Results of the microarray with those of qRT-PCR following insulin infusion
| Fold Change |
||||
|---|---|---|---|---|
| Probe Set ID | Gene Symbol | Microarray (4-h clamp) | qRT-PCR (4-h clamp) | qRT-PCR (3-h clamp) |
| 216598_s_at | CCL2 | 3.78 | 64.3±21.4* | 1.7±0.2* |
| 214038_at | CCL8 | 3.05 | 116.6±49.5* | 4.8±1.5* |
| 202284_s_at | CDKN1A | 3.91 | 39.4±16.6* | 2.2±0.4* |
| 209774_x_at | CXCL2 | 3.80 | 82.0±39.9* | 1.6±0.2* |
| 201693_s_at | EGR1a | 6.09 | 174.2±121.0 | 3.4±0.8* |
| 201329_s_at | ETS2a | 2.56 | 5.0±1.3* | 1.3±0.2 |
| 209189_at | FOS | 7.00 | 203.9±93.4 | 2.7±0.7* |
| 207574_s_at | GADD45Ba | 6.42 | 70.2±18.2* | 2.9±1.8 |
| 202948_at | IL1R1 | 2.30 | 3.9±0.8* | 1.4±0.1* |
| 201473_at | JUNB | 4.06 | 14.4±8.8 | 2.1±0.3* |
| 202393_s_at | KLF10 | 2.30 | 5.4±1.2* | 1.7±0.2* |
| 202431_s_at | MYC | 4.79 | 43.1±15.5* | 3.5±1.1* |
| 203574_at | NFIL3 | 5.38 | 8.7±2.6* | 1.7±0.1* |
| 211302_s_at | PDE4Ba | 3.97 | 8.9±2.6* | 1.7±0.2* |
| 204803_s_at | RRADa | 3.80 | 14.3±4.4* | 2.5±0.7 |
| 201739_at | SGK | 1.63 | 3.6±0.9* | 1.5±0.2* |
| 203887_s_at | THBDa | 7.69 | 8.4±5.0 | 1.9±0.3* |
| 201531_at | ZFP36 | 4.34 | 54.4±17.4* | 1.7±0.3 |
| 209357_at | CITED2 | −2.25 | −2.5±0.1* | −2.1±0.1* |
| 206765_at | KCNJ2 | −1.79 | −2.0±0.1* | −1.6±0.1* |
More than one probe set was significantly altered for this gene on the microarray. The probe set with the greatest fold change in the microarray analysis is represented here (data taken directly from Supplementary Tables 1 and 2). Significance for the genes selected for qRT-PCR was assessed looking at each dataset separately and using Student's paired t-test.
P < 0.05 vs. control 18S ribosomal RNA (used for qRT-PCR analysis).
QRT-PCR analysis performed on control saline infusion samples
To verify that the changes observed during the insulin clamp were due to insulin action on the muscle and not due to the biopsy procedure per se, we performed QRT-PCR analysis on the muscle biopsies from subjects who received saline infusion. For this study, we analyzed the same genes that were used to validate the insulin infusion microarray analysis. The results of the saline infusion study are presented in Table 4. Of the 20 genes examined, three genes were significantly altered compared with the basal state. ETS2 and RRAD mRNA levels were significantly increased during the saline infusion, by 1.24 ± 0.03 and 1.78 ± 0.09, respectively (both P < 0.05), but the changes were quantitatively much less than the fold increases observed in the insulin infusion group (Table 3). Likewise, the significant decrease in EGR1 mRNA expression in the control saline infusion (–3.57 ± 2.30 fold change) was contrary to the significant increase observed during insulin infusion (Table 3).
Table 4.
mRNA expression determined by qRT-PCR during saline infusion
| Probe Set ID | Gene Symbol | Fold Change: Saline Infusion |
|---|---|---|
| 216598_s_at | CCL2 | −1.09±0.22 |
| 214038_at | CCL8 | 1.02±0.67 |
| 202284_s_at | CDKN1A | 1.10±0.14 |
| 209774_x_at | CXCL2 | −1.69±0.83 |
| 201693_s_at | EGR1 | −3.57±2.30* |
| 201329_s_at | ETS2 | 1.23±0.03* |
| 209189_at | FOS | −2.94±2.60 |
| 207574_s_at | GADD45B | −1.28±0.30 |
| 202948_at | IL1R1 | 1.60±0.60 |
| 201473_at | JUNB | −1.02±0.40 |
| 202393_s_at | KLF10 | 2.45±0.66 |
| 202431_s_at | MYC | 1.00±0.41 |
| 203574_at | NFIL3 | −1.25±0.30 |
| 211302_s_at | PDE4B | 1.38±0.31 |
| 204803_s_at | RRAD | 1.78±0.09* |
| 201739_at | SGK | −1.28±0.16 |
| 203887_s_at | THBD | 2.33±1.68 |
| 201531_at | ZFP36 | −1.56±0.73 |
| 209357_at | CITED2 | 1.15±0.26 |
| 206765_at | KCNJ2 | −1.12±0.25 |
P < 0.05 vs. control 18S ribosomal RNA, using Student's paired t-test.
DISCUSSION
The present study was undertaken in part to test the hypothesis that short-term exposure (4 h) to physiological hyperinsulinemia in normal, healthy subjects without a family history of diabetes would induce a low-grade inflammatory response independently of glycemic status. To test this hypothesis subjects received an 80 mU·m−2·min−1 euglycemic hyperinsulinemic clamp with vastus lateralis muscle biopsies performed before, during, and at the end of the insulin clamp. The concentration of insulin (~560 pmol/l) achieved during the insulin clamp was within the high physiological range, as described elsewhere (32), and was similar to that observed during an oral glucose/meal tolerance test, although we infused insulin for 4 h, which is longer than the usual tissue exposure to insulin following a meal in normal, healthy subjects. Therefore, we believe that the results in the present study have relevance to the effects of physiological hyperinsulinemia on muscle mRNA expression and may provide insights into the etiology of insulin resistance in type 2 diabetes and obesity (11).
The uniqueness of the present study compared with previous studies (3, 15, 21, 25, 28, 37, 40, 44) is the inclusion of a control saline infusion group and validation of the results in a separate group of individuals. In our study, we felt that it was important to demonstrate that the observed changes in mRNA expression during the insulin infusion clamp were independent of the biopsy procedure per se, which could have led to increased mRNA expression of some genes involved in inflammation. The results of the saline infusion study clearly demonstrate that the genes altered during the euglycemic hyperinsulinemic clamp were due to insulin and independent of the muscle biopsy procedure.
The results of the present study demonstrate that a number of genes involved in inflammation were upregulated. Thrombomodulin, which is dysregulated during inflammation, had the largest fold change (7.69) from the microarray analysis. A number of inflammatory chemokines (CCL8, CXCL2, and CCL2) and cytokines (interleukin-1 receptor type I and interferon-γ-inducible protein-16) were also upregulated by insulin. Chemokine, cc motif ligand 2 (CCL2) and chemokine, cc motif ligand 2 (CCL8), also known as monocyte chemoattractant protein-1 and -2, are members of the CC chemokine family and promote monocyte chemotaxis to sites of inflammation. These results are consistent with those of a previous study that identified a number of inflammatory genes (including THBD, CCL2, and CXCL2) that were upregulated following insulin treatment (21). Rome et al. (37) also reported that a number of immune-response genes were increased following a euglycemic hyperinsulinemic clamp, but those genes did not overlap with our study (37). QRT-PCR confirmed the increased mRNA expression of inflammation genes originally identified by microarray analysis. Furthermore, the control saline infusion demonstrated that the increased expression of inflammatory genes in response to the insulin infusion was due to the action of insulin on the muscle and was not secondary to the muscle biopsy per se. Taken collectively, these results provide evidence that in skeletal muscle insulin augments the expression of multiple genes that are involved in inflammation. This inflammation response may then provide positive feedback to cause/enhance insulin resistance.
Several candidate gene and microarray studies have identified genes whose expression was altered following an insulin infusion (3, 15, 21, 25, 28, 37, 40, 44). We examined some of these previously published microarray datasets (21, 28, 37) and found a number of genes that changed in parallel with our insulin-regulated genes. The majority of genes whose expression was altered by insulin coded for proteins involved in transcription regulator binding and activity. Other groups have reported similar findings concerning the effect of insulin on the mRNA expression of transcription factors (21, 37). Hansen et al. (21) described seven transcription genes (ATF3, BHLHB2, HES1, KLF10, JUNB, FOS, and FOSB) whose mRNA expression was upregulated and in common with those in the present study. Of particular note, we found that increased mRNA expression of HES1 and BHLHB2 was replicated in the majority of the datasets analyzed (21, 28, 37). Reduced insulin-mediated glucose disposal in skeletal muscle is a common metabolic feature of type 2 diabetes (4, 10, 14). In the present study, we observed an inverse relationship between BHLHB2 expression and whole body (primarily reflects muscle) glucose Rd. Moreover, we observed that the mRNA expression of HES1 and BHLHB2 was increased in muscle tissue of type 2 diabetic subjects (27, 29). Collectively, the HES1 and BHLHB2 mRNA expression results suggest that these two genes may be involved in the insulin-resistant phenotype observed in type 2 diabetes. However, the exact mechanism of action of the HES1 and BHLHB2 genes in skeletal muscle requires further investigation.
Oxidative stress has been implicated in the development of insulin resistance and microvascular/macrovascular complications in patients with type 2 diabetes (9). Metallothioneins (MTs) are cysteine-rich, low-molecular-weight intracellular proteins that have been shown to regulate the metabolism of metals and to be efficient scavengers of reactive oxygen species (ROS) (6, 38). In the present study, functional analysis using DAVID identified a group of MT mRNAs, whose expression is increased following insulin stimulation. Furthermore, analysis of the Parikh et al. (28) dataset identified a significant increase in expression of the mRNA for MT1H during a 2-h euglycemic hyperinsulinemic clamp. We also observed an increased mRNA expression of MT1H in muscle tissue of type 2 diabetic subjects (27), although opposite findings have been reported elsewhere (40). Another study found no differences in MT mRNA levels when type 2 diabetic groups were compared with control groups (38). Taken together, these results suggest that insulin activates the MT antioxidant defense system in normal, healthy controls, but further studies are needed to establish what role, if any, altered MT mRNA expression plays in the pathogenesis of human type 2 diabetes and its associated complications.
Insulin modulates intracellular metabolism by regulating pathways involved in protein, glycogen, and fat metabolism. In skeletal muscle, insulin stimulates glucose uptake, glycogen synthesis, and glucose oxidation (14). In the present study, we show that insulin increased the expression of a number of mRNAs in the phosphoinositol (PI) 3-kinase (metabolic) and MAP kinase (mitogenic) pathways (Table 2). In addition to the insulin receptor IRS2, regulatory/catalytic subunits of PI 3-kinase and hexokinase II, in particular, the ras-related associated with diabetes gene (RRAD) was increased following insulin stimulation and similar findings have been reported by other groups (15, 25). RRAD is overexpressed in the skeletal muscle of type 2 diabetes subjects (35, 40), and has been implicated in the pathogenesis of insulin resistance in type 2 diabetes. RRAD is involved in the classical MAP kinase pathway, which is an integral component of the insulin-signaling cascade and has been implicated in the development of atherosclerosis (22). We hypothesize that the increase in RRAD observed by previous investigators and in the present study may represent a response to the compensatory hyperinsulinemia that occurs as the β-cell attempts to offset the insulin resistance. RRAD may provide a link between insulin resistance, hyperinsulinemia, and atherosclerosis (11).
Insulin, as well as insulin-like growth factor, serum, and glucocorticoids, stimulate SGK activity by a mechanism requiring the participation and activation of PI 3-kinase (24, 31). We demonstrate in the present study that muscle mRNA expression of SGK is significantly increased by insulin in normal healthy subjects and that its expression was negatively correlated with basal EGP. To our knowledge, this result has not been replicated in any other microarray study. However, it has been shown elsewhere that SGK transcription is markedly enhanced in diabetic nephropathy (24). Interestingly, the SGK gene has been localized to chromosome 6q23, a major locus associated with fasting insulin concentrations and insulin resistance in Mexican Americans from the San Antonio Family Diabetes Study (16). These results suggest a potential role for SGK in insulin resistance in type 2 diabetes and other insulin resistant states.
cAMP-responsive element binding protein (CREB)/p300-interacting transactivator, with Glu/Asp-rich carboxy-terminal domain 2 (CITED2) also is located at 6q23, and in the present study was downregulated by insulin. CITED2 interacts with the CREB-binding protein (CBP)/p300, TFAP2, Lhx2, and nuclear receptors, such as peroxisome proliferator-activated receptor and estrogen receptor, to function as a transcriptional modulator (41). Another chromosomal region that has been linked with insulin resistance, type 2 diabetes and obesity is 10q24–25 (17). The peroxisome proliferative activated receptor-γ, coactivator-related 1 (PPRC1) is located at 10q24.32 and was found to be significantly upregulated by insulin in the present study. PPRC1 was first identified in database searches for sequences with similarities to peroxisome proliferator-activated receptor (PPAR)γ, coactivator-1 (PGC1) (2). PGC1 is a transcriptional regulator that activates (in conjunction with nuclear respiratory factor-1 and PPARα), PPARγ, hepatocyte nuclear factor 4, and other transcription factors which are key metabolic regulators. PPRC1 is a growth-regulated coactivator that may coordinate the activities of multiple transcription factors required for cell growth (2). The exact role of the CITED2 and PPRC1 genes remains unclear, and further studies will be needed to understand their role in insulin signaling.
In summary, we have demonstrated that a sustained physiological increase in the plasma insulin concentration for as little as 4 h in nonexercised subjects alters the levels of multiple muscle mRNAs, including inflammatory factors, which may be involved in the development of insulin resistance in type 2 diabetes mellitus. Of note, several of these genes are localized to chromosomal regions that previously have been shown to be associated with type 2 diabetes, insulin resistance, obesity, and atherosclerosis.
Supplementary Material
ACKNOWLEDGMENTS
We thank John Kincaid, Jim King, Norma Diaz, and Tricia Wolfe for outstanding nursing assistance, as well as the volunteers who took part in this study. All microarray studies were carried out in the Microarray Core Facility of the University of Texas Health Science Center at San Antonio, San Antonio, TX, under the direction of Drs. Dawn K. Coletta and Christopher P. Jenkinson.
REFERENCES
- 1.Adams JM, 2nd, Pratipanawatr T, Berria R, Wang E, DeFronzo RA, Sullards MC, Mandarino LJ. Ceramide content is increased in skeletal muscle from obese insulin-resistant humans. Diabetes. 2004;53:25–31. doi: 10.2337/diabetes.53.1.25. [DOI] [PubMed] [Google Scholar]
- 2.Andersson U, Scarpulla RC. Pgc-1-related coactivator, a novel, serum-inducible coactivator of nuclear respiratory factor 1-dependent transcription in mammalian cells. Mol Cell Biol. 2001;21:3738–3749. doi: 10.1128/MCB.21.11.3738-3749.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Asmann YW, Stump CS, Short KR, Coenen-Schimke JM, Guo Z, Bigelow ML, Nair KS. Skeletal muscle mitochondrial functions, mitochondrial DNA copy numbers, and gene transcript profiles in type 2 diabetic and nondiabetic subjects at equal levels of low or high insulin and euglycemia. Diabetes. 2006;55:3309–3319. doi: 10.2337/db05-1230. [DOI] [PubMed] [Google Scholar]
- 4.Bajaj M, DeFronzo RA. Metabolic and molecular basis of insulin resistance. J Nucl Cardiol. 2003;10:311–323. doi: 10.1016/s1071-3581(03)00520-8. [DOI] [PubMed] [Google Scholar]
- 5.Bajaj M, Suraamornkul S, Romanelli A, Cline GW, Mandarino LJ, Shulman GI, DeFronzo RA. Effect of a sustained reduction in plasma free fatty acid concentration on intramuscular long-chain fatty Acyl-CoAs and insulin action in type 2 diabetic patients. Diabetes. 2005;54:3148–3153. doi: 10.2337/diabetes.54.11.3148. [DOI] [PubMed] [Google Scholar]
- 6.Beattie JH, Wood AM, Newman AM, Bremner I, Choo KH, Michalska AE, Duncan JS, Trayhurn P. Obesity and hyperleptinemia in metallothionein (-I and -II) null mice. Proc Natl Acad Sci USA. 1998;95:358–363. doi: 10.1073/pnas.95.1.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berria R, Wang L, Richardson DK, Finlayson J, Belfort R, Pratipanawatr T, De Filippis EA, Kashyap S, Mandarino LJ. Increased collagen content in insulin-resistant skeletal muscle. Am J Physiol Endocrinol Metab. 2006;290:E560–E565. doi: 10.1152/ajpendo.00202.2005. [DOI] [PubMed] [Google Scholar]
- 8.Bonadonna RC, Del Prato S, Bonora E, Saccomani MP, Gulli G, Natali A, Frascerra S, Pecori N, Ferrannini E, Bier D, Cobelli C, DeFronzo RA. Roles of glucose transport and glucose phosphorylation in muscle insulin resistance of NIDDM. Diabetes. 1996;45:915–925. doi: 10.2337/diab.45.7.915. [DOI] [PubMed] [Google Scholar]
- 9.Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. 2005;54:1615–1625. doi: 10.2337/diabetes.54.6.1615. [DOI] [PubMed] [Google Scholar]
- 10.Cusi K, Maezono K, Osman A, Pendergrass M, Patti ME, Pratipanawatr T, DeFronzo RA, Kahn CR, Mandarino LJ. Insulin resistance differentially affects the PI 3-kinase- and MAP kinase-mediated signaling in human muscle. J Clin Invest. 2000;105:311–320. doi: 10.1172/JCI7535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DeFronzo RA, Ferrannini E. Insulin resistance. A multifaceted syndrome responsible for NIDDM, obesity, hypertension, dyslipidemia, and atherosclerotic cardiovascular disease. Diabetes Care. 1991;14:173–194. doi: 10.2337/diacare.14.3.173. [DOI] [PubMed] [Google Scholar]
- 12.DeFronzo RA, Gunnarsson R, Bjorkman O, Olsson M, Wahren J. Effects of insulin on peripheral and splanchnic glucose metabolism in non-insulin-dependent (type II) diabetes mellitus. J Clin Invest. 1985;76:149–155. doi: 10.1172/JCI111938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.DeFronzo RA, Tobin R, Andres R. The glucose clamp technique. A method for quantifying insulin secretion and resistance. Am J Physiol Endocrinol Metab Gastrointest Physiol. 1979;237:E214–E223. doi: 10.1152/ajpendo.1979.237.3.E214. [DOI] [PubMed] [Google Scholar]
- 14.DeFronzo RA. Lilly lecture 1987. The triumvirate: beta-cell, muscle, liver. A collusion responsible for NIDDM. Diabetes. 1988;37:667–687. doi: 10.2337/diab.37.6.667. [DOI] [PubMed] [Google Scholar]
- 15.Ducluzeau PH, Perretti N, Laville M, Andreelli F, Vega N, Riou JP, Vidal H. Regulation by insulin of gene expression in human skeletal muscle and adipose tissue. Evidence for specific defects in type 2 diabetes. Diabetes. 2001;50:1134–1142. doi: 10.2337/diabetes.50.5.1134. [DOI] [PubMed] [Google Scholar]
- 16.Duggirala R, Blangero J, Almasy L, Arya R, Dyer TD, Williams KL, Leach RJ, O'Connell P, Stern MP. A major locus for fasting insulin concentrations and insulin resistance on chromosome 6q with strong pleiotropic effects on obesity-related phenotypes in nondiabetic Mexican Americans. Am J Hum Genet. 2001;68:1149–1164. doi: 10.1086/320100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Duggirala R, Blangero J, Almasy L, Dyer TD, Williams KL, Leach RJ, O'Connell P, Stern MP. Linkage of type 2 diabetes mellitus and of age at onset to a genetic location on chromosome 10q in Mexican Americans. Am J Hum Genet. 1999;64:1127–1140. doi: 10.1086/302316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eady JJ, Wortley GM, Wormstone YM, Hughes JC, Astley SB, Foxall RJ, Doleman JF, Elliott RM. Variation in gene expression profiles of peripheral blood mononuclear cells from healthy volunteers. Physiol Genomics. 2005;22:402–411. doi: 10.1152/physiolgenomics.00080.2005. [DOI] [PubMed] [Google Scholar]
- 19.Golay A, Felber JP, Jequier E, DeFronzo RA, Ferrannini E. Metabolic basis of obesity and noninsulin-dependent diabetes mellitus. Diabetes Metab Rev. 1988;4:727–747. doi: 10.1002/dmr.5610040803. [DOI] [PubMed] [Google Scholar]
- 20.Goodpaster BH, Kelley DE. Skeletal muscle triglyceride: marker or mediator of obesity-induced insulin resistance in type 2 diabetes mellitus? Curr Diab Rep. 2002;2:216–222. doi: 10.1007/s11892-002-0086-2. [DOI] [PubMed] [Google Scholar]
- 21.Hansen L, Gaster M, Oakeley EJ, Brusgaard K, Damsgaard Nielsen EM, Beck-Nielsen H, Pedersen O, Hemmings BA. Expression profiling of insulin action in human myotubes: induction of inflammatory and pro-angiogenic pathways in relationship with glycogen synthesis and type 2 diabetes. Biochem Biophys Res Commun. 2004;323:685–695. doi: 10.1016/j.bbrc.2004.08.146. [DOI] [PubMed] [Google Scholar]
- 22.Hsueh WA, Law RE. Insulin signaling in the arterial wall. Am J Cardiol. 1999;84:21–24. doi: 10.1016/s0002-9149(99)00353-7. [DOI] [PubMed] [Google Scholar]
- 23.Krssak M, Falk Petersen K, Dresner A, DiPietro L, Vogel SM, Rothman DL, Roden M, Shulman GI. Intramyocellular lipid concentrations are correlated with insulin sensitivity in humans: a 1H NMR spectroscopy study. Diabetologia. 1999;42:113–116. doi: 10.1007/s001250051123. [DOI] [PubMed] [Google Scholar]
- 24.Lang F, Cohen P. Regulation and physiological roles of serum- and glucocorticoid-induced protein kinase isoforms. Sci STKE. 2001;108:RE17. doi: 10.1126/stke.2001.108.re17. [DOI] [PubMed] [Google Scholar]
- 25.Laville M, Auboeuf D, Khalfallah Y, Vega N, Riou JP, Vidal H. Acute regulation by insulin of phosphatidylinositol-3-kinase, Rad, Glut 4, and lipoprotein lipase mRNA levels in human muscle. J Clin Invest. 1996;98:43–49. doi: 10.1172/JCI118775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mandarino LJ, Printz RL, Cusi KA, Kinchington P, O'Doherty RM, Osawa H, Sewell C, Consoli A, Granner DK, DeFronzo RA. Regulation of hexokinase II and glycogen synthase mRNA, protein, and catalytic activity in human skeletal muscle in vivo. Am J Physiol Endocrinol Metab. 1995;269:E701–E708. doi: 10.1152/ajpendo.1995.269.4.E701. [DOI] [PubMed] [Google Scholar]
- 27.Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E, Houstis N, Daly MJ, Patterson N, Mesirov JP, Golub TR, Tamayo P, Spiegelman B, Lander ES, Hirschhorn JN, Altshuler D, Groop LC. PGC-1 a-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 28.Parikh H, Carlsson E, Chutkow WA, Johansson LE, Storgaard H, Poulsen P, Saxena R, Ladd C, Schulze PC, Mazzini MJ, Jensen CB, Krook A, Björnholm M, Tornqvist H, Zierath JR, Ridderstråle M, Altshuler D, Lee RT, Vaag A, Groop LC, Mootha VK. TXNIP regulates peripheral glucose metabolism in humans. PLoS Med. 2007;4:e158. doi: 10.1371/journal.pmed.0040158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Patti ME, Butte AJ, Crunkhorn S, Cusi K, Berria R, Kashyap S, Miyazaki Y, Kohane I, Costello M, Saccone R, Landaker EJ, Goldfine AB, Mun E, DeFronzo R, Finlayson J, Kahn CR, Mandarino LJ. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: potential role of PGC1 and NRF1. Proc Natl Acad Sci USA. 2003;100:8466–8471. doi: 10.1073/pnas.1032913100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pendergrass M, Koval J, Vogt C, Yki-Jarvinen H, Iozzo P, Pipek R, Ardehali H, Printz R, Granner D, DeFronzo RA, Mandarino LJ. Insulin-induced hexokinase II expression is reduced in obesity and NIDDM. Diabetes. 1998;47:387–394. doi: 10.2337/diabetes.47.3.387. [DOI] [PubMed] [Google Scholar]
- 31.Perrotti N, He RA, Phillips SA, Haft CR, Taylor SI. Activation of serum- and glucocorticoid-induced protein kinase (Sgk) by cyclic AMP and insulin. J Biol Chem. 2001;276:9406–9412. doi: 10.1074/jbc.M007052200. [DOI] [PubMed] [Google Scholar]
- 32.Polonsky KS, Given BD, Van Cauter E. Twenty-four-hour profiles and pulsatile patterns of insulin secretion in normal and obese subjects. J Clin Invest. 1988;81:442–448. doi: 10.1172/JCI113339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Radich JP, Mao M, Stepaniants S, Biery M, Castle J, Ward T, Schimmack G, Kobayashi S, Carleton M, Lampe J, Linsley PS. Individual-specific variation of gene expression in peripheral blood leukocytes. Genomics. 2004;83:980–988. doi: 10.1016/j.ygeno.2003.12.013. [DOI] [PubMed] [Google Scholar]
- 34.Reaven GM. Pathophysiology of insulin resistance in human disease. Physiol Rev. 1995;75:473–486. doi: 10.1152/physrev.1995.75.3.473. [DOI] [PubMed] [Google Scholar]
- 35.Reynet C, Kahn CR. Rad: a member of the Ras family overexpressed in muscle of type II diabetic humans. Science. 1993;262:1441–1444. doi: 10.1126/science.8248782. [DOI] [PubMed] [Google Scholar]
- 36.Richardson DK, Kashyap S, Bajaj M, Cusi K, Mandarino SJ, Finlayson J, DeFronzo RA, Jenkinson CP, Mandarino LJ. Lipid infusion decreases the expression of nuclear encoded mitochondrial genes and increases the expression of extracellular matrix genes in human skeletal muscle. J Biol Chem. 2005;280:10290–10297. doi: 10.1074/jbc.M408985200. [DOI] [PubMed] [Google Scholar]
- 37.Rome S, Clement K, Rabasa-Lhoret R, Loizon E, Poitou C, Barsh GS, Riou JP, Laville M, Vidal H. Microarray profiling of human skeletal muscle reveals that insulin regulates approximately 800 genes during a hyperinsulinemic clamp. J Biol Chem. 2003;278:18063–18068. doi: 10.1074/jbc.M300293200. [DOI] [PubMed] [Google Scholar]
- 38.Scheede-Bergdahl C, Penkowa M, Hidalgo J, Olsen DB, Schjerling P, Prats C, Boushel R, Dela F. Metallothionein-mediated antioxidant defense system and its response to exercise training are impaired in human type 2 diabetes. Diabetes. 2005;54:3089–3094. doi: 10.2337/db12-wd10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shepherd PR, Kahn BB. Glucose transporters and insulin action - implications for insulin resistance and diabetes mellitus. N Engl J Med. 1999;341:248–257. doi: 10.1056/NEJM199907223410406. [DOI] [PubMed] [Google Scholar]
- 40.Sreekumar R, Halvatsiotis P, Schimke JC, Nair KS. Gene expression profile in skeletal muscle of type 2 diabetes and the effect of insulin treatment. Diabetes. 2002;51:1913–1920. doi: 10.2337/diabetes.51.6.1913. [DOI] [PubMed] [Google Scholar]
- 41.Tien ES, Davis JW, Vanden Heuvel JP. Identification of the CREB-binding protein/p300-interacting protein CITED2 as a peroxisome proliferator-activated receptor alpha coregulator. J Biol Chem. 2004;279:24053–24063. doi: 10.1074/jbc.M401489200. [DOI] [PubMed] [Google Scholar]
- 42.Vestergaard H, Lund S, Larsen FS, Bjerrum OJ, Pedersen O. Glycogen synthase and phosphofructokinase protein and mRNA levels in skeletal muscle from insulin-resistant patients with non-insulin-dependent diabetes mellitus. J Clin Invest. 1993;91:2342–2350. doi: 10.1172/JCI116466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Whitney AR, Diehn M, Popper SJ, Alizadeh AA, Boldrick JC, Relman DA, Brown PO. Individuality and variation in gene expression patterns in human blood. Proc Natl Acad Sci USA. 2003;100:1896–1901. doi: 10.1073/pnas.252784499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang X, Pratley RE, Tokraks S, Bogardus C, Permana PA. Microarray profiling of skeletal muscle tissues from equally obese, non-diabetic insulin-sensitive and insulin-resistant Pima Indians. Diabetologia. 2002;45:1584–1593. doi: 10.1007/s00125-002-0905-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.