

Background: The epigenetic mechanism underlying E4BP4-dependent repression of hepatic Fgf21 during refeeding is unknown.
Results: E4BP4 interacts with G9a, and knockdown of G9a by shRNA abolishes suppression of Fgf21 by refeeding in vivo.
Conclusion: G9a is a co-repressor required for E4BP4-dependent repression of Fgf21 expression.
Significance: G9a is a critical histone methyltransferase in E4BP4-dependent repression of Fgf21 during refeeding.
Keywords: Histone Methylation, Lipid Metabolism, Liver, Metabolic Diseases, Transcription Repressor, Circadian Rhythm
Abstract
The liver responds to fasting-refeeding cycles by reprogramming expression of metabolic genes. Fasting potently induces one of the key hepatic hormones, fibroblast growth factor 21 (FGF21), to promote lipolysis, fatty acid oxidation, and ketogenesis, whereas refeeding suppresses its expression. We previously reported that the basic leucine zipper transcription factor E4BP4 (E4 binding protein 4) represses Fgf21 expression and disrupts its circadian oscillations in cultured hepatocytes. However, the epigenetic mechanism for E4BP4-dependent suppression of Fgf21 has not yet been addressed. Here we present evidence that histone methyltransferase G9a mediates E4BP4-dependent repression of Fgf21 during refeeding by promoting repressive histone modification. We find that Fgf21 expression is up-regulated in E4bp4 knock-out mouse liver. We demonstrate that the G9a-specific inhibitor BIX01294 abolishes suppression of the Fgf21 promoter activity by E4BP4, whereas overexpression of E4bp4 leads to increased levels of dimethylation of histone 3 lysine 9 (H3K9me2) around the Fgf21 promoter region. Furthermore, we also show that E4BP4 interacts with G9a, and knockdown of G9a blocks repression of Fgf21 promoter activity and expression in cells overexpressing E4bp4. A G9a mutant lacking catalytic activity, due to deletion of the SET domain, fails to inhibit the Fgf21 promoter activity. Importantly, acute hepatic knockdown by adenoviral shRNA targeting G9a abolishes Fgf21 repression by refeeding, concomitant with decreased levels of H3K9me2 around the Fgf21 promoter region. In summary, we show that G9a mediates E4BP4-dependent suppression of hepatic Fgf21 by enhancing histone methylation (H3K9me2) of the Fgf21 promoter.
Introduction
During the normal fasting-refeeding cycles, the liver responds to nutrient influx and deprivation by turning on and off corresponding metabolic gene expression (1). Fasting activates catabolic events, such as lipolysis, fatty acid oxidation, and proteolysis, whereas feeding promotes anabolic pathways, including lipogenesis and glycogenesis. FGF21 is a hepatic hormone highly induced by food starvation or ketogenic diet and suppressed by refeeding (2). As an adaptive response to fasting, elevated levels of FGF21 promote lipolysis in the white fat tissue and ketogenesis in the liver (3). Several nuclear receptors, including peroxisome proliferator-activated receptor α (PPARα),2 thyroid hormone receptor β, retinoid X receptor, and RAR-related orphan receptor α have been identified as transcription activators for Fgf21 in the liver (2–6). The critical role of PPARα has been highlighted by abolishment of Fgf21 induction in the liver PPARα knockout mice after prolonged fasting or bezofibrate (PPARα agonist) treatment (7, 8). So far, few repressors of Fgf21 gene expression are known. Our laboratory previously reported that the basic leucine zipper transcription factor E4BP4 represses Fgf21 upon insulin stimulation and is critical for circadian oscillations of Fgf21 (9). However, the epigenetic program underlying the E4BP4-dependent repression of Fgf21 remains unknown.
E4BP4 is a basic leucine zipper transcription factor initially identified as adenoviral E4 promoter-binding repressor and an IL3-induced factor in B-lymphocytes (10, 11). In E4bp4 knock-out mice, it has been shown that E4BP4 is critical for NK cell development and IgE expression in B cells (12–14). In addition to its role in the immune system, E4bp4 is also highly expressed in the liver, adipose tissue, and the suprachiasmatic nuclei of the hypothalamus (15). Hepatic E4bp4 mRNA displays a potent cycling in the liver with peak expression at circadian time 0 and trough expression at circadian time 12 h (15). As a circadian output oscillator, E4BP4 may regulate circadian oscillations of a subset of circadian out genes, such as Mdr1 (multidrug-resistant gene 1) (16), PPARγ (17), and Cyp7a (18). In Drosophila, the E4bp4 homologue vrille functions as a key negative regulator of the fly circadian clock (19, 20). E4BP4 actively represses target gene expression primarily through its binding to the D-box elements of target promoters (21). The TATA box-binding protein (TBP)-binding protein D1 was postulated as a potential repression cofactor of E4BP4 based upon its interaction with E4BP4 (22). Despite the potent repression of targets by E4BP4, epigenetic changes and histone-modifying enzymes involved in its transcriptional silencing remain largely unknown.
A subset of transcriptional repression is characterized by a repressive mark, dimethylation at lysine 9 of histone H3 (H3K9me2), around the promoter region of target genes (23). H3K9me2 is catalyzed by a group of histone methyltransferases, including G9a and G9a-like protein (GLP), members of the highly conserved SET (Su (var) 3–9-enhancer of zeste-trithorax) domain family (24). G9a and GLP exist predominantly in a heterodimeric complex to function as a functional H3K9 methyltransferase in vivo (25). Global deletion of G9a in mice drastically reduced levels of H3K9me2 and resulted in embryonic lethality, highlighting the consequences of global perturbation of gene repression during embryogenesis (26). G9a-associated transcription repression might be also involved in the postdevelopmental stage and participate in a short term gene regulation. Indeed, G9a-dependent methyltransferase activity has been found to be involved in the expression of Th2-associated cytokines (27), terminal differentiation of B-lymphocytes (28), bile acid metabolism (29), cocaine-induced neuronal plasticity (30), tumor growth and metastasis (31–33), and the interferon response (34). No previous studies have reported the role of G9a in lipid metabolism in the liver.
Previously, we identified Fgf21 as a direct circadian target of E4BP4 in hepatocytes (9). In the present study, we demonstrate that E4BP4 is an in vivo repressor of Fgf21 transcription during fasting-refeeding cycles. At the molecular level, we provide evidence that E4BP4 interacts with histone methyltransferase G9a to repress Fgf21 expression. Manipulation of G9a expression or inhibition of its enzymatic activity alters Fgf21 transcription in cultured hepatocytes. Crucially, depletion of G9a expression by shRNA knockdown in the mouse liver abolishes the hepatic Fgf21 oscillations during fasting-refeeding cycles.
EXPERIMENTAL PROCEDURES
Plasmids and Adenovirus Generation
Both E4BP4 overexpression and shRNA vectors were reported before (9). The adenovirus for Ad-E4bp4 (overexpression), Ad-shE4bp4 (knockdown), and Ad-shG9a (knockdown) were generated using the pAdEasy system (Agilent) and pAd-Block-it system (Invitrogen). The shRNA targeting sequence for mouse E4bp4 is 5′-ggaattcattccggacgagaa. The shRNA targeting sequence for mouse G9a is 5′-gcctgtactatgatgctgact. The HA-G9a expression vector is a generous gift from Dr. Kenneth Wright (Moffitt Cancer Center, University of South Florida). The HA-G9a-ΔSET mutant (Δ995–1159aa) was generated via QuikChange mutagenesis (Agilent) and confirmed by sequencing.
Animal Experiments
All animal care and use procedures were in accordance with guidelines of the University of Michigan Institutional Animal Care and Use Committee. All mice used in this study were of the C57 Bl/6 strain. Both male wild-type (WT) and E4bp4−/− mice age 8–10 weeks were maintained on 12-h/12-h light/dark with free access to food and water. Both WT and E4bp4−/− mice (n = 4–5 mice/group) were then sacrificed at Zeitgeber time 6 h, and both serum and liver tissues were collected for analysis. Serum FGF21 was measured by ELISA according to the manufacturer's instructions (R&D Systems). Serum level of β-hydroxybutyrate (ketone body) was measured by a commercial kit (Pointe Scientific).
Adenoviral Injection
WT C57BL/6J male mice (between 8 and 10 weeks old) were maintained on a 12-h/12-h light/dark cycle with free access to standard diet and water. For adenoviral injections, 1 × 1012 plaque-forming units (pfu)/recombinant adenovirus were administrated via tail vein injection. For each virus (Ad-shLacz or Ad-shG9a), a group of 4–5 mice were injected with the same dose treatment. 14 days after injection, mice were sacrificed around Zeitgeber time 8 h following either overnight fasting or refeeding for 16 h. Both serum and liver tissues were harvested for protein analysis.
Primary Hepatocyte Isolation
Primary mouse hepatocytes (PMHs) were isolated by a two-step collagenase digestion with 100 units/ml collagenase in HBSS at pH 7.4. After dissection, the liver was placed in DMEM and carefully pulled apart to release hepatocytes. Hepatocytes in DMEM were passed through a 100 μm cell strainer and then spun at 50 × g for 1 min. The pellet was resuspended in DMEM and then spun at 50 × g for 10 min in a Percoll gradient to remove dead hepatocytes. The viable cells were washed with DMEM at 50 × g for 10 min and checked by trypan blue staining. PMHs were cultured on collagen-coated plates in 5% FBS and DMEM. The adenoviral transduction was performed within 6 h after seeding. After overnight incubation, cells were switched to fresh serum-free medium and incubated for another 24 h before harvest.
Cell Culture and Cell Synchronization
Both 293T and mouse hepatoma Hepa1c1c-7 cell lines were maintained in minimum essential medium supplemented with 10% fetal bovine serum at 37 °C under 5% CO2. The protocol for in vitro synchronization study was described previously. Briefly, the freshly seeded Hepa1c1c-7 cells were firstly transduced with Ad-shLacz or Ad-shG9a for 16 h. 50% horse serum shock was performed 24 h later. Cells then were collected for mRNA analysis at 4-h intervals between the 24 and 60 h time points postsynchronization.
Luciferase Reporter Assay
Cells were plated in a 24-well plate overnight before transfection with mFgf21-WT or -D-box deletion mutant promoter luciferase reporter alongside either overexpression or shRNA vectors using Lipofectamine 2000 (Invitrogen). 48 h post-transfection, cells were lysed for luciferase activity assay measurement on a BioTek Synergy 2 microplate reader. The β-galactosidase construct was also co-transfected in each well for normalizing luciferase activity.
Immunoprecipitation (IP) and Immunoblotting (IB)
The standard immunoprecipitation method has been described previously (35). For detecting the protein interaction between FLAG-E4BP4 and HA-G9a, 293T cells were first transfected with both expression plasmids at a 1:1 ratio and then lysed in FLAG-IP buffer 48 h later. Cell lysates were then incubated with FLAG-M2-agarose beads (Sigma) overnight at 4 °C. The beads were washed five times in FLAG-IP buffer and eluted in 30 μl of 2× SDS loading buffer. For IP of endogenous proteins, liver tissues or cell pellets were lysed in ice-cold radioimmune precipitation assay buffer supplemented with 1× protease inhibitor and 50 mm NaF and incubated on ice for 20 min. Protein lysates were cleared by centrifugation at 14,000 rpm at 4 °C for 10 min. The supernatants were collected and quantified using the Bio-Rad protein assay kit. Cell lysates were then incubated with specific antibodies overnight at 4 °C. The protein complex was captured by adding 30 μl of Protein A-Sepharose beads and incubating at 4 °C for 1 h. The beads were washed five times in radioimmune precipitation assay buffer and eluted in 30 μl of 2× SDS loading buffer. Western blotting was performed to detect the presence of targeted proteins.
Chromatin Immunoprecipitation (ChIP)
The ChIP assay in culture cells was performed as described before (36). PMHs were infected with either Ad-GFP or Ad-E4pb4 and harvested 48 h later for ChIP assay. Liver ChIP assay was performed according the protocol described before (37, 38). The frozen liver tissues were minced in the presence of liquid nitrogen and cross-linked in 1% of formaldehyde-PBS buffer. The resulted nuclei pellets were then sonicated to generate soluble chromatin materials with sizes between 1 and 2 kb. The chromatin materials were immunoprecipitated with the following antibodies at 4 °C overnight: anti-G9a (Bethyl Laboratory) and anti-H3K9me2 (Cell Signaling). The resulting DNA fragments were purified and subjected to PCR analysis using primers encompassing the D-box region of the mouse Fgf21 promoter. The sequences of the PCR primers were as follows: for mFgf21–5′UTR, 5′-agtccttgctcagggttcct-3′ (forward) and 5′-acaggtgctctccagatgct-3′ (reverse); for mFgf21-TSS, 5′-tggtatttctgcgttcacca-3′ (forward) and 5′-atgggtcaggttcagactgg-3′ (reverse); for mFgf21-3′UTR, 5′-tttgctccacagcagtcttg-3′ (forward) and 5′-tcagaaatctgcctgcctct-3′ (reverse); for mFgf21-intron3, 5′-ttctgggggttgaaacaaag-3′ (forward) and 5′-aagccagcctggtctacaaa-3′ (reverse); for 18 S RNA, 5′-ttgacggaagggcaccaccag-3′ (forward) and 5′-gcaccaccacccacggaatgg-3′ (reverse).
cDNA Synthesis and Q-PCR
Total cellular RNA extraction and reverse transcription were described previously (9). 18 S ribosomal RNA was used as the housekeeping control to calculate the -fold change for each transcript. The primer sequences used in this study were as follows: for mFgf21, 5′-gctgctggaggacggttaca-3′ (forward) and 5′- cacaggtccccaggatgttg-3′ (reverse); for mE4bp4, 5′-cggagcttgaatcgcgcccc-3′ (forward) and 5′-gggttatcgtggttctgctccctg-3′ (reverse); for mG9a, 5′-tgcctatgtggtcagctcag-3′ (forward) and 5′-ggttcttgcagcttctccag-3′ (reverse).
Statistical Analysis
All of the luciferase assays were done in triplicates. Luciferase readings were normalized to β-galactosidase activity for each well. Three individual replicates were performed for each treatment in FGF21 ELISA. The results were indicated as mean ± S.E. To compare two groups, p < 0.05 was considered statistically significant for Student's t test.
RESULTS
E4BP4 Represses Hepatic Fgf21 Expression in Vivo
We previously established that E4BP4 represses Fgf21 via directly binding to a D-box cis element of its promoter in the mouse hepatoma Hepa1c1c-7 cells (9). In the mouse liver, we similarly detected direct binding of E4BP4 protein to the promoter of Fgf21 by ChIP assay, suggesting E4BP4 might regulate Fgf21 transcription in whole animals. To assess the in vivo role of E4BP4 in Fgf21 regulation, we examined the Fgf21 expression in E4bp4−/− mice. The loss of E4BP4 expression was confirmed by the lack of E4BP4 protein in the liver (Fig. 1A). In comparison with 3-month-old WT male mice on regular chow, serum FGF21 levels in male E4bp4−/− mice were significantly higher (Fig. 1B), consistent with the higher levels of Fgf21 mRNA in the liver (Fig. 1C). Therefore, we established E4BP4 as a physiological regulator of Fgf21 expression in the liver.
FIGURE 1.
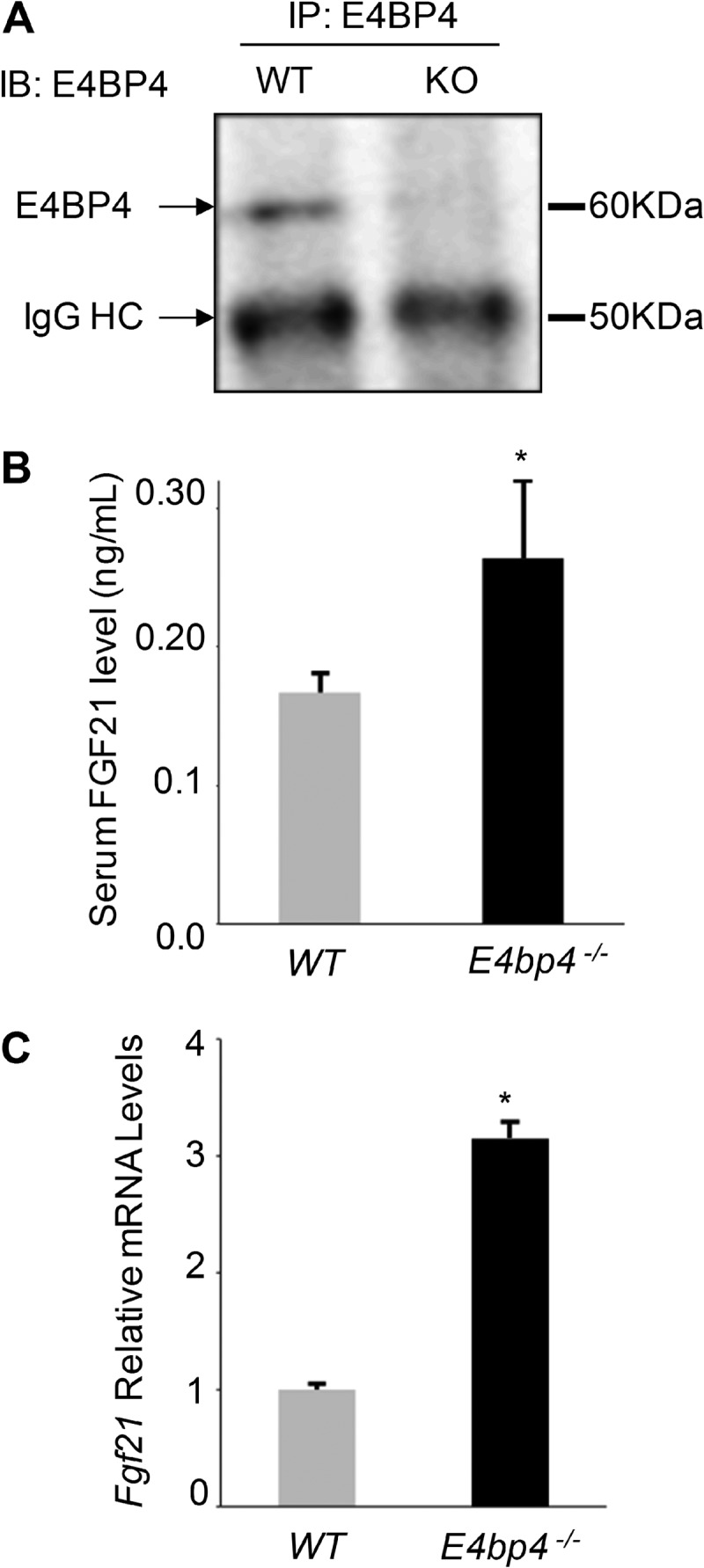
E4BP4 suppresses Fgf21 gene expression in vivo. A, loss of E4BP4 protein expression in the liver of E4pb4−/− mice. Pooled protein lysates (about 500 μg) from three mice (WT or E4pb4−/−) were immunoprecipitated by anti-E4BP4 (A9 from Santa Cruz Biotechnology, Inc.). The presence of E4BP4 protein was detected by immunoblotting with anti-E4BP4 (H300 from Santa Cruz Biotechnology, Inc.). HC, heavy chain. B, serum levels of FGF21 are elevated in E4pb4−/− mice. Serum samples of 3-month E4pb4−/− male mice fed regular chow were collected for FGF21 measurement. *, p < 0.05 by Student's t test. C, mRNA levels of Fgf21 are increased in the liver of E4bp4 knock-out mice. Liver tissues from the same cohort of mice shown in B were harvested for mRNA measurement. Data were plotted as mean ± S.E. (error bars) (n = 4–5). *, p < 0.05 by Student's t test.
Because it was reported that circadian transcription repressors, including REV-ERBα and PER1, function by recruiting histone deacetylases (HDACs) as co-repressors (39, 40), we hypothesized that E4BP4-mediated repression of Fgf21 also requires a corepressor protein or a repression complex to achieve the optimal effect. To that end, we first examined the role of HDACs in E4BP4-mediated FGF21 repression. To our surprise, neither trichostatin A (inhibitor of class I and II HDACs) nor nicotinamide (inhibitor of class III HDACs) could block the E4BP4-dependent repression of the Fgf21 promoter-driven luciferase construct (Fgf21-luc) activity (Fig. 2, A and B), suggesting that HDACs may not be required for E4BP4 repression function. In contrast, a histone methyltransferase inhibitor, BIX01294 (41), completely reversed E4BP4-dependent repression on Fgf21-luc (Fig. 2C). BIX01294 is a reversible specific inhibitor of G9a, which creates a repressive milieu for transcription by enhancing H3K9me2 of target promoters. We therefore examined the level of H3K9me2 on the Fgf21 promoter when E4BP4 level is elevated. 48 h post-transduction with either Ad-E4bp4 or Ad-GFP control, we assayed the histone modification level of the Fgf21 promoter in Hepa1 cells by ChIP using anti-H3K9me2 antibody. Compared with Ad-GFP control, Ad-E4bp4-transduced cells showed a drastic increase in the level of H3K9 dimethylation around the 5′-untranslated region (UTR) of the Fgf21 promoter (19-fold) and transcription start site (TSS) (5-fold). In contrast, there was no change in the H3K9me2 level around the 3′-UTR region (Fig. 2D). Together, our results suggest that histone methyltransferase G9a is a potential corepressor protein involved in E4BP4-dependent suppression of Fgf21.
FIGURE 2.

Effects of inhibitors of histone-modifying enzymes on E4BP4-mediated suppression of the Fgf21 promoter. A, trichostatin A (TSA; class I and II HDAC inhibitor) does not affect E4BP4-dependent suppression of Fgf21 promoter activity. 24 h postcotransfection with Fgf21-luc plus E4bp4 expression vector versus GFP control vector, Hepa1 cells were exposed to 100 μm trichostatin A overnight prior to the luciferase assay. The luciferase levels were normalized to an internal control of β-galactosidase, and the value of the GFP control group was set as 1. Data were plotted as mean ± S.E. (error bars) (n = 3). *, p < 0.05 by Student's t test. B, nicotinamide (NAM; class III HDAC inhibitor) shows no impact on repression of the Fgf21 promoter activity by E4BP4. Hepa1 cells were similarly transfected as in A and then subjected to 20 mm nicotinamide overnight before luciferase assay. Data were plotted as mean ± S.E. (n = 3). *, p < 0.05 by Student's t test. C, BIX01294, a specific inhibitor of histone methyltransferase G9a, abolishes E4BP4-dependent suppression of the Fgf21 promoter activity. Transfected Hepa1 cells were subjected to 10 μm BIX01294 treatment overnight before the luciferase assay. Data were plotted as mean ± S.E. (n = 3). *, p < 0.05 by Student's t test. D, overexpression of E4BP4 increases the level of H3K9me2 on the 5′-UTR and TSS region around the mouse Fgf21 promoter. 48 h after transduction by either Ad-GFP or Ad-E4bp4, Hepa1 cells were harvested for a ChIP assay with antibody against H3K9me2. The -fold increase was calculated with the level of the Ad-GFP control group set as 1. Primers used for the ChIP assay are indicated. The overexpression of E4BP4 was determined by immunoblotting with anti-E4BP4.
As a major histone methyltransferase targeting H3K9, G9a has mainly been studied for its epigenetic effects on stem cell differentiation, cell fate determination, and heterochromatin formation (24). Very little is known about its function in metabolic regulation. Previous work has shown that G9a acts as a corepressor protein for the nuclear receptor SHP (small heterodimer protein) to repress Cyp7a expression in the bile acid biosynthesis pathway (29). To test whether G9a is involved in E4BP4-dependent repression, we first tested whether these two proteins form a protein complex by overexpressing both G9a and E4BP4 in 293T cells and performing IP with antibodies. Indeed, we detected a strong interaction between FLAG-tagged E4BP4 and HA-tagged G9a (Fig. 3A). In contrast, no specific interaction was detected between E4BP4 and LSD1 (lysine demethylase 1) (Fig. 3B). To map the region of E4BP4 that interacts with G9a, we generated an E4BP4 truncation mutant without its C-terminal repression domain R-del (Δ-R). Such a mutant no longer interacts with G9a protein, indicating that the C-terminal region is required for interaction of E4BP4 with G9a (Fig. 3C). Based upon our previous finding that E4BP4 is required for insulin-mediated repression of Fgf21 (9), we asked whether insulin could stimulate interaction of E4BP4 with G9a. When both proteins were transiently overexpressed in 293T cells via transfection, treatment of insulin did not impact the E4BP4-G9a complex formation, indicating that insulin signaling is not required for E4BP4-G9a interaction (Fig. 3D). To determine whether these two proteins interact with each other in vivo, we tested both Hepa1 cells and PMHs but failed to detect protein-protein interaction between the endogenous E4BP4 and G9a proteins (data not shown), possibly due to relative low abundance of G9a.
FIGURE 3.

E4BP4 interacts with the histone methyltransferase G9a. A, E4BP4 and G9a interact with each other in vitro. 293T cells were transfected with FLAG-E4bp4 expression vector in the presence or absence of full-length HA-G9a. 48 h post-transfection, protein lysates were used for IP with anti-FLAG antibody, and the presence of G9a was detected with immunoblotting with anti-HA antibody. B, E4BP4 does not interact with LSD1 in vitro. The potential interaction between FLAG-E4BP4 and HA-LSD1 was similarly assayed in transiently transfected 293T cells. IP was performed with anti-FLAG antibody. The presence of HA-LSD1 was detected by immunoblotting with anti-HA antibody. NS, nonspecific signal. C, the C-terminal repression domain of E4BP4 is required for interaction with G9a. The HA-G9a expression vector was co-transfected with GFP, FLAG-E4bp4-WT, or FLAG-E4bp4-Δ-R (amino acids 1–380) expression vector. IP was performed as usual with anti-FLAG antibody. The presence of HA-G9a was detected by immunoblotting with anti-HA antibody. D, insulin has no effect on interaction between E4BP4 and G9a. Interactions between FLAG-E4BP4 and HA-G9a were tested in the presence of insulin treatment. 293T cells were first transfected with FLAG-E4bp4 and HA-G9a vectors. 24 h later, cells were switched to serum-free medium and treated with 100 nm insulin for 16 h before IP with anti-FLAG antibody. The presence of HA-G9a was detected by immunoblotting with anti-HA.
G9a Represses Fgf21 Expression in Hepatocytes
So far, our results suggest that the C-terminal region of E4BP4 can interact with G9a in cells, prompting us to ask whether G9a is involved in E4BP4-mediated repression. We first asked whether manipulation of G9a expression affects the Fgf21 promoter activity and mRNA expression. Overexpression of G9a-WT via transient transfection suppressed the Fgf21 promoter as efficiently as overexpression of E4bp4 (Fig. 4A). Moreover, G9a repression is dependent on its enzymatic activity because we did not detect any repression of the Fgf21 promoter activity in cells expressing the G9a-ΔSET enzyme-dead mutant with deletion of its SET domain, which has been shown to be critical for G9a-mediated H3K9 dimethylation (42–44) (Fig. 4B). The expression of both G9a-WT and -ΔSET mutant was confirmed by immunoblotting with HA antibody (data not shown). In addition, suppression of G9a enzymatic activity by BIX01294 led to a dose-dependent activation of the Fgf21-luc reporter in 293T cells (Fig. 4C). To test the role of G9a on the endogenous Fgf21 expression, we used adenoviral shG9a to knock down expression of G9a in freshly isolated PMHs. In Ad-shG9a-transduced PMHs, mRNA G9a expression was reduced by about 80% in comparison with control shLacz, whereas Fgf21 mRNA was elevated by 7-fold, indicating that G9a plays a significant repressive role on Fgf21 transcription (Fig. 4D). We previously showed that depletion of E4bp4 expression leads to dysregulated circadian oscillations of Fgf21 mRNA in synchronized Hepa1 cells (9). To examine the role of G9a in Fgf21 oscillations, we used Ad-shG9a to deplete G9a and synchronized Hepa1 cells for mRNA measurement. Between 24 and 48 h, Ad-Lacz-transduced control cells displayed normal oscillations of Fgf21 mRNA. However, in the case of G9a shRNA knockdown, the Fgf21 mRNA oscillated at higher amplitude and peaked at both 32 and 44 h (Fig. 4E). To test whether G9a is present on the endogenous Fgf21 promoter, we performed a ChIP assay with anti-G9a in mouse liver tissues from refed mice. Indeed, we detected clear G9a binding to the Fgf21 promoter during refeeding (Fig. 4F). Taken together, our data support a direct role of G9a in down-regulating the promoter activity and mRNA expression of Fgf21 in hepatocytes.
FIGURE 4.

G9a represses Fgf21 transcription in hepatocytes. A, G9a inhibits the Fgf21 promoter activity as efficiently as E4BP4. Hepa1 cells were transfected with Fgf21-luc along with GFP, FLAG-E4bp4, or HA-G9a expression vector. 48 h later, cells were harvested for the luciferase assay and the β-gal assay as a transfection efficiency control. The data were plotted as mean ± S.E. (n = 3). *, p < 0.05 by Student's t test. B, the inhibitory effect of G9a on Fgf21-luc activity depends on its SET domain. Hepa1 cells were transfected with Fgf21-luc along with GFP, G9a-WT, or G9a-ΔSET expression vector and lysed 48 h later for the luciferase assay. The data were plotted as mean ± S.E. (n = 3). *, p < 0.05 by Student's t test. C, inhibition of G9a by BIX01294 causes dose-dependent activation of Fgf21-luc activity. Data were plotted as mean ± S.E. (error bars) (n = 3). *, p < 0.05. D, G9a knockdown elevates Fgf21 mRNA in PMHs. PMHs were harvested 48 h after transduction by either Ad-shLacz or Ad-shG9a, and the mRNAs were quantified by QPCR. Data were plotted as mean ± S.E. (n = 4). *, p < 0.05 by Student's t test. E, G9a knockdown enhanced circadian oscillations of Fgf21 mRNA in synchronized Hepa1 cells. After depletion of G9a expression by adenoviral knockdown, Hepa1 cells were synchronized by serum shock and then harvested at the indicated time points for RNA isolation. The expression of Fgf21 mRNA was measured by QPCR and normalized to internal control 18 S RNA. Data were plotted as mean ± S.E. (n = 3). F, occupancy of G9a on the Fgf21 promoter in the liver. G9a binding to the Fgf21 promoter in the refed mouse livers was detected by a ChIP assay with anti-G9a antibody.
E4BP4-dependent Suppression of Fgf21 Expression Requires G9a
To test whether G9a is a required co-repressor for E4BP4 to repress Fgf21, we compared the abilities of G9a to repress Fgf21-luc-WT and D-box 1 deletion mutant (Fgf21-luc-ΔD1), which was shown to be resistant to E4BP4-mediated suppression (9). G9a suppressed the luciferase activity of Fgf21-luc-WT but not -ΔD1 mutant (Fig. 5A), indicating that G9a repression requires a functional E4BP4-binding site. Further, depletion of endogenous E4bp4 expression by Ad-shE4bp4 abolished the G9a-mediated repression on Fgf21-luc (Fig. 5B). Reciprocally, knockdown of G9a blocked E4BP4-mediated repression of Fgf21-luc in Hepa1 cells overexpressing E4bp4 (Fig. 5C), consistent with the elevated endogenous Fgf21 mRNA in those cells (Fig. 5D). Thus, we demonstrate the evidence that G9a is essential for E4BP4-dependent suppression of Fgf21.
FIGURE 5.

E4BP4 represses Fgf21 expression and promoter activity via G9a. A, G9a repression of Fgf21 requires a functional E4BP4-binding site. Hepa1 cells were transfected with either Fgf21-luc-WT or Fgf21-luc-ΔD1 mutant (D-box 1 deletion) along with G9a expression vector. 48 h later, cells were harvested for the luciferase assay and the β-gal assay as a transfection efficiency control. Data were plotted as mean ± S.E. (error bars) (n = 3). *, p < 0.05 by Student's t test. B, knockdown of E4bp4 abrogates G9a-dependent suppression of the Fgf21 promoter activity in hepatocytes. Hepa1 cells were cotransfected with Fgf21-WT-luc and G9a expression vectors after transduction by either Ad-shLacz or Ad-shE4bp4. 48 h later, cells were harvested for the luciferase assay and the β-gal assay as a transfection efficiency control. Data were plotted as mean ± S.E. (n = 3). *, p < 0.05 by Student's t test. C, knockdown of G9a abrogates E4BP4-dependent suppression of Fgf21 promoter activity in hepatocytes. Hepa1 cells were cotransfected with Fgf21-WT-luc and E4bp4 expression vector after transduction of either Ad-shLacz or Ad-shG9a. 48 h later, cells were harvested for the luciferase assay and the β-gal assay as a transfection efficiency control. Data were plotted as mean ± S.E. (n = 3). *, p < 0.05 by Student's t test. D, depletion of G9a blocks E4BP4-dependent repression of Fgf21 mRNA expression in hepatocytes. Hepa1 cells were transduced with the following paired adenoviruses: Ad-GFP plus Ad-shLacz versus Ad-E4bp4 plus Ad-shLacz; Ad-GFP plus Ad-shG9a versus Ad-E4bp4 plus Ad-shG9a. Cells were then synchronized by serum shock and harvested 36 h postsynchronization. Data were plotted as mean ± S.E. (n = 3). *, p < 0.05 by Student's t test.
G9a Is Required for Refeeding-induced Repression of Fgf21 in the Liver
Finally, we sought to test whether G9a regulates Fgf21 expression in vivo. We injected mice with Ad-shG9a control and studied its effect on metabolic parameters and hepatic Fgf21 expression during a fasting-refeeding cycle. Knockdown of G9a in the liver was confirmed by QPCR analysis (Fig. 6A). G9a knockdown had no effect on body weight and plasma glucose levels (data not shown) but significantly increased β-hydroxybutyrate, a ketone body, during refeeding (Fig. 6B). More importantly, G9a knockdown abolished the refeeding-induced down-regulation of Fgf21 (Fig. 6C) in comparison with Ad-shLacz control. Because G9a is necessary for maintaining H3K9me2, a repressive histone mark on the chromatin, we determined how acute knockdown of G9a alters the levels of H3K9me2 around the Fgf21 promoter during refeeding. Indeed, we observed a significant decrease in H3K9me2 around the 5′-UTR region and TSS of the Fgf21 promoter in the refed liver tissues after G9a shRNA knockdown (Fig. 6D), in parallel with elevation of Fgf21 expression. However, a similar H3K9me2 level was found around the exon 2 region of the Fgf21 gene. In summary, we concluded that G9a depletion leads to increased Fgf21 expression and reduced dimethylation of H3K9 of its promoter in the liver during refeeding.
FIGURE 6.

In vivo role of G9a in regulating hepatic Fgf21 expression. A, depletion of G9a in mouse liver tissues injected with either Ad-shLacz or Ad-shG9a. The mRNA levels of G9a were detected by QPCR. Data were plotted as mean ± S.E. (error bars) (n = 5). **, p < 0.01 by Student's t test. B, G9a knockdown elevated the serum level of the ketone body in mice during refeeding. Shown is the serum β-hydroxybutyrate level in mice injected with Ad-shLacz or Ad-shG9a and subjected to a fasting-refeeding regimen. Levels of serum β-hydroxybutyrate are shown as mean ± S.E. (n = 5). *, p < 0.05 by Student's t test. C, abolishment of refeeding-induced suppression of Fgf21 by G9a knockdown in the mouse liver. Liver tissues after injection of either Ad-shLacz or Ad-shG9a were harvested during a fasting and refeeding cycle. mRNA of Fgf21 was extracted and assayed by QPCR. Data are plotted as mean ± S.E. (n = 5). **, p < 0.01 by Student's t test. D, G9a knockdown reduced dimethylation of H3K9 of the Fgf21 promoter in the refed mouse liver. Levels of H3K9me2 around the Fgf21 promoter after adenoviral knockdown of G9a were measured by a ChIP assay in refed mouse liver tissues. PCR primers amplifying various regions of the Fgf21 promoter are indicated. Data were shown as mean ± S.E. (n = 5). *, p < 0.05 by Student's t test.
DISCUSSION
In this report, we uncovered the novel role of histone methyltransferase G9a in regulating the expression of Fgf21, an important metabolic regulator. In response to refeeding and insulin stimulation, E4BP4 recruits G9a to dimethylate the promoter region of the Fgf21 gene and represses its transcription in the mouse liver, whereas loss of G9a-dependent H3K9me2 facilitates transcription of Fgf21 by PPARα during fasting (Fig. 7).
FIGURE 7.

The working model of G9a-dependent repression of Fgf21 by E4BP4. E4BP4 is a potent repressor for the Fgf21 promoter in response to insulin stimulation and refeeding. Such potent repression is achieved through the recruitment of E4BP4-associated histone methyltransferase G9a to the Fgf21 promoter. Enzymatic activity of G9a enhances the repressive transcription mark of H3K9me2 around the Fgf21 promoter. During the fasting state, PPARα activates Fgf21 transcription while the E4BP4-G9a complex is disrupted and dissociated from the Fgf21 promoter.
Regulation of Fgf21 Expression during Normal and Diseased States
Given the multiple effects of FGF21 on carbohydrate and lipid metabolism, elucidation of regulation pathways of Fgf21 induction is important for identifying novel therapeutic targets during pathogenesis of obesity and diabetes. In transgenic mice, overexpression of Fgf21 improves insulin sensitivity and lowers glucose concentration (45). However, Fgf21 global knockout did not show a diabetic phenotype, although mice displayed a defect in lipid metabolism upon ketogenic diet treatment (46). In diabetic and obese conditions, both Fgf21 mRNA and serum FGF21 levels are paradoxically elevated, indicative of development of peripheral FGF21 resistance in the context of obesity (47–49). Nuclear receptors, including PPARα and farnesoid X receptor, are important activators of Fgf21 in response to increased levels of fatty acids and bile acid during high fat and low carbohydrate feeding (50). ATF4 induces Fgf21 expression upon amino acid deprivation (51), whereas liver X receptor represses Fgf21 expression during a cholesterol-rich diet (52). Our work demonstrates that, by inhibiting G9a enzyme via either shRNA or a small chemical compound (BIX01294), Fgf21 expression is potently induced in hepatocytes, indicating G9a as a novel epigenetic modifying enzyme for Fgf21 expression. Although so far there is no study directly showing that G9a contributes to obesity or diabetes, loss of JHDM2a, a demethylase of H3K9me2, promotes obesity and metabolic syndrome in mice (53, 54), suggesting that this particular histone modification and associated modifying enzymes are involved in the pathogenesis of obesity. FGF21, like other insulin-responsive genes, might undergo a dynamic epigenetic modification during fasting-feeding cycles in order to achieve a robust amplification. It can be envisioned that the promoter of Fgf21 is capable of being turned off or on efficiently in response to hormone action. We hypothesize that such control requires concerted activities of transcription factors, histone-modifying enzymes, and chromatin-remodeling enzymes. Our future work will use epigenetic approaches to discover specific components and their activities on the Fgf21 promoter during the switch between fasting and feeding cycles.
E4BP4 Couples Food Intake with Suppression of Metabolic Genes
As an IL3-inducible factor initially identified in lymphocytes, E4BP4 is also a first degree circadian output oscillator and controls circadian cycling of a number of circadian target genes (55). In cultured hepatocytes, E4bp4 mRNA expression is diurnal and anti-phase to that of Fgf21 and is highly induced by insulin treatment (9), raising the possibility that E4BP4 may combine both circadian and nutritional signals to control metabolism. It would be interesting to test whether circadian regulation of E4BP4 in the liver is impaired in the insulin-deficient condition, such as in liver-specific insulin receptor knock-out mice. Induction of Fgf21 is associated with fatty acid oxidation, lipolysis, and ketogenesis during fasting. During refeeding, E4BP4 might repress Fgf21 expression as a circadian regulator to coordinate lipid metabolism with food intake during the dark phase of circadian night. During fasting, we suspect that disruption of the E4BP4-G9a complex and dissociation from the Fgf21 promoter is also required for elevated Fgf21 expression besides PPARα-mediated transactivation. It is likely that E4BP4 is suppressed during fasting via either transcriptional repression or enhanced protein degradation.
Histone Modifiers in Circadian Regulation and Cycles of Fasting and Refeeding
Epigenetic modification has emerged as a novel regulatory mechanism in circadian-dependent transcription. The NAD-dependent histone deacetylase SIRT1 interacts with not only the BMAL1-CLOCK complex but also the PER2 protein, suggesting that SIRT1 is an integral part of the molecular clock (56, 57). SIRT1 in fact plays a positive role in regulating Fgf21 gene expression by enhancing PPARα activity in hepatocytes (58). However, we did not detect any effects of SIRT1 inhibition by nicotinamide on the E4BP4-dependent repression of Fgf21, indicating that E4BP4 represses Fgf21 independently of SIRT1. Several studies have already explored the role of histone methylation in circadian-associated transcription. For example, MLL (mixed lineage leukemia 1) and EZH2 (enhancer of zeste 2), two enzymes targeting histone methylation, have been identified to interact with the core clock proteins and modulate circadian transcription (59, 60). Throughout the literature, the epigenetic function of G9a has been associated with maintaining stem cell differentiation and heterochromatin formation. Very little is known about the biological function of G9a in the postdifferentiation state. Our report is the first to report that G9a regulates gene oscillations of a metabolic regulator in the liver via interaction with a known circadian protein. The widely available small molecule inhibitors of G9a with high specificity make G9a an ideal target for treating circadian-relevant or metabolic diseases. Because FGF21 has been shown to have diverse functions in white adipose tissue, brown adipose tissue, and the central nervous system (61–63), it will be of great interest to determine whether G9a suppresses Fgf21 expression in a similar fashion in other tissues in future studies.
Acknowledgments
We thank Dr. Steven Kliewer (University of Texas Southwestern) for providing the Fgf21-luc plasmids and Dr. Kenneth Wright (Moffitt Cancer Center) for the HA-G9a expression construct.
This work was supported, in whole or in part, by National Institutes of Health Grant K99/R00 DK 077449 (to L. Y.). Part of this work was also supported by a pilot grants (to L. Y.) from the Michigan Diabetes Research Center (P600K020572) and Michigan Obesity and Nutrition Research Center (DK089503).
- PPARα
- peroxisome proliferator-activated receptor α
- H3K9
- histone 3 lysine 9
- H3K9me2
- dimethylation of H3K9
- GLP
- G9a-like protein
- PMH
- primary mouse hepatocyte
- IP
- immunoprecipitation
- IB
- immunoblot
- HDAC
- histone deacetylase
- TSS
- transcription start site
- Ad
- adenoviral
- QPCR
- quantitative PCR.
REFERENCES
- 1. Vollmers C., Gill S., DiTacchio L., Pulivarthy S. R., Le H. D., Panda S. (2009) Time of feeding and the intrinsic circadian clock drive rhythms in hepatic gene expression. Proc. Natl. Acad. Sci. U.S.A. 106, 21453–21458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Badman M. K., Pissios P., Kennedy A. R., Koukos G., Flier J. S., Maratos-Flier E. (2007) Hepatic fibroblast growth factor 21 is regulated by PPARα and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab. 5, 426–437 [DOI] [PubMed] [Google Scholar]
- 3. Inagaki T., Dutchak P., Zhao G., Ding X., Gautron L., Parameswara V., Li Y., Goetz R., Mohammadi M., Esser V., Elmquist J. K., Gerard R. D., Burgess S. C., Hammer R. E., Mangelsdorf D. J., Kliewer S. A. (2007) Endocrine regulation of the fasting response by PPARα-mediated induction of fibroblast growth factor 21. Cell Metab. 5, 415–425 [DOI] [PubMed] [Google Scholar]
- 4. Adams A. C., Astapova I., Fisher F. M., Badman M. K., Kurgansky K. E., Flier J. S., Hollenberg A. N., Maratos-Flier E. (2010) Thyroid hormone regulates hepatic expression of fibroblast growth factor 21 in a PPARα-dependent manner. J. Biol. Chem. 285, 14078–14082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Estall J. L., Ruas J. L., Choi C. S., Laznik D., Badman M., Maratos-Flier E., Shulman G. I., Spiegelman B. M. (2009) PGC-1α negatively regulates hepatic FGF21 expression by modulating the heme/Rev-Erb(α) axis. Proc. Natl. Acad. Sci. U.S.A. 106, 22510–22515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang Y., Solt L. A., Burris T. P. (2010) Regulation of FGF21 expression and secretion by retinoic acid receptor-related orphan receptor α. J. Biol. Chem. 285, 15668–15673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Oishi K., Uchida D., Ishida N. (2008) Circadian expression of FGF21 is induced by PPARα activation in the mouse liver. FEBS Lett. 582, 3639–3642 [DOI] [PubMed] [Google Scholar]
- 8. Lundäsen T., Hunt M. C., Nilsson L. M., Sanyal S., Angelin B., Alexson S. E., Rudling M. (2007) PPARα is a key regulator of hepatic FGF21. Biochem. Biophys. Res. Commun. 360, 437–440 [DOI] [PubMed] [Google Scholar]
- 9. Tong X., Muchnik M., Chen Z., Patel M., Wu N., Joshi S., Rui L., Lazar M. A., Yin L. (2010) Transcriptional repressor E4-binding protein 4 (E4BP4) regulates metabolic hormone fibroblast growth factor 21 (FGF21) during circadian cycles and feeding. J. Biol. Chem. 285, 36401–36409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cowell I. G., Skinner A., Hurst H. C. (1992) Transcriptional repression by a novel member of the bZIP family of transcription factors. Mol. Cell. Biol. 12, 3070–3077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang W., Zhang J., Kornuc M., Kwan K., Frank R., Nimer S. D. (1995) Molecular cloning and characterization of NF-IL3A, a transcriptional activator of the human interleukin-3 promoter. Mol. Cell. Biol. 15, 6055–6063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gascoyne D. M., Long E., Veiga-Fernandes H., de Boer J., Williams O., Seddon B., Coles M., Kioussis D., Brady H. J. (2009) The basic leucine zipper transcription factor E4BP4 is essential for natural killer cell development. Nat. Immunol. 10, 1118–1124 [DOI] [PubMed] [Google Scholar]
- 13. Kamizono S., Duncan G. S., Seidel M. G., Morimoto A., Hamada K., Grosveld G., Akashi K., Lind E. F., Haight J. P., Ohashi P. S., Look A. T., Mak T. W. (2009) Nfil3/E4bp4 is required for the development and maturation of NK cells in vivo. J. Exp. Med. 206, 2977–2986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kashiwada M., Levy D. M., McKeag L., Murray K., Schröder A. J., Canfield S. M., Traver G., Rothman P. B. (2010) IL-4-induced transcription factor NFIL3/E4BP4 controls IgE class switching. Proc. Natl. Acad. Sci. U.S.A. 107, 821–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mitsui S., Yamaguchi S., Matsuo T., Ishida Y., Okamura H. (2001) Antagonistic role of E4BP4 and PAR proteins in the circadian oscillatory mechanism. Genes Dev. 15, 995–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Murakami Y., Higashi Y., Matsunaga N., Koyanagi S., Ohdo S. (2008) Circadian clock-controlled intestinal expression of the multidrug-resistance gene mdr1a in mice. Gastroenterology 135, 1636–1644.e3 [DOI] [PubMed] [Google Scholar]
- 17. Takahashi S., Inoue I., Nakajima Y., Seo M., Nakano T., Yang F., Kumagai M., Komoda T., Awata T., Ikeda M., Katayama S. (2010) A promoter in the novel exon of hPPARγ directs the circadian expression of PPARγ. J. Atheroscler. Thromb. 17, 73–83 [DOI] [PubMed] [Google Scholar]
- 18. Duez H., van der Veen J. N., Duhem C., Pourcet B., Touvier T., Fontaine C., Derudas B., Baugé E., Havinga R., Bloks V. W., Wolters H., van der Sluijs F. H., Vennström B., Kuipers F., Staels B. (2008) Regulation of bile acid synthesis by the nuclear receptor Rev-erbα. Gastroenterology 135, 689–698 [DOI] [PubMed] [Google Scholar]
- 19. Glossop N. R., Houl J. H., Zheng H., Ng F. S., Dudek S. M., Hardin P. E. (2003) VRILLE feeds back to control circadian transcription of Clock in the Drosophila circadian oscillator. Neuron 37, 249–261 [DOI] [PubMed] [Google Scholar]
- 20. Cyran S. A., Buchsbaum A. M., Reddy K. L., Lin M. C., Glossop N. R., Hardin P. E., Young M. W., Storti R. V., Blau J. (2003) vrille, Pdp1, and dClock form a second feedback loop in the Drosophila circadian clock. Cell 112, 329–341 [DOI] [PubMed] [Google Scholar]
- 21. Ueda H. R., Hayashi S., Chen W., Sano M., Machida M., Shigeyoshi Y., Iino M., Hashimoto S. (2005) System-level identification of transcriptional circuits underlying mammalian circadian clocks. Nat. Genet. 37, 187–192 [DOI] [PubMed] [Google Scholar]
- 22. Cowell I. G., Hurst H. C. (1996) Protein-protein interaction between the transcriptional repressor E4BP4 and the TBP-binding protein Dr1. Nucleic Acids Res. 24, 3607–3613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hublitz P., Albert M., Peters A. H. (2009) Mechanisms of transcriptional repression by histone lysine methylation. Int. J. Dev. Biol. 53, 335–354 [DOI] [PubMed] [Google Scholar]
- 24. Shinkai Y., Tachibana M. (2011) H3K9 methyltransferase G9a and the related molecule GLP. Genes Dev. 25, 781–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tachibana M., Ueda J., Fukuda M., Takeda N., Ohta T., Iwanari H., Sakihama T., Kodama T., Hamakubo T., Shinkai Y. (2005) Histone methyltransferases G9a and GLP form heteromeric complexes and are both crucial for methylation of euchromatin at H3-K9. Genes Dev. 19, 815–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tachibana M., Sugimoto K., Nozaki M., Ueda J., Ohta T., Ohki M., Fukuda M., Takeda N., Niida H., Kato H., Shinkai Y. (2002) G9a histone methyltransferase plays a dominant role in euchromatic histone H3 lysine 9 methylation and is essential for early embryogenesis. Genes Dev. 16, 1779–1791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lehnertz B., Northrop J. P., Antignano F., Burrows K., Hadidi S., Mullaly S. C., Rossi F. M., Zaph C. (2010) Activating and inhibitory functions for the histone lysine methyltransferase G9a in T helper cell differentiation and function. J. Exp. Med. 207, 915–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gyory I., Wu J., Fejér G., Seto E., Wright K. L. (2004) PRDI-BF1 recruits the histone H3 methyltransferase G9a in transcriptional silencing. Nat Immunol 5, 299–308 [DOI] [PubMed] [Google Scholar]
- 29. Fang S., Miao J., Xiang L., Ponugoti B., Treuter E., Kemper J. K. (2007) Coordinated recruitment of histone methyltransferase G9a and other chromatin-modifying enzymes in SHP-mediated regulation of hepatic bile acid metabolism. Mol. Cell. Biol. 27, 1407–1424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Maze I., Covington H. E., 3rd, Dietz D. M., LaPlant Q., Renthal W., Russo S. J., Mechanic M., Mouzon E., Neve R. L., Haggarty S. J., Ren Y., Sampath S. C., Hurd Y. L., Greengard P., Tarakhovsky A., Schaefer A., Nestler E. J. (2010) Essential role of the histone methyltransferase G9a in cocaine-induced plasticity. Science 327, 213–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kondo Y., Shen L., Ahmed S., Boumber Y., Sekido Y., Haddad B. R., Issa J. P. (2008) Down-regulation of histone H3 lysine 9 methyltransferase G9a induces centrosome disruption and chromosome instability in cancer cells. PLoS One 3, e2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen M. W., Hua K. T., Kao H. J., Chi C. C., Wei L. H., Johansson G., Shiah S. G., Chen P. S., Jeng Y. M., Cheng T. Y., Lai T. C., Chang J. S., Jan Y. H., Chien M. H., Yang C. J., Huang M. S., Hsiao M., Kuo M. L. (2010) H3K9 histone methyltransferase G9a promotes lung cancer invasion and metastasis by silencing the cell adhesion molecule Ep-CAM. Cancer Res. 70, 7830–7840 [DOI] [PubMed] [Google Scholar]
- 33. Dong C., Wu Y., Yao J., Wang Y., Yu Y., Rychahou P. G., Evers B. M., Zhou B. P. (2012) G9a interacts with Snail and is critical for Snail-mediated E-cadherin repression in human breast cancer. J. Clin. Invest. 122, 1469–1486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fang T. C., Schaefer U., Mecklenbrauker I., Stienen A., Dewell S., Chen M. S., Rioja I., Parravicini V., Prinjha R. K., Chandwani R., MacDonald M. R., Lee K., Rice C. M., Tarakhovsky A. (2012) Histone H3 lysine 9 di-methylation as an epigenetic signature of the interferon response. J. Exp. Med. 209, 661–669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yin L., Joshi S., Wu N., Tong X., Lazar M. A. E3 ligases Arf-bp1 and Pam mediate lithium-stimulated degradation of the circadian heme receptor Rev-erb α. Proc. Natl. Acad. Sci. U.S.A. 107, 11614–11619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yin L., Wu N., Curtin J. C., Qatanani M., Szwergold N. R., Reid R. A., Waitt G. M., Parks D. J., Pearce K. H., Wisely G. B., Lazar M. A. (2007) Rev-erbα, a heme sensor that coordinates metabolic and circadian pathways. Science 318, 1786–1789 [DOI] [PubMed] [Google Scholar]
- 37. Schmidt D., Wilson M. D., Ballester B., Schwalie P. C., Brown G. D., Marshall A., Kutter C., Watt S., Martinez-Jimenez C. P., Mackay S., Talianidis I., Flicek P., Odom D. T. Five-vertebrate ChIP-seq reveals the evolutionary dynamics of transcription factor binding. Science 328, 1036–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Thomas A. M., Hart S. N., Kong B., Fang J., Zhong X. B., Guo G. L. Genome-wide tissue-specific farnesoid X receptor binding in mouse liver and intestine. Hepatology 51, 1410–1419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yin L., Lazar M. A. (2005) The orphan nuclear receptor Rev-erbα recruits the N-CoR/histone deacetylase 3 corepressor to regulate the circadian Bmal1 gene. Mol. Endocrinol. 19, 1452–1459 [DOI] [PubMed] [Google Scholar]
- 40. Naruse Y., Oh-hashi K., Iijima N., Naruse M., Yoshioka H., Tanaka M. (2004) Circadian and light-induced transcription of clock gene Per1 depends on histone acetylation and deacetylation. Mol. Cell. Biol. 24, 6278–6287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kubicek S., O'Sullivan R. J., August E. M., Hickey E. R., Zhang Q., Teodoro M. L., Rea S., Mechtler K., Kowalski J. A., Homon C. A., Kelly T. A., Jenuwein T. (2007) Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase. Mol. Cell 25, 473–481 [DOI] [PubMed] [Google Scholar]
- 42. Nishio H., Walsh M. J. (2004) CCAAT displacement protein/cut homolog recruits G9a histone lysine methyltransferase to repress transcription. Proc. Natl. Acad. Sci. U.S.A. 101, 11257–11262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tachibana M., Sugimoto K., Fukushima T., Shinkai Y. (2001) Set domain-containing protein, G9a, is a novel lysine-preferring mammalian histone methyltransferase with hyperactivity and specific selectivity to lysines 9 and 27 of histone H3. J. Biol. Chem. 276, 25309–25317 [DOI] [PubMed] [Google Scholar]
- 44. Estève P. O., Patnaik D., Chin H. G., Benner J., Teitell M. A., Pradhan S. (2005) Functional analysis of the N and C terminus of mammalian G9a histone H3 methyltransferase. Nucleic Acids Res. 33, 3211–3223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kharitonenkov A., Shiyanova T. L., Koester A., Ford A. M., Micanovic R., Galbreath E. J., Sandusky G. E., Hammond L. J., Moyers J. S., Owens R. A., Gromada J., Brozinick J. T., Hawkins E. D., Wroblewski V. J., Li D. S., Mehrbod F., Jaskunas S. R., Shanafelt A. B. (2005) FGF-21 as a novel metabolic regulator. J. Clin. Invest. 115, 1627–1635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Badman M. K., Koester A., Flier J. S., Kharitonenkov A., Maratos-Flier E. (2009) Fibroblast growth factor 21-deficient mice demonstrate impaired adaptation to ketosis. Endocrinology 150, 4931–4940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chavez A. O., Molina-Carrion M., Abdul-Ghani M. A., Folli F., Defronzo R. A., Tripathy D. (2009) Circulating fibroblast growth factor-21 is elevated in impaired glucose tolerance and type 2 diabetes and correlates with muscle and hepatic insulin resistance. Diabetes Care 32, 1542–1546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dushay J., Chui P. C., Gopalakrishnan G. S., Varela-Rey M., Crawley M., Fisher F. M., Badman M. K., Martinez-Chantar M. L., Maratos-Flier E. (2010) Increased fibroblast growth factor 21 in obesity and nonalcoholic fatty liver disease. Gastroenterology 139, 456–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhang X., Yeung D. C., Karpisek M., Stejskal D., Zhou Z. G., Liu F., Wong R. L., Chow W. S., Tso A. W., Lam K. S., Xu A. (2008) Serum FGF21 levels are increased in obesity and are independently associated with the metabolic syndrome in humans. Diabetes 57, 1246–1253 [DOI] [PubMed] [Google Scholar]
- 50. Cyphert H. A., Ge X., Kohan A. B., Salati L. M., Zhang Y., Hillgartner F. B. (2012) Activation of the farnesoid X receptor induces hepatic expression and secretion of fibroblast growth factor 21. J. Biol. Chem. 287, 25123–25138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. De Sousa-Coelho A. L., Marrero P. F., Haro D. (2012) Activating transcription factor 4-dependent induction of FGF21 during amino acid deprivation. Biochem. J. 443, 165–171 [DOI] [PubMed] [Google Scholar]
- 52. Uebanso T., Taketani Y., Yamamoto H., Amo K., Tanaka S., Arai H., Takei Y., Masuda M., Yamanaka-Okumura H., Takeda E. (2012) Liver X receptor negatively regulates fibroblast growth factor 21 in the fatty liver induced by cholesterol-enriched diet. J. Nutr. Biochem. 23, 785–790 [DOI] [PubMed] [Google Scholar]
- 53. Inagaki T., Tachibana M., Magoori K., Kudo H., Tanaka T., Okamura M., Naito M., Kodama T., Shinkai Y., Sakai J. (2009) Obesity and metabolic syndrome in histone demethylase JHDM2a-deficient mice. Genes Cells 14, 991–1001 [DOI] [PubMed] [Google Scholar]
- 54. Tateishi K., Okada Y., Kallin E. M., Zhang Y. (2009) Role of Jhdm2a in regulating metabolic gene expression and obesity resistance. Nature 458, 757–761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cowell I. G. (2002) E4BP4/NFIL3, a PAR-related bZIP factor with many roles. BioEssays 24, 1023–1029 [DOI] [PubMed] [Google Scholar]
- 56. Nakahata Y., Kaluzova M., Grimaldi B., Sahar S., Hirayama J., Chen D., Guarente L. P., Sassone-Corsi P. (2008) The NAD+-dependent deacetylase SIRT1 modulates CLOCK-mediated chromatin remodeling and circadian control. Cell 134, 329–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Asher G., Gatfield D., Stratmann M., Reinke H., Dibner C., Kreppel F., Mostoslavsky R., Alt F. W., Schibler U. (2008) SIRT1 regulates circadian clock gene expression through PER2 deacetylation. Cell 134, 317–328 [DOI] [PubMed] [Google Scholar]
- 58. Vilà-Brau A., De Sousa-Coelho A. L., Mayordomo C., Haro D., Marrero P. F. (2011) Human HMGCS2 regulates mitochondrial fatty acid oxidation and FGF21 expression in HepG2 cell line. J. Biol. Chem. 286, 20423–20430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Katada S., Sassone-Corsi P. (2010) The histone methyltransferase MLL1 permits the oscillation of circadian gene expression. Nat. Struct. Mol. Biol. 17, 1414–1421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Etchegaray J. P., Yang X., DeBruyne J. P., Peters A. H., Weaver D. R., Jenuwein T., Reppert S. M. (2006) The polycomb group protein EZH2 is required for mammalian circadian clock function. J. Biol. Chem. 281, 21209–21215 [DOI] [PubMed] [Google Scholar]
- 61. Muise E. S., Azzolina B., Kuo D. W., El-Sherbeini M., Tan Y., Yuan X., Mu J., Thompson J. R., Berger J. P., Wong K. K. (2008) Adipose fibroblast growth factor 21 is up-regulated by peroxisome proliferator-activated receptor γ and altered metabolic states. Mol. Pharmacol. 74, 403–412 [DOI] [PubMed] [Google Scholar]
- 62. Fisher F. M., Kleiner S., Douris N., Fox E. C., Mepani R. J., Verdeguer F., Wu J., Kharitonenkov A., Flier J. S., Maratos-Flier E., Spiegelman B. M. (2012) FGF21 regulates PGC-1α and browning of white adipose tissues in adaptive thermogenesis. Genes Dev. 26, 271–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sarruf D. A., Thaler J. P., Morton G. J., German J., Fischer J. D., Ogimoto K., Schwartz M. W. (2010) Fibroblast growth factor 21 action in the brain increases energy expenditure and insulin sensitivity in obese rats. Diabetes 59, 1817–1824 [DOI] [PMC free article] [PubMed] [Google Scholar]