

Background: The nuclear export of myocardin (Mycd) family members is unclear.
Results: CRM1 binding to Mycd is weaker than to MRTF-A and is under the control of multiple inhibitory mechanisms.
Conclusion: Compared with Mycd, MRTF-A is much more likely to be exported from the nucleus.
Significance: Our findings provide new insight into the functional regulation of Mycd family members.
Keywords: Cell Biology, Nuclear Transport, Protein/Protein Interactions, Smooth Muscle, Transcription Coactivators, CRM1, G-actin, MRTF-A/B, Myocardin, Serum Response Factor
Abstract
Myocardin (Mycd), a key factor in smooth muscle cell differentiation, is constitutively located in the nucleus, whereas myocardin-related transcription factors A and B (MRTF-A/B) reside mostly in the cytoplasm and translocate to the nucleus in a Rho-dependent manner. Here, we investigated the nuclear export of Mycd family members. They possess two leucine-rich sequences: L1 in the N terminus and L2 in the Gln-rich domain. Although L2 (but not L1) served as a CRM1-binding site for Mycd, CRM1-mediated nuclear export did not affect its subcellular localization. Serum response factor (SRF) competitively inhibited Mycd/CRM1 interaction. Furthermore, such interaction was autonomously inhibited. The N terminus of Mycd bound intramolecularly to Mycd, resulting in masking L2. In contrast, the CRM1-binding affinity of MRTF-A was much higher than that of Mycd because both L1 and L2 of MRTF-A served as functional CRM1-binding sites, and the autoinhibition observed in the Mycd/CRM1 interaction was absent in the MRTF-A/CRM1 interaction. Additionally, because the SRF-binding affinity of MRTF-A was lower than that of Mycd, the inhibitory effect of SRF on the MRTF-A/CRM1 interaction was weak. Thus, MRTF-A is much more likely to be exported from the nucleus. These differences could be the reason for the distinct subcellular localization of Mycd and MRTF-A.
Introduction
The myocardin (Mycd)2 family members, Mycd, myocardin-related transcription factor (MRTF) A (MAL/MKL1), and MRTF-B (MAL16/MKL2), bind to serum response factor (SRF) and function as its specific coactivator (1, 2). The expression of Mycd is restricted to cardiac and smooth muscles (3), but MRTF-A and MRTF-B are ubiquitously expressed (1, 4, 5). Two isoforms of Mycd (cardiac cell-type Mycd and smooth muscle cell (SMC)-type Mycd) arise by alternative mRNA splicing (6). Initiation of translation from the first ATG codon in exon 1 produces a cardiac cell-type Mycd protein. However, in SMCs, translation initiation from the second ATG codon in exon 4 gives rise to an SMC-Mycd protein that lacks the N-terminal 79 amino acids. In this study, we refer to cardiac cell-type Mycd and SMC-Mycd as Mycd WT and SMC-Mycd/Mycd ΔN79, respectively. Although Mycd plays a critical role in the differentiation of cardiac cell and SMC lineages (3, 7), Mycd knock-out mice die by embryonic day 10.5 due to a lack of vascular SMCs (VSMCs), but their heart development is unaffected (8). MRTF-A and MRTF-B expressed in the heart (2, 9) may compensate for the loss of Mycd under these conditions, and SMC gene expression is eliminated by suppression of Mycd function (10, 11). These findings suggest that Mycd is specifically required for VSMC differentiation. In contrast, MRTF-A and MRTF-B are involved in multiple cell functions in a variety of non-muscle cells. They activate the expression of several genes encoding cytoskeletal proteins in a Rho signaling-dependent manner (1) and are involved in transforming growth factor β1-induced epithelial-mesenchymal transition (12). The MRTF-A/SRF signaling pathway is also critical for tumor cell migration and metastasis (13). Furthermore, a null mutation of MRTF-A results in the failure of mammary myoepithelial cell to differentiate during lactation (14), and MRTF-B null mice die at about embryonic day 13.5 from abnormalities in SMCs within the aortic arch (15).
The N termini of Mycd family members (which possess three actin-binding RPEL motifs) are involved in their subcellular localization. Mycd appears to be constitutively located in the nucleus (3), whereas MRTF-A and MRTF-B reside mainly in the cytoplasm and transiently translocate to the nucleus in response to a signaling-induced decrease in monomeric actin (G-actin) (1, 9, 16). Moreover, the RPEL motifs of MRTF-A and MRTF-B have a much higher affinity for G-actin compared with Mycd (17). These properties are also closely related to the subcellular localization of Mycd family members (17–20). Guettler et al. (17) published a model for such actin dynamics-dependent regulation of MRTF-A nucleocytoplasmic shuttling. The basis of this model is as follows: G-actin binding to the RPEL motifs of MRTF-A inhibits its nuclear import at high G-actin concentrations, thereby facilitating CRM1-mediated nuclear export. However, at the time the model was published, the nuclear import machinery had not yet been identified. Recently, we (21) and Treisman and co-workers (22) found that the nuclear import of Mycd family members is mediated by importin α/β1 heterodimers. In cultured VSMCs, the importin α1/β1 heterodimer plays a critical role in the nuclear import of Mycd, and the expression of importins α1 and β1 and Mycd is closely related to the VSMC phenotype (21). We further established that the N-terminal basic domain of Mycd family members, which is also known as B2 (1), functions as a binding site for the importin α/β1 heterodimer (21). Actin dynamics does not affect the interaction between Mycd and the importin α1/β1 heterodimer and the nuclear localization of Mycd, but G-actin significantly suppresses the interaction between MRTF-A/B and importin α1/β1 and affects the nuclear import of MRTF-A/B (21). Furthermore, Treisman and co-workers (22) argued that the importin α/β1 heterodimer interacts with a bipartite nuclear localization signal including the N-terminal basic domain (B2) plus another N-terminal basic domain (B3) in the RPEL motifs, and they proposed that a similar competitive inhibition is exerted by G-actin. We have demonstrated, however, that even in the absence of G-actin, Mycd has a higher binding affinity for importin α/β1 compared with MRTF-A and MRTF-B and that their serum-induced nuclear import also correlates with their binding affinities for the importin α/β1 heterodimers (21). We have therefore come to the conclusion that the constitutive nuclear localization of Mycd is due to its strong binding affinity for the importin α/β1 heterodimer even when the concentration of G-actin is high.
It has been demonstrated that the nuclear export of some proteins depends on a nuclear export signal (NES) that consists of a leucine-rich sequence (23) and that CRM1 mediates the nuclear export of NES-containing proteins (24). In this process, Ran-GTP is required for CRM1 to bind its cargo protein (25), and leptomycin B (LMB) specifically blocks CRM1 binding to the NES of cargo proteins (26). There is substantial circumstantial evidence to suggest that CRM1 may mediate the nuclear export of Mycd family members (19–21). However, in contrast to their nuclear import mechanism, their nuclear export mechanism has remained obscure, and it is still unclear why Mycd is constitutively accumulated in the nucleus, but MRTF-A is located primarily in the cytoplasm. In this study, we therefore investigated the regulation of the nuclear export of Mycd family members to further understand the differences in their subcellular localization. Our findings have led us to conclude that Mycd is less likely to interact with CRM1 compared with MRTF-A. This is the first report to characterize the inhibitory mechanism for the nuclear export of Mycd, and our findings provide new insight into the functional regulation of Mycd family members and their related physiological events.
EXPERIMENTAL PROCEDURES
Reagents and Antibodies
The following commercially available primary antibodies were used in this study: anti-FLAG M2-agarose, anti-FLAG (catalog number F7425), anti-α-smooth muscle actin (clone 1A4), and anti-α-tubulin (clone DM1A) antibodies (Sigma-Aldrich); anti-HA affinity matrix and anti-HA (clone 3F10) antibodies (Roche Applied Science); anti-histone H2B, anti-c-Myc, anti-Mycd, anti-MRTF-A, and anti-SRF antibodies (Santa Cruz Biotechnology, Santa Cruz, CA); and anti-DYKDDDDK (anti-FLAG) antibody (TransGenic, Inc., Kobe, Japan). Protein A-Sepharose was purchased from GE Healthcare. Secondary antibodies were conjugated to Alexa Fluor 568 (Molecular Probes, Eugene, OR).
Plasmids
Construction of the expression plasmids for FLAG-tagged Mycd family members, β-actin R62D (unpolymerized mutant), and Myc-tagged SRF has been described previously (11, 21). In brief, the cDNAs of mouse full-length Mycd WT (GenBankTM accession number AF384055), MRTF-A (accession number NM_153049), and MRTF-B (accession number AF532598) were inserted into mammalian expression plasmid pCS2+ with the indicated tag. The expression plasmid for SMC-Mycd/Mycd ΔN79 (6) was similarly constructed. The cDNA of human β-actin (accession number NM_001101) was amplified and inserted into the same expression plasmid. The expression plasmid for unpolymerized mutant β-actin (β-actin R62D) was constructed by PCR-mediated mutagenesis. Similarly, the cDNA of human CRM1 (accession number BC032847) was amplified by RT-PCR and inserted into the same expression plasmid with an HA tag. A series of expression plasmids for truncated and/or mutated derivatives of Mycd family members were constructed by PCR-mediated methods, and their sequences were confirmed. Introduction of mutations in L1 and/or L2 of Mycd WT was performed as reported previously (20); the L1 sequence (LQLRL) and/or the L2 sequence (LFLQL) was changed to LQARL (L22A) and/or LFLQA (L295A), respectively. Introduction of mutations in L1 (L21A for MRTF-A and L46A for MRTF-B) and/or L2 (L307A for MRTF-A and L328A for MRTF-B) of MRTF-A/B was similarly performed. The Mycd derivative with CBmut carries a mutated central basic domain (CB) in which all of the lysine residues in the CB have been changed to alanine. A bacterial expression plasmid for the GST fusion protein of the FLAG-tagged constitutively active form of human Ran (RanQ69L) was kindly provided by Drs. Sekimoto and Yoneda (Osaka University Graduate School of Medicine) (27).
Cell Culture and Transfection
COS-7 cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum. The preparation of primary cultured rat aortic VSMCs was described in our previous report (21). Transfection of the indicated expression plasmids was performed using TransIT-LT1 (PanVera Corp., Madison, WI) according to the manufacturer's instructions. The transfected cells were then cultured under the indicated conditions for 24 or 48 h.
Knockdown of SRF Using siRNA
An siRNA against rat SRF (Rn_Srf_7084) and a scrambled siRNA for control experiments were purchased from Sigma-Aldrich. VSMCs were transfected with the indicated siRNAs using Lipofectamine RNAiMAX (Invitrogen).
Immunocytochemistry
Cells were fixed with 4% formaldehyde for 30 min; permeabilized; and blocked with 0.1% Triton X-100, 10% normal goat serum, and 0.2% bovine serum albumin in phosphate-buffered saline for 1 h at room temperature. The cells were then incubated with the indicated primary antibodies for 1 h, followed by the specified secondary antibodies with Hoechst 33258 for 1 h at room temperature. Fluorescent images were collected using a BIOREVO BZ-9000 fluorescence microscope (Keyence Corp., Osaka, Japan). The expression patterns of Mycd derivatives were categorized into three groups: nuclear localization, diffuse distribution in the nucleus and cytoplasm (defined as equivalent immunostaining intensities of the target molecules in the cytoplasm and nucleus), and cytoplasmic localization. In each experiment (n = at least three independent experiments), 100–200 cells were analyzed. The proportion of FLAG-positive cells exhibiting the respective expression patterns (mean ± S.E.) is presented.
Protein/Protein Interaction Analyses
A GST-FLAG-RanQ69L protein was expressed in Escherichia coli and purified with glutathione-Sepharose 4B (GE Healthcare) according to the manufacturer's instructions. The recombinant protein thus obtained was associated with GTP as described (27) with slight modifications. In brief, purified GST-FLAG-RanQ69L protein was incubated for 1 h in phosphate-buffered saline containing 2 mm GTP and 1 mm 2-mercaptoethanol on ice. Other proteins were prepared using the TnT SP6 high-yield expression system based on an optimized wheat germ extract (Promega) according to the manufacturer's instructions. We preliminarily confirmed that there was no significant protein cross-reaction between each of the antibodies against Mycd family members, SRF, and nuclear import/export proteins and this wheat germ extract for in vitro translation and checked the expression levels of in vitro translated proteins by immunoblotting (IB) using the specified antibodies (data not shown). The composition of the immunoprecipitation (IP) buffer in this study was as follows: 20 mm Tris-HCl (pH 7.5), 0.5% Nonidet P-40, 150 mm NaCl, 5 mm MgCl2, 1 mm EDTA, 50 mm NaF, 10 mm β-glycerophosphate, and protease inhibitors (cOmplete Mini, Roche Applied Science). The IP buffer mixtures (total of 500 μl) containing approximately equal amounts of FLAG- or HA-tagged Mycd, MRTF-A, or MRTF-B (5–20 μl) and defined amounts of the indicated proteins (HA-tagged CRM1 protein, 10 μl; SRF protein, 15 μl; β-actin R62D protein, 10 or 20 μl; and GTP-bound GST-FLAG-RanQ69L protein, 3 μl) were subjected to IP analyses. The IP buffer mixtures were first incubated with a control gel or control immunoglobulin G-bound protein A-Sepharose beads for 1 h to clear nonspecific interactions and then incubated with either the control gel or the specified affinity gel (antibody) for 6 h at 4 °C. The protein/protein interaction was also examined using cell extracts. Whole cell extracts were prepared from COS-7 cells transfected with the indicated expression plasmids according to previously described methods (11). In brief, cells were incubated for 30 min at 0 °C in lysis buffer; the salt concentration was then decreased to a physiological state; and the resulting whole cell extracts were further incubated for 30 min in the presence of 2 mm GTP. Equal amounts (400 μg of protein) of the whole cell extracts thus obtained were subjected to IP analysis as described above. Proteins in the immunoprecipitates were detected by IB using the specified antibodies. Target proteins were detected with a SuperSignal chemiluminescence detection kit (Pierce). In the immunoblot analysis, 3.3% of the input proteins and 22.2% of the immunoprecipitated proteins were loaded on the input and IP lanes, respectively. Quantification of the respective immunoblot signal intensities was performed with NIH Image software. These interaction analyses were repeated at least three times. Representative data are shown.
Preparation of Cytoplasmic and Nuclear Fractions
The subcellular proteomes were extracted from cultured VSMCs using the ProteoExtract subcellular proteome extraction kit (Calbiochem) according to the manufacturer's instructions. The cytoplasmic and nuclear fractions obtained were analyzed by IB using the indicated antibodies. In this analysis, histone H2B and α-tubulin were used as loading controls for the nuclear and cytoplasmic fractions, respectively. Quantification of the respective immunoblot signal intensities was performed with NIH Image software. These analyses were repeated at least three times. Representative data are shown.
Promoter Assays
COS-7 cells were transfected with a luciferase reporter gene carrying the SM22α promoter (SM22P-Luc) (11), pSV-β-gal (Promega), and the indicated expression plasmids for MRTF-A derivatives and then cultured for 48 h. The cell extracts were prepared using passive lysis buffer (Promega) according to the manufacturer's instructions and assayed for luciferase activity with a luciferase assay kit (Promega).
RESULTS
Interaction between CRM1 and Mycd
Mycd WT possesses two NES-like Leu-rich sequences: the N-terminal Leu-rich sequence in the first RPEL motif (L1) extending from amino acids 18 to 22 and the Leu-rich sequence in the Gln-rich domain (L2) extending from amino acids 291 to 295. They are well conserved among Mycd family members (Fig. 1A). To determine whether these sites are functional CRM1-binding sites, we examined the interaction in vitro between CRM1 and Mycd WT or each truncated Mycd (Mycd ΔN128 and Mycd N128) (Fig. 1B). Mycd ΔN128 and Mycd N128 lack the N-terminal region from amino acids 1 to 128 (N128) and the C-terminal region from amino acids 129 to the C-terminal end, respectively (Fig. 1A, left). We found that Mycd WT and each truncated Mycd bound to CRM1 in the presence of GTP-bound GST-FLAG-RanQ69L but with different binding affinities (Fig. 1B). When GTP-bound GST-FLAG-RanQ69L was absent, no such binding occurred (Fig. 1C). We also confirmed the Mycd/CRM1 interaction using the whole cell extracts from COS-7 cells coexpressing FLAG-tagged Mycd WT and HA-tagged CRM1 (data not shown). The order of Mycd CRM1-binding affinities was as follows: Mycd ΔN128 (786.3 ± 73.1%) > Mycd N128 (164.8 ± 25.7%) > Mycd WT (100%) (Fig. 1B, left). Furthermore, to assess whether L1 or L2 functions as a CRM1-binding site in Mycd WT, we introduced mutations into L1 (L22A) and/or L2 (L295A) of Mycd WT and examined their CRM1 binding in vitro (Fig. 2). Although the CRM1-binding affinity of Mycd L22A was comparable with that of Mycd WT (Fig. 2, A, lanes IP1 and IP2; and B), the CRM1-binding affinities of Mycd L295A and Mycd L22A/L295A were markedly reduced (decreases to 31.3 ± 5.1% (Mycd L295A) and 32.1 ± 7.4% (Mycd L22A/L295A) of the original binding affinity) (Fig. 2, A, lanes IP3 and IP4; and B). These results indicate that L2 functions as the CRM1-binding site in Mycd WT, but L1 does not. We then analyzed the subcellular localization of exogenously expressed Mycd WT and Mycd L295A in COS-7 cells. In the vast majority of cells expressing Mycd WT, the protein was observed primarily in the nucleus (90.9 ± 2.1%), and cells expressing Mycd L295A did not show any significant difference in its subcellular localization compared with cells expressing Mycd WT (data not shown). This evidence suggests that the CRM1-mediated nuclear export system does not affect the subcellular localization of Mycd WT.
FIGURE 1.

Interaction between CRM1 and Mycd WT. A, left, schematic representation of Mycd WT and truncated Mycd isoforms. The two Leu-rich sequences are indicated by black vertical lines: L1 in the first RPEL motif and L2 in the Gln-rich domain (Q). NB, N-terminal basic domain; CC, coiled-coil domain; TA, transactivation domain. Right, sequences of L1 and L2 (gray letters) in Mycd WT and MRTF-A/B orthologs from different species (mouse (m) and human (h)). B and C, interaction between CRM1 and Mycd WT or each truncated Mycd. Mixtures of HA-tagged CRM1 and each indicated FLAG-tagged Mycd with (B) or without (C) GTP-bound GST-FLAG-RanQ69L were immunoprecipitated with a control gel or anti-HA affinity gel, and the immunoprecipitates thus obtained were analyzed by IB using the indicated antibodies (left). The positions of molecular mass markers are between the panels in kilodaltons. Control experiments using the control gel did not show any significant signals on immunoblots (data not shown). Quantification of the IP analysis is shown (B, right). The respective IP/IB signal intensities were quantified as described under “Experimental Procedures.” The percentages indicate relative binding affinities for CRM1 normalized by the affinity of Mycd WT, which was set at 100%. Results are means ± S.E. of three independent experiments (error bars).
FIGURE 2.
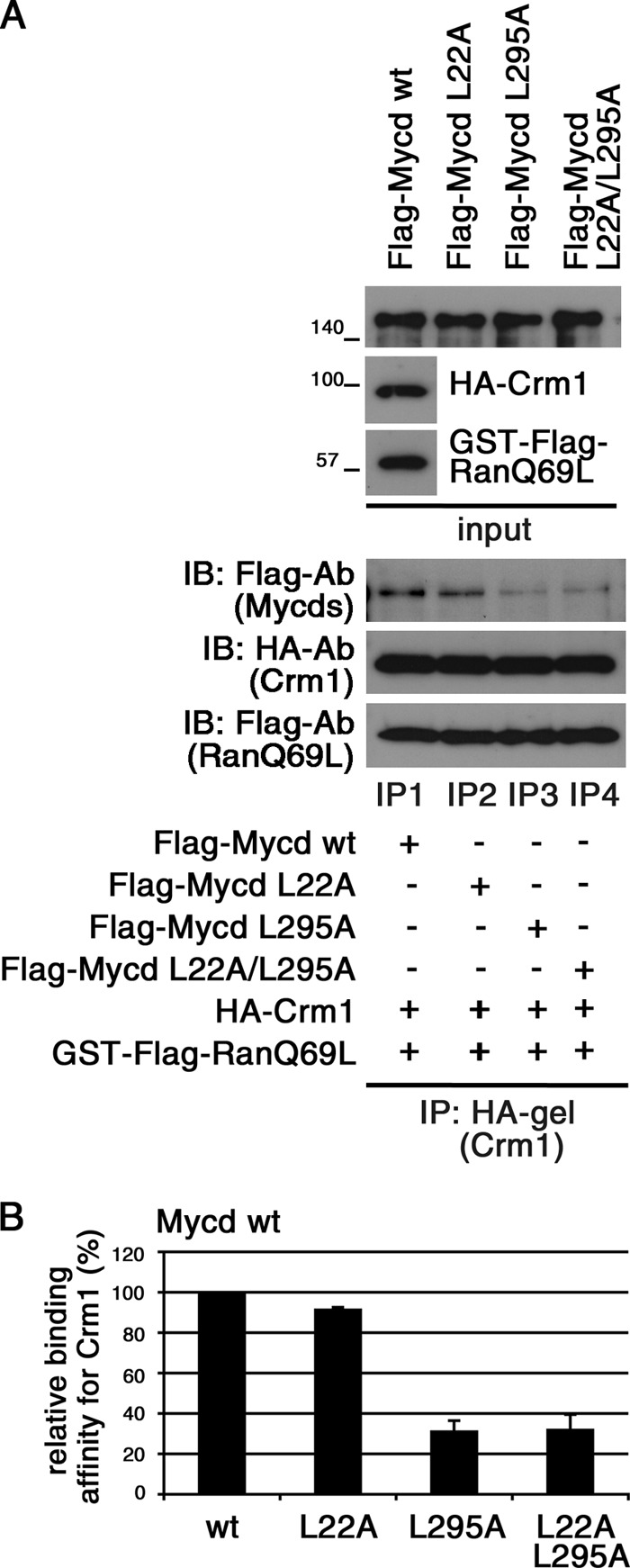
Effects of mutations in L1 (L22A) and/or L2 (L295A) on binding affinity of Mycd WT for CRM1. A, mixtures of HA-tagged CRM1, GTP-bound GST-FLAG-RanQ69L, and each indicated FLAG-tagged Mycd were subjected to IP analyses as described in the legend for Fig. 1. B, the respective IP/IB signal intensities were quantified as described under “Experimental Procedures.” The percentages indicate relative binding affinities for CRM1 normalized by the affinity of Mycd WT (wt), which was set at 100%. Results are means ± S.E. of three independent experiments (error bars).
Critical Roles of L1 and L2 of MRTF-A in Its CRM1-mediated Nuclear Export
We addressed the functional role of two Leu-rich sequences (L1 and L2) of MRTF-A in the interaction with CRM1 in vitro (Fig. 3). Introduction of a mutation in either L1 (MRTF-A L21A) or L2 (MRTF-A L307A) resulted in a moderate reduction in CRM1 binding (decreases to 50.5 ± 8.3% (MRTF-A L21A) and 64.5 ± 17.2% (MRTF-A L307A) of the original binding affinity) (Fig. 3, A, lanes IP2 and IP3; and B). Furthermore, MRTF-A L21A/L307A, in which both of the two Leu-rich sequences were mutated, exhibited a severe reduction in CRM1 binding (a decrease to 23.3 ± 6.2% of the original binding affinity) (Fig. 3, A, lane IP4; and B). These results indicate that unlike in the case of Mycd WT, both L1 and L2 serve as functional CRM1-binding sites.
FIGURE 3.
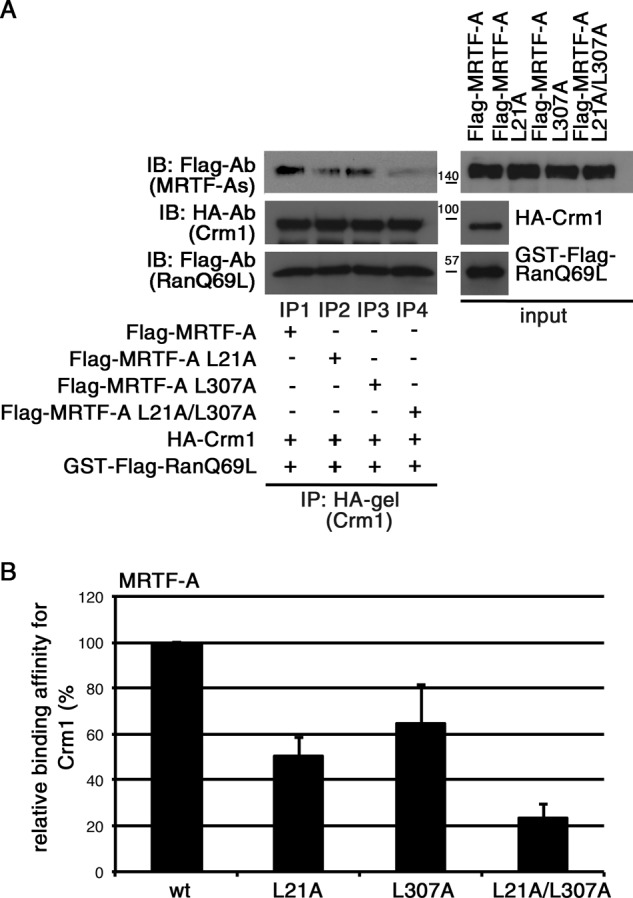
Effects of mutations in L1 (L21A) and/or L2 (L307A) on binding affinity of MRTF-A for CRM1. A, mixtures of HA-tagged CRM1, GTP-bound GST-FLAG-RanQ69L, and each indicated FLAG-tagged MRTF-A were subjected to IP analyses as described in the legend for Fig. 1. B, the respective IP/IB signal intensities were quantified as described under “Experimental Procedures.” The percentages indicate relative binding affinities for CRM1 normalized by the affinity of wild-type MRTF-A (wt), which was set at 100%. Results are means ± S.E. of three independent experiments (error bars).
To assess the roles of L1 and L2 in the nuclear export of MRTF-A in cultured cells, we analyzed the subcellular localization of exogenously expressed wild-type MRTF-A and mutants in COS-7 cells (Fig. 4). In the majority of cells expressing wild-type MRTF-A, the protein was observed primarily in the cytoplasm (67.2 ± 8.0%). Treatment with LMB markedly increased the percentage of cells showing a nuclear accumulation of MRTF-A (from 10.0 ± 3.5 to 57.8 ± 2.4%) (data not shown). The expression of MRTF-A L21A or MRTF-A L307A resulted in reduced percentages of cells with cytoplasmic localization (46.7 ± 1.4% for MRTF-A L21A and 58.9 ± 3.0% for MRTF-A L307A). In contrast, in cells expressing MRTF-A with mutations in both L1 and L2 (MRTF-A L21A/L307A), it was distributed primarily in the nucleus (65.4 ± 3.4%). These results indicate that both L1 and L2 play a critical role in the CRM1-mediated nuclear export of MRTF-A. A similar marked increase in the nuclear localization of MRTF-A L21A/L307A was also observed when cells were restimulated with serum (data not shown). In accordance with the subcellular localization of MRTF-A mutants (Fig. 4), enhancement of SM22α promoter activity by MRTF-A L21A or MRTF-A L307A was moderate, but that by MRTF-A L21A/L307A was more potent (data not shown).
FIGURE 4.

Effect of mutation in L1 (L21A) and/or L2 (L307A) on subcellular localization of MRTF-A. A, COS-7 cells were transfected with expression plasmids for the indicated FLAG-tagged MRTF-A under serum-stimulated conditions. For the final 20 h, the cells were cultured under serum-starved conditions. The cells were then stained with anti-DYKDDDDK (FLAG) antibody (red) and Hoechst 33258 (blue). Representative images from at least three independent experiments are shown. Scale bar = 20 μm. B, the images were quantified as described under “Experimental Procedures.” N, nuclear accumulation; NC, diffuse distribution in the nucleus and cytoplasm; C, cytoplasmic localization. Statistical differences were calculated using Student's t test. *, p < 0.05 versus wild-type MRTF-A.
We addressed the relationship between the MRTF-A/CRM1 interaction and actin dynamics and found a novel function of G-actin in the MRTF-A/CRM1 interaction (Fig. 5). In the presence of G-actin (β-actin R62D), CRM1 binding to MRTF-A was dose-dependently suppressed (Fig. 5A). The interaction between MRTF-A L307A and CRM1 was inhibited by β-actin R62D, but that between MRTF-A L21A and CRM1 was not (Fig. 5, B and C), indicating that CRM1 binding to only L1 is inhibited by G-actin.
FIGURE 5.

Effects of G-actin on interaction between CRM1 and MRTF-A. The indicated proteins were mixed with 10 μl (+) and 20 μl (++) of β-actin R62D, and their interactions were analyzed by the same procedures as described in the legend for Fig. 1 (upper panels). The respective IP/IB signal intensities were quantified as described under “Experimental Procedures.” The percentages indicate relative binding affinities for CRM1 normalized by the respective affinities of wild-type MRTF-A (A), MRTF-A L21A (the L1 mutant) (B), and MRTF-A L307A (the L2 mutant) (C) in the absence of β-actin R62D, which were set at 100% (lower panels). Results are means ± S.E. of three independent experiments (error bars).
We also addressed the roles of L1 and/or L2 in the nuclear export of MRTF-B in cultured cells (Fig. 6). Introduction of mutations in both L1 (L46A) and L2 (L328A) of MRTF-B more significantly decreased the percentage of cells showing a cytoplasmic localization of MRTF-B L46A/L328A (46.5 ± 6.2% for MRTF-B L46A/L328A and 88.5 ± 0.8% for wild-type MRTF-B). However, unlike MRTF-A with mutations in both L1 and L2 (MRTF-A L21A/L307A), a marked increase in the nuclear accumulation of MRTF-B L46A/L328A was not observed. This is probably due to lower nuclear import activity of MRTF-B (21). There was no significant difference in the subcellular localization of wild-type MRTF-B in the cells with LMB and that of MRTF-B L46A/L328A in the cells without LMB. Nuclear accumulation of wild-type MRTF-B was still low even in the cells with LMB. These results clearly indicate that MRTF-B is unlikely to be imported into the nucleus. In conclusion, these immunocytochemical data suggest that both L1 and L2 play a functional role in the CRM1-mediated nuclear export of MRTF-B.
FIGURE 6.

Effects of mutations in L1 (L46A) and/or L2 (L328A) on subcellular localization of MRTF-B. COS-7 cells were transfected with expression plasmids for the indicated FLAG-tagged MRTF-B and cultured under the same conditions as described in the legend for Fig. 4. For the last 2 h, the cells were incubated with vehicle or LMB (5 ng/ml). The cells were then stained with anti-DYKDDDDK (FLAG) antibody and Hoechst 33258. The images were quantified as described under “Experimental Procedures.” N, nuclear accumulation; NC, diffuse distribution in the nucleus and cytoplasm; C, cytoplasmic localization. Statistical differences were calculated using Student's t test. *, p < 0.05 versus wild-type MRTF-B.
Inhibitory Effects of SRF on Mycd Family Member/CRM1 Interaction
It has been reported that the SRF-binding region of Mycd extends from CB to the Gln-rich domain (10). The functional CRM1-binding site of Mycd (L2) is located within this SRF-binding region. These findings led to the hypothesis of a competitive binding model in which Mycd-SRF complex formation inhibits the Mycd/CRM1 interaction. We first examined this possibility with an in vitro binding assay. When SRF was present, the interaction between CRM1 and Mycd WT was markedly reduced, whereas a significant interaction between CRM1 and SRF was not observed. In contrast, however, the presence of Myc-tagged G-actin (β-actin R62D) did not decrease the Mycd WT/CRM1 interaction (data not shown). Thus, such a specific effect of SRF would be due to the formation of an Mycd WT-SRF complex but not the formation of a complex containing CRM1 and SRF. Because the other Mycd family members, MRTF-A and MRTF-B, also interact with SRF (28), we compared the CRM1-binding affinities of Mycd family members and the inhibitory effects of SRF on their binding affinities (Fig. 7). Their CRM1-binding affinities varied, and they were found to decrease in the following order: MRTF-A (207.8 ± 12.1%) > Mycd WT (100%) > MRTF-B (34.5 ± 2.2%) (Fig. 7A, left, lanes IP1, IP2, and IP3; and right, black bars). The extent of the inhibitory effects of SRF also varied. The hierarchy of this inhibition was Mycd WT > MRTF-B > MRTF-A (Fig. 7A, left, lanes IP1′, IP2′, and IP3′; and right, white bars). Unlike in the case of Mycd WT and MRTF-B, the CRM1-binding affinity of MRTF-A was only modestly reduced in the presence of SRF. These results suggest that the SRF-binding affinities of Mycd family members are not similar. Further assessment revealed that their relative binding affinities for SRF were decreased in the following order: Mycd WT (100%) > MRTF-B (41.7 ± 2.4%) > MRTF-A (30. 0 ± 2.9%) (Fig. 7B). This order shows a good correlation with the hierarchy of the inhibitory effects of SRF.
FIGURE 7.

Effects of SRF on interactions between CRM1 and Mycd family members. A, the binding affinities of Mycd family members for CRM1 and the inhibitory effects of SRF on their interactions were compared. Mixtures of HA-tagged CRM1, GTP-bound GST-FLAG-RanQ69L, and each of the indicated FLAG-tagged Mycd family members with or without Myc-tagged SRF were subjected to IP analyses as described in the legend for Fig. 1 (left). The respective IP/IB signal intensities were quantified as described under “Experimental Procedures.” The percentages indicate relative binding affinities for CRM1 normalized by the affinity of Mycd WT in the absence of SRF, which was set at 100% (right). Results are means ± S.E. of three independent experiments (error bars). The reductions in binding affinities for CRM1 by SRF were calculated as follows: 19.5 ± 3.9% for Mycd WT, 57.4 ± 5.0% for MRTF-B, and 88.1 ± 1.5% for MRTF-A. Each of the binding affinities in the absence of SRF was set at 100%. B, differences in the binding affinities of Mycd family members for SRF. Mixtures of Myc-tagged SRF and each of the indicated FLAG-tagged Mycd family members were immunoprecipitated with anti-Myc antibody and protein A-Sepharose. The immunoprecipitates thus obtained were analyzed by IB using the indicated antibodies (left). In control experiments using a control antibody, no significant signals were observed on immunoblots (data not shown). The respective IP/IB signal intensities were quantified as described under “Experimental Procedures.” The percentages indicate relative binding affinities for SRF normalized by the affinity of Mycd WT, which was set at 100% (right). Results are means ± S.E. of three independent experiments (error bars).
Role of N Termini of Mycd WT and MRTF-A in Their Interactions with CRM1
The CRM1-binding affinity of Mycd WT was low compared with that of Mycd ΔN128 (Fig. 1B). This finding led us to speculate that Mycd N128 may prevent the interaction between CRM1 and Mycd WT by masking L2. To assess this possibility, we first examined the interaction between HA-tagged Mycd N128 and FLAG-tagged Mycd WT or Mycd ΔN128 in vitro (Fig. 8A). Mycd ΔN128 clearly bound to Mycd N128 (lane IP2), whereas the binding of Mycd WT to Mycd N128 was weak (23.2 ± 1.7% of the binding affinity of Mycd ΔN128) (lane IP1). Similar binding profiles were also observed when FLAG-tagged Mycd WT or Mycd ΔN128 was immunoprecipitated with the anti-FLAG M2 affinity gel (data not shown). These findings suggest that Mycd N128 binds intramolecularly to Mycd WT and that this self-association represses the interaction between CRM1 and Mycd WT in an autoinhibitory manner. In contrast, this type of intramolecular association of MRTF-A would be unlikely. This is because the N terminus of MRTF-A extending from amino acids 1 to 125 (MRTF-A N125), which corresponds to Mycd N128, scarcely interacted with MRTF-A ΔN125, which corresponds to Mycd ΔN128 (Fig. 8B). We then performed domain mapping to identify a binding site for Mycd N128 in Mycd ΔN128 using several deletion mutants derived from Mycd ΔN128 (Fig. 8C). Mycd ΔN128/ΔCB, Mycd ΔN128/ΔQ, and Mycd ΔN128/ΔSAP lack CB, the Gln-rich domain, and the SAP domain, respectively. The Mycd N128-binding affinities of Mycd ΔN128, Mycd ΔN128/ΔQ, and Mycd ΔN128/ΔSAP were equivalent (lanes IP1, IP3, and IP4), whereas that of Mycd ΔN128/ΔCB was significantly reduced (a decrease to 28.3 ± 1.3 of the original binding affinity) (lane IP2). These results indicate that CB plays a critical role in the interaction between Mycd N128 and Mycd ΔN128. Furthermore, introduction of mutations at all of the lysine residues in CB (Mycd ΔN128/CBmut) partially disrupted the interaction with Mycd N128 (a decrease to 43.0 ± 7.3% of the original binding affinity) (data not shown). Because CB is known to be necessary for interaction with SRF (3), we speculated a competitive binding of SRF and Mycd N128 to Mycd ΔN128. We examined this possibility with an in vitro binding assay (Fig. 8D). SRF markedly suppressed the interaction between Mycd ΔN128 and Mycd N128 (a decrease to 18.1 ± 5.9% of the binding affinity of Mycd ΔN128 for Mycd N128 in the absence of SRF) (lanes IP1 and IP2). In this assay, the Mycd ΔN128/SRF interaction was detected (lane IP4), but the Mycd N128/SRF interaction was not (lane IP2). Thus, this suppressive effect would be due to the formation of a complex containing Mycd ΔN128 and SRF.
FIGURE 8.

Possibility of self-association of Mycd WT and MRTF-A. A and B, differences in the binding affinities of Mycd WT and Mycd ΔN128 for Mycd N128 (A) and the less significant possibility of MRTF-A self-association (B). Mixtures of the indicated FLAG-tagged and HA-tagged Mycd or MRTF-A were subjected to IP analyses. C, mapping of the functional domain in Mycd WT for the interaction with Mycd N128. Mixtures of HA-tagged Mycd N128 and each indicated FLAG-tagged Mycd were subjected to IP analyses as described in the legend for Fig. 1. The respective IP/IB signal intensities were quantified as described under “Experimental Procedures.” The percentages indicated at the top of the gels indicate relative binding affinities for Mycd N128 normalized by the affinity of Mycd ΔN128, which was at 100%. Results are means ± S.E. of three independent experiments. D, effect of SRF on Mycd WT self-association. The binding of Mycd ΔN128 to Mycd N128 was examined in the absence or presence of SRF (left). Mixtures of the indicated tagged proteins with or without Myc-tagged SRF were subjected to IP analysis. The relative binding affinities of Mycd ΔN128 for Mycd N128 are indicated at the top of the gels; affinity in the absence of SRF was set at 100%. The interaction between SRF and Mycd ΔN128 is shown (right). Mixtures of the indicated tagged proteins were immunoprecipitated with anti-Myc antibody and protein A-Sepharose. The resulting immunoprecipitates were analyzed by IB using the indicated antibodies.
To investigate whether Mycd N128 functionally affects the interaction between CRM1 and Mycd WT, we examined the interaction between CRM1 and Mycd ΔN128 or Mycd ΔN128/ΔCB in the absence or presence of Mycd N128 (Fig. 9A). In the absence of Mycd N128, both Mycd ΔN128 and Mycd ΔN128/ΔCB similarly interacted with CRM1 (upper panel, lanes IP1 and IP3). On the other hand, in the presence of Mycd N128, the CRM1-binding affinity of Mycd ΔN128 was reduced (a decrease to 55.1 ± 3.3% of the affinity without Mycd N128) (upper panel, lane IP2), but that of Mycd ΔN128/ΔCB was not (upper panel, lane IP4). No significant interaction between CRM1 and Mycd N128 was detected in these protein/protein interaction analyses (upper middle panel, lanes IP1, IP2, IP3, and IP4). This is thought to be due to the lower binding affinity of Mycd N128 for CRM1 (Fig. 1B). These results suggest that Mycd N128 binds intramolecularly to Mycd via CB and that this self-association inhibits the interaction between CRM1 and Mycd WT.
FIGURE 9.
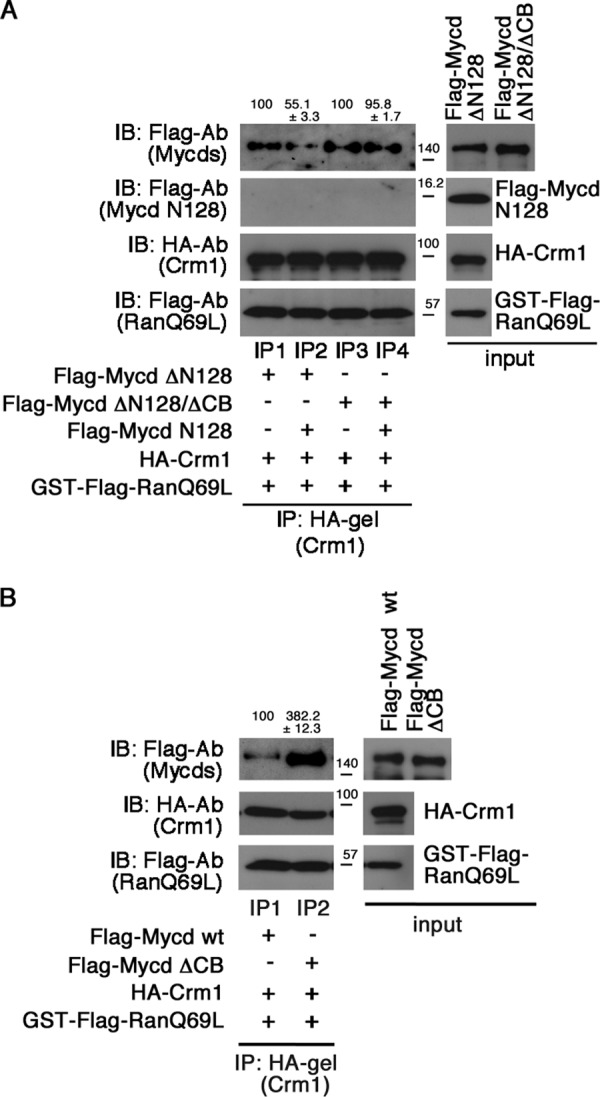
Autoinhibition of Mycd WT/CRM1 interaction by N-terminal region of Mycd WT (Mycd N128). A, inhibitory effect of Mycd N128 on the interaction between CRM1 and Mycd ΔN128. The CRM1-binding affinities of Mycd ΔN128 and Mycd ΔN128/ΔCB were examined in the absence or presence of Mycd N128. Mixtures of HA-tagged CRM1, GTP-bound GST-FLAG-RanQ69L, and each indicated FLAG-tagged Mycd were subjected to IP analyses as described in the legend for Fig. 1. The respective IP/IB signal intensities were quantified as described under “Experimental Procedures.” The percentages at the top of the gels indicate relative binding affinities for CRM1 normalized by the respective affinities of Mycd ΔN128 and Mycd ΔN128/ΔCB in the absence of Mycd N128 (−), which were set at 100%. Results are means ± S.E. of three independent experiments. B, effect of the CB deletion on the Mycd WT/CRM1 interaction. Mixtures of HA-tagged CRM1, GTP-bound GST-FLAG-RanQ69L, and each indicated FLAG-tagged Mycd were subjected to IP analyses. The respective IP/IB signal intensities were quantified. The percentages at the top of the gels indicate relative binding affinities for CRM1 normalized by the affinity of Mycd WT, which was set at 100%. Results are means ± S.E. of three independent experiments.
Regulatory Domains in Mycd WT for Suppression of Mycd WT/CRM1 Interaction
To gain further insight into the regulation of the Mycd WT/CRM1 interaction, we investigated the roles of CB in this interaction. We examined the effect of CB deletion on the interaction between CRM1 and Mycd WT (Fig. 9B). The CRM1-binding affinity of Mycd ΔCB was markedly increased (382.2 ± 12.3% of the affinity of Mycd WT) (lanes IP1 and IP2). This result indicates that CB plays a critical role in the suppression of the Mycd WT/CRM1 interaction and suggests that this suppression is due to self-association as described above.
Differences in Regulation of CRM1 Binding to Mycd Isoforms
To further address the function of the N-terminal regions of Mycd WT and SMC-Mycd/Mycd ΔN79, we first compared the CRM1-binding affinities of Mycd WT, SMC-Mycd/Mycd ΔN79, and Mycd ΔN128 (Fig. 10A). The order of the CRM1-binding affinities was Mycd ΔN128 (738.8 ± 143.7%) > SMC-Mycd/Mycd ΔN79 (513.6 ± 38.2%) > Mycd WT (100%), suggesting that a short N-terminal region of SMC-Mycd/Mycd ΔN79 (Mycd N80–128) weakly inhibits the interaction between SMC-Mycd/Mycd ΔN79 and CRM1. To confirm this prediction, we examined the interaction between CRM1 and Mycd ΔN128 in the absence or presence of Mycd N80–128 or Mycd N128 (Fig. 10B). The inhibitory effect of Mycd N80–128 on CRM1 binding to Mycd ΔN128 (lanes IP1 and IP2) was modest compared with that of Mycd N128 (lanes IP4 and IP5). We further investigated the binding affinities of Mycd N128 and Mycd N80–128 for Mycd ΔN128 and found that their relative binding affinities were as follows: Mycd N80–128 (20.2 ± 5.0%) < Mycd N128 (100%) (Fig. 10C). These results indicate that the difference in these inhibitory effects of the N-terminal regions of Mycd isoforms depends on their binding affinity for Mycd ΔN128.
FIGURE 10.
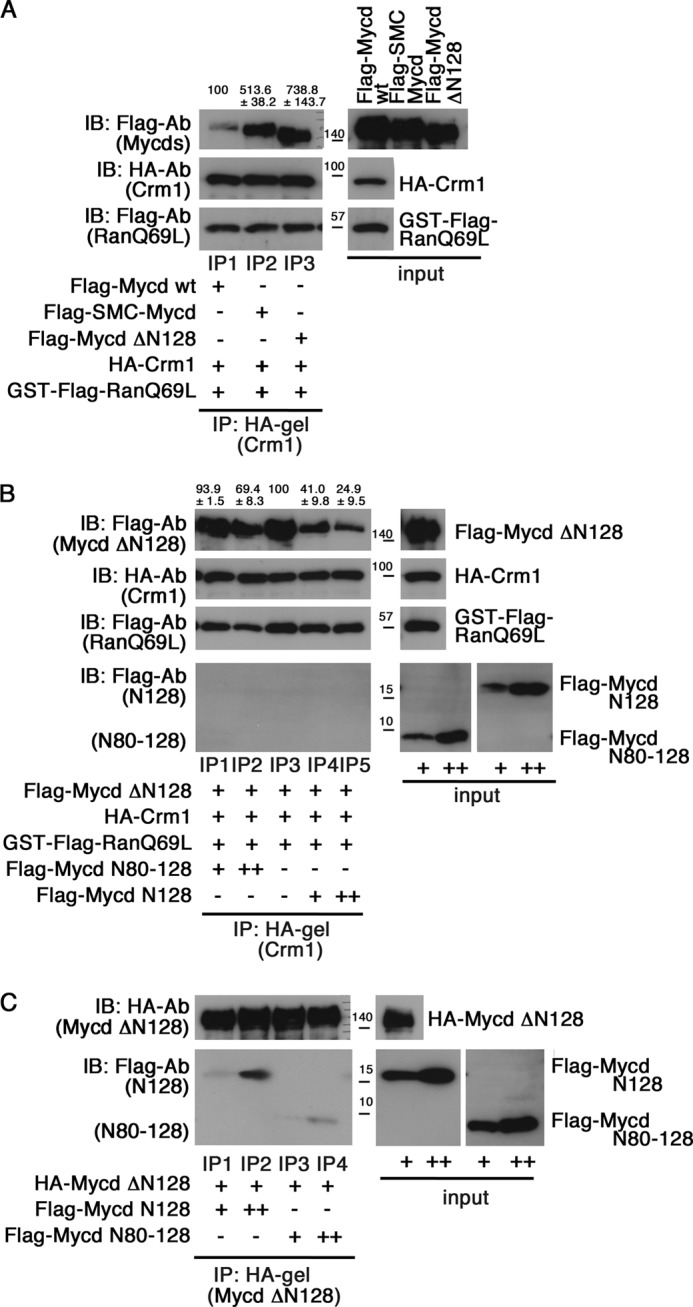
Differential regulation of CRM1 binding to Mycd isoforms. A, comparison of the binding affinities of Mycd WT, SMC-Mycd/Mycd ΔN79, and Mycd ΔN128 for CRM1. Mixtures of the indicated tagged proteins were subjected to IP analyses as described in the legend for Fig. 1. The resulting immunoprecipitates were analyzed by IB using the indicated antibodies. The respective IP/IB signal intensities were quantified as described under “Experimental Procedures.” The percentages at the top of the gels indicate relative binding affinities for CRM1 normalized by the affinity of Mycd WT, which was at 100%. Results are means ± S.E. of three independent experiments. B, differences in the inhibitory effects of the N-terminal regions of Mycd isoforms on the interaction between CRM1 and Mycd ΔN128. Mixtures of the indicated tagged proteins without or with 5 μl (+) and 10 μl (++) of the respective FLAG-tagged N-terminal regions of Mycd WT (Mycd N128) and SMC-Mycd/Mycd ΔN79 (Mycd N80–128) were subjected to IP analyses. The percentages at the top of the gels indicate relative binding affinities for CRM1 normalized by the affinity of Mycd ΔN128 without the N-terminal regions of the Mycd proteins (lane IP3), which was at 100%. Results are means ± S.E. of three independent experiments. C, differences in the binding affinities of the respective N-terminal regions of Mycd isoforms for Mycd ΔN128. Mixtures of HA-tagged Mycd ΔN128 and different doses (+ and ++) of FLAG-tagged Mycd N128 or Mycd N80–128 were subjected to IP analyses.
To address a functional role of the two distinct mechanisms for the inhibition of the Mycd/CRM1 interaction (competitive inhibition by SRF and inhibition by Mycd self-association) in the regulation of Mycd isoform subcellular localization, we first examined the subcellular localization of endogenous SMC-Mycd/Mycd ΔN79 in cultured VSMCs with knockdown of endogenous SRF. The expression level of SRF protein was markedly decreased in VSMCs transfected with SRF siRNA, but not in VSMCs transfected with a control scrambled siRNA (Fig. 11A). To examine the role of SRF in the nuclear localization of SMC-Mycd/Mycd ΔN79, we biochemically analyzed the levels of SMC-Mycd/Mycd ΔN79 in the nuclear and cytoplasmic fractions of VSMCs transfected with the control or SRF siRNA (Fig. 11B). In VSMCs transfected with the control siRNA, SMC-Mycd/Mycd ΔN79 was located exclusively in the nucleus (96.7 ± 1.5%), and SRF was detected in the nuclear fraction. In contrast, in VSMCs transfected with SRF siRNA, the proportion of cytoplasmic SMC-Mycd/Mycd ΔN79 was slightly but significantly increased in close correlation with the down-regulation of SRF (21.2 ± 6.1% for SRF siRNA and 3.3 ± 1.5% for control siRNA), implying that the interaction between SMC-Mycd/Mycd ΔN79 and SRF plays a partial role in the nuclear localization of this Mycd isoform. These results were in good agreement with the subcellular localization of exogenously expressed SMC-Mycd/Mycd ΔN79, but exogenously expressed Mycd WT was absolutely accumulated in the nucleus even in VSMCs with knockdown of endogenous SRF (data not shown). Furthermore, knockdown of SRF markedly reduced the nuclear localization of exogenously expressed Mycd ΔN128 (45.7 ± 4.5% for SRF siRNA and 83.2 ± 4.4% for control siRNA). We also performed similar analyses to examine the subcellular localization of endogenous MRTF-A in VSMCs transfected with SRF siRNA (Fig. 11C). Knockdown of endogenous SRF barely affected the subcellular localization of MRTF-A. These results suggest that the capability of the Mycd isoforms self-association is closely related to the effects of knockdown of SRF on their nuclear localization.
FIGURE 11.
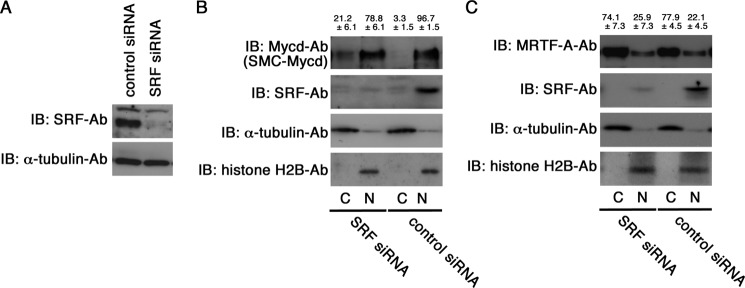
Effect of siRNA-mediated knockdown of endogenous SRF on subcellular localization of SMC-Mycd/Mycd ΔN79 and MRTF-A in VSMCs. A, VSMCs were transfected with the indicated siRNAs and cultured for 2 days. Whole cell lysates were then subjected to IB using the indicated antibodies. The level of α-tubulin served as a loading control. B and C, effect of knockdown of endogenous SRF on the subcellular localization of endogenous SMC-Mycd/Mycd ΔN79 (B) and MRTF-A (C) in VSMCs. VSMCs were transfected with the indicated siRNAs and cultured for 2 days. Their nuclear (N) and cytoplasmic (C) fractions (2% of the respective fractions) were analyzed by IB using the indicated antibodies as described under “Experimental Procedures.” Representative results from at least three independent experiments are shown. The respective IB signal intensities in the nuclear and cytoplasmic fractions were quantified, and the sum of their intensities in respective cells was set at 100%. The percentages of the intensities of the nuclear and cytoplasmic fractions were calculated, and they are indicated at the top of the gels. Results are means ± S.E. of three independent experiments.
DISCUSSION
The novel findings from this study are as follows. 1) L2 (but not L1) is a CRM1-binding site of Mycd WT. However, L2 is not involved in the regulation of the subcellular localization of Mycd WT. In contrast, both L1 and L2 serve as a functional NES of MRTF-A. Although a signaling-induced decrease in G-actin promotes the nuclear accumulation of MRTF-A, the MRTF-A/CRM1 interaction itself is inhibited by G-actin. In this case, G-actin suppresses CRM1 binding to L1, but not to L2. 2) SRF inhibits the interactions between CRM1 and Mycd family members. 3) The differences in the binding affinities of Mycd family members for SRF and CRM1 are closely related to the SRF inhibitory effects on the interactions between CRM1 and Mycd family members. 4) Mycd N128 binds intramolecularly to Mycd WT via CB, resulting in inhibition of the Mycd WT/CRM1 interaction by masking L2. This intramolecular association is attenuated by SRF. In contrast, such autoinhibition is moderate in the interaction between SMC-Mycd/Mycd ΔN79 and CRM1 and is absent in the MRTF-A/CRM1 interaction. Thus, we conclude that these multiple inhibitory mechanisms would isolate Mycd isoforms from the CRM1-mediated nuclear export system. Fig. 12 schematically summarizes the findings of this study combined with the findings of our previous work: the nuclear localization of Mycd is determined by its high binding affinity for the importin α/β1 heterodimer (21) and its molecular design to be isolated from CRM1-mediated nuclear export. In contrast, MRTF-A is much more likely to be exported from the nucleus because of its high binding affinity for CRM1 and a lack of the inhibitory regulations observed in the Mycd/CRM1 interaction as described above.
FIGURE 12.

Summary of differential regulation of subcellular localization between Mycd and MRTF-A. Our current findings combined with our previous ones are schematically represented. The nuclear accumulation of Mycd is determined by its high binding affinity for the importin α/β1 heterodimer and its molecular design for exclusion from CRM1-mediated nuclear export (autoinhibition by self-association and protection by SRF binding). In contrast, the binding affinity of MRTF-A for the importin α/β1 heterodimer is lower than that of Mycd, and this interaction is competitively inhibited by G-actin. Thus, MRTF-A is less likely to be imported to the nucleus. In contrast, MRTF-A is much more likely to be exported from the nucleus by CRM1 because of its high binding affinity for CRM1, its low binding affinity for SRF, and absence of autoinhibition. These differences may cause a distinct subcellular localization of Mycd and MRTF-A. NB, N-terminal basic domain; Q, Gln-rich domain; CC, coiled-coil domain; TA, transactivation domain.
NES of Mycd Family Members and Regulation of Their Interaction with CRM1
In Mycd WT, L2 (but not L1) serves as a CRM1-binding site (Fig. 2B). However, L2 is a functional NES only in artificial Mycd molecules such as Mycd ΔN128 and its derivatives. In Mycd WT, L2 is masked by Mycd N128. Therefore, the CRM1-mediated nuclear export system does not affect the nuclear localization of Mycd WT. We discussed this molecular event above. In contrast to Mycd WT, both L1 and L2 of MRTF-A serve as the functional NES (Figs. 3 and 4), and MRTF-A exhibits a strong binding affinity for CRM1 (Fig. 7A). This may be one reason why MRTF-A resides mostly in the cytoplasm. Although the MRTF-A/CRM1 interaction itself is suppressed by G-actin (Fig. 5), the nuclear localization of MRTF-A is inhibited under conditions in which the concentration of G-actin is increased. We (21) and Pawlowski et al. (22) have demonstrated that the interaction between MRTF-A and importin α/β1 is severely inhibited by G-actin, and Mouilleron et al. (30) recently proposed a model for actin dynamics-dependent competitive binding of importin α/β1 and G-actin to the MRTF-A RPEL motifs. Based on these findings, the nuclear accumulation of MRTF-A induced by depletion of the G-actin pool is critically dependent on the promotion of nuclear import rather than the modulation of nuclear export.
Role of N-terminal Region of Mycd in Interaction with CRM1
We proposed a novel function of the N-terminal region of Mycd besides the critical role in nuclear import (21). Because the basic amino acids in CB play a partial role in the interaction between Mycd N128 and Mycd ΔN128 (data not shown), such a molecular event is likely to occur as a result of an ionic bonding interaction. We speculate that the acidic amino acid(s) in Mycd N128, which we have not yet identified, are the bonding partner(s). The acidic amino acids within the N-terminal 79 amino acid residues of Mycd WT may play a critical role because the N-terminal region of SMC-Mycd/Mycd ΔN79 (Mycd N80–128) exhibits significantly lower binding affinity for Mycd ΔN128 (Fig. 10C). In our previous study (21), we concluded that CB is not the binding site for the importin α/β1 heterodimer and does not play a vital role in the nuclear import of Mycd WT, whereas it is critically involved in the nuclear import of Mycd ΔN128 in an importin α/β1-independent manner. These findings suggest that CB performs distinctly different functions in the regulation of Mycd subcellular localization. The tertiary structures of the respective Mycd molecules probably determine the various CB functions. A structural biology approach is necessary to elucidate these points.
Ransom et al. (31) identified a rare human sequence variation in Mycd in a patient with congenital heart disease: a missense mutation at codon 259 resulting in a lysine-to-arginine substitution at codon 259 (K259R). The N-terminal region of this Mycd mutant acts in an autoinhibitory fashion to bind Mycd, resulting in severe suppression of SRF binding to Mycd. Our present data suggest that the self-association of Mycd WT is easily dissociated under conditions in which SRF is present (Fig. 8D). Thus, Mycd WT self-association is limited to conditions in which the expression level of SRF is low. Knockdown of endogenous SRF barely affects the subcellular localization of Mycd WT but moderately affects the nuclear localization of SMC-Mycd/Mycd ΔN79 (Fig. 11). As shown below, such heterogeneous effects are due to the different effects of self-association in masking the CRM1-binding site. Self-association may serve as a way to exclude Mycd isoforms from the CRM1-mediated nuclear export system when the expression level of SRF is reduced; Mycd isoforms not bound to SRF may undergo self-association to prevent CRM1 binding to L2. This model could explain the different effects of SRF knockdown on the subcellular localization of Mycd WT, SMC-Mycd/Mycd ΔN79, and Mycd ΔN128 (Fig. 11). In the case of SMC-Mycd/Mycd ΔN79, the capability of self-association is low (Fig. 10). Thus, CRM1 is more likely to interact with SMC-Mycd/Mycd ΔN79 in SRF-depleted cells, resulting in a slight increase in its cytoplasmic localization. On the basis of these findings, we speculate that such distinct inhibitory mechanisms would play a significant role in keeping Mycd proteins in the nucleus.
This work was supported by Grant-in-aid for Scientific Research 23590332 from the Ministry of Education, Science, Sports and Culture of Japan (to K. H.).
- Mycd
- myocardin
- SRF
- serum response factor
- MRTF
- myocardin-related transcription factor
- SMC
- smooth muscle cell
- VSMC
- vascular SMC
- NES
- nuclear export signal(s)
- LMB
- leptomycin B
- CB
- central basic domain
- IB
- immunoblotting
- IP
- immunoprecipitation.
REFERENCES
- 1. Miralles F., Posern G., Zaromytidou A. I., Treisman R. (2003) Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell 113, 329–342 [DOI] [PubMed] [Google Scholar]
- 2. Wang D. Z., Li S., Hockemeyer D., Sutherland L., Wang Z., Schratt G., Richardson J. A., Nordheim A., Olson E. N. (2002) Potentiation of serum response factor activity by a family of myocardin-related transcription factors. Proc. Natl. Acad. Sci. U.S.A. 99, 14855–14860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wang D., Chang P. S., Wang Z., Sutherland L., Richardson J. A., Small E., Krieg P. A., Olson E. N. (2001) Activation of cardiac gene expression by myocardin, a transcriptional cofactor for serum response factor. Cell 105, 851–862 [DOI] [PubMed] [Google Scholar]
- 4. Ma Z., Morris S. W., Valentine V., Li M., Herbrick J. A., Cui X., Bouman D., Li Y., Mehta P. K., Nizetic D., Kaneko Y., Chan G. C., Chan L. C., Squire J., Scherer S. W., Hitzler J. K. (2001) Fusion of two novel genes, RBM15 and MKL1, in the t(1;22)(p13;q13) of acute megakaryoblastic leukemia. Nat. Genet. 28, 220–221 [DOI] [PubMed] [Google Scholar]
- 5. Mercher T., Coniat M. B., Monni R., Mauchauffe M., Nguyen Khac F., Gressin L., Mugneret F., Leblanc T., Dastugue N., Berger R., Bernard O. A. (2001) Involvement of a human gene related to the Drosophila spen gene in the recurrent t(1;22) translocation of acute megakaryocytic leukemia. Proc. Natl. Acad. Sci. U.S.A. 98, 5776–5779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Creemers E. E., Sutherland L. B., Oh J., Barbosa A. C., Olson E. N. (2006) Coactivation of MEF2 by the SAP domain proteins myocardin and MASTR. Mol. Cell 23, 83–96 [DOI] [PubMed] [Google Scholar]
- 7. Wang Z., Wang D. Z., Pipes G. C., Olson E. N. (2003) Myocardin is a master regulator of smooth muscle gene expression. Proc. Natl. Acad. Sci. U.S.A. 100, 7129–7134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Li S., Wang D. Z., Wang Z., Richardson J. A., Olson E. N. (2003) The serum response factor coactivator myocardin is required for vascular smooth muscle development. Proc. Natl. Acad. Sci. U.S.A. 100, 9366–9370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Du K. L., Chen M., Li J., Lepore J. J., Mericko P., Parmacek M. S. (2004) Megakaryoblastic leukemia factor-1 transduces cytoskeletal signals and induces smooth muscle cell differentiation from undifferentiated embryonic stem cells. J. Biol. Chem. 279, 17578–17586 [DOI] [PubMed] [Google Scholar]
- 10. Wang Z., Wang D. Z., Hockemeyer D., McAnally J., Nordheim A., Olson E. N. (2004) Myocardin and ternary complex factors compete for SRF to control smooth muscle gene expression. Nature 428, 185–189 [DOI] [PubMed] [Google Scholar]
- 11. Hayashi K., Nakamura S., Nishida W., Sobue K. (2006) Bone morphogenetic protein-induced MSX1 and MSX2 inhibit myocardin-dependent smooth muscle gene transcription. Mol. Cell. Biol. 26, 9456–9470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Morita T., Mayanagi T., Sobue K. (2007) Dual roles of myocardin-related transcription factors in epithelial-mesenchymal transition via slug induction and actin remodeling. J. Cell Biol. 179, 1027–1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Medjkane S., Perez-Sanchez C., Gaggioli C., Sahai E., Treisman R. (2009) Myocardin-related transcription factors and SRF are required for cytoskeletal dynamics and experimental metastasis. Nat. Cell Biol. 11, 257–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li S., Chang S., Qi X., Richardson J. A., Olson E. N. (2006) Requirement of a myocardin-related transcription factor for development of mammary myoepithelial cells. Mol. Cell. Biol. 26, 5797–5808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Oh J., Richardson J. A., Olson E. N. (2005) Requirement of myocardin-related transcription factor-B for remodeling of branchial arch arteries and smooth muscle differentiation. Proc. Natl. Acad. Sci. U.S.A. 102, 15122–715127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hinson J. S., Medlin M. D., Lockman K., Taylor J. M., Mack C. P. (2007) Smooth muscle cell-specific transcription is regulated by nuclear localization of the myocardin-related transcription factors. Am. J. Physiol. Heart Circ. Physiol. 292, H1170–H1180 [DOI] [PubMed] [Google Scholar]
- 17. Guettler S., Vartiainen M. K., Miralles F., Larijani B., Treisman R. (2008) RPEL motifs link the serum response factor cofactor MAL but not myocardin to Rho signaling via actin binding. Mol. Cell. Biol. 28, 732–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Posern G., Treisman R. (2006) Actin' together: serum response factor, its cofactors and the link to signal transduction. Trends Cell Biol. 16, 588–596 [DOI] [PubMed] [Google Scholar]
- 19. Vartiainen M. K., Guettler S., Larijani B., Treisman R. (2007) Nuclear actin regulates dynamic subcellular localization and activity of the SRF cofactor MAL. Science 316, 1749–1752 [DOI] [PubMed] [Google Scholar]
- 20. Muehlich S., Wang R., Lee S. M., Lewis T. C., Dai C., Prywes R. (2008) Serum-induced phosphorylation of the serum response factor coactivator MKL1 by the extracellular signal-regulated kinase 1/2 pathway inhibits its nuclear localization. Mol. Cell. Biol. 28, 6302–6313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nakamura S., Hayashi K., Iwasaki K., Fujioka T., Egusa H., Yatani H., Sobue K. (2010) Nuclear import mechanism for myocardin family members and their correlation with vascular smooth muscle cell phenotype. J. Biol. Chem. 285, 37314–37323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pawłowski R., Rajakylä E. K., Vartiainen M. K., Treisman R. (2010) An actin-regulated importin α/β-dependent extended bipartite NLS directs nuclear import of MRTF-A. EMBO J. 29, 3448–3458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nakielny S., Dreyfuss G. (1997) Nuclear export of proteins and RNAs. Curr. Opin. Cell Biol. 9, 420–429 [DOI] [PubMed] [Google Scholar]
- 24. Moroianu J. (1998) Distinct nuclear import and export pathways mediated by members of the karyopherin β family. J. Cell. Biochem. 70, 231–239 [PubMed] [Google Scholar]
- 25. Yoneda Y., Hieda M., Nagoshi E., Miyamoto Y. (1999) Nucleocytoplasmic protein transport and recycling of Ran. Cell Struct. Funct. 24, 425–433 [DOI] [PubMed] [Google Scholar]
- 26. Yashiroda Y., Yoshida M. (2003) Nucleo-cytoplasmic transport of proteins as a target for therapeutic drugs. Curr. Med. Chem. 10, 741–748 [DOI] [PubMed] [Google Scholar]
- 27. Hieda M., Tachibana T., Yokoya F., Kose S., Imamoto N., Yoneda Y. (1999) A monoclonal antibody to the COOH-terminal acidic portion of Ran inhibits both the recycling of Ran and nuclear protein import in living cells. J. Cell Biol. 144, 645–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Settleman J. (2003) A nuclear MAL-function links Rho to SRF. Mol. Cell 11, 1121–1123 [DOI] [PubMed] [Google Scholar]
- 29. Deleted in proof.
- 30. Mouilleron S., Langer C. A., Guettler S., McDonald N. Q., Treisman R. (2011) Structure of a pentavalent G-actin*MRTF-A complex reveals how G-actin controls nucleocytoplasmic shuttling of a transcriptional coactivator. Sci. Signal. 4, ra40. [DOI] [PubMed] [Google Scholar]
- 31. Ransom J. F., King I. N., Garg V., Srivastava D. (2008) A rare human sequence variant reveals myocardin autoinhibition. J. Biol. Chem. 283, 35845–35852 [DOI] [PMC free article] [PubMed] [Google Scholar]