

Background: Endothelin-converting enzymes (ECEs) degrade β-amyloid (Aβ) peptide.
Results: ECE inhibition produces, in addition to extracellular Aβ accumulation, intracellular Aβ accumulation within endosomal/lysosomal and autophagic vesicles.
Conclusion: An intracellular pool of Aβ is regulated by ECE activity at the sites of production.
Significance: ECE dysfunction may cause intraneuronal Aβ accumulation, which is associated with neurotoxicity early in AD progression.
Keywords: Alzheimer Disease, Amyloid, Amyloid Precursor Protein, Autophagy, Endosomes, Metalloprotease, Protein Degradation, Proteolytic Enzymes, Endothelin-converting Enzyme
Abstract
Impairments in Aβ removal are increasingly being considered as a possible cause for the abnormal Aβ build-up typical of Alzheimer disease. Of particular interest is a pool of Aβ that accumulates intraneuronally and may contribute to neuronal toxicity. The mechanism for intraneuronal accumulation, however, is not well understood and is commonly attributed to impaired removal of extracellular Aβ by neurons. Based on the intracellular distribution of the well established Aβ degrading enzymes, ECE-1 and ECE-2, we tested whether impairments in their catalytic activity could lead to intracellular Aβ accumulation. Using SH-SY5Y cells overexpressing wild-type amyloid precursor protein and pharmacological inhibition of endogenous ECE activity, we found that ECEs participate in the degradation of at least two distinct pools of Aβ; one destined for secretion and the other being produced and degraded within the endosomal-autophagic-lysosomal pathways. Although ECE-1 regulates both pools of Aβ, ECE-2 regulates mainly the intracellular pool of the peptide. Consistent with this result, ECE-2 was found to co-localize with markers of the endosomal/lysosomal pathway but not with a trans-Golgi network marker. Furthermore, ECE-2 was detected in autophagic vesicles in cells treated with chloroquine. Under these conditions, ECE inhibition produced significantly higher elevations in intracellular Aβ than chloroquine treatment alone. This study highlights the existence of Aβ clearance mechanisms by ECEs at intracellular sites of production. Alterations in ECE activity may be considered as a cause for increased intraneuronal Aβ in Alzheimer disease.
Introduction
One of the main pathological findings in Alzheimer disease (AD)2 is the abnormal accumulation of β-amyloid peptide (Aβ) in the CNS, in different soluble states of aggregation or as extracellular insoluble deposits known as amyloid plaques (1). The accumulation is progressive and widely accepted as an important contributor to the neuronal dysfunction and ultimate neuronal loss characteristic of the disease (2). Early in the disease progression, a pool of Aβ accumulates intraneuronally before extracellular amyloid plaque formation (3–6), and mounting evidence points to intracellular Aβ (iAβ) as a possible cause of the neuronal toxicity typical of AD (7–14).
As in the case of extracellular Aβ (eAβ), the mechanism of iAβ accumulation is unknown. Based on the evidence that Aβ is constitutively secreted to the extracellular space following the sequential cleavage of amyloid precursor protein (APP) by β-secretase and γ-secretase, it has been assumed that iAβ originates from internalization of secreted Aβ. However, the ultimate γ-secretase cleavage for the release of Aβ is largely conducted at the endosome (15), thus requiring intracellular trafficking of Aβ before secretion. At the same time, Aβ-degrading enzymes are active in multiple compartments other than the lysosome and the extracellular space (16). If Aβ degradation is an important contributor to the steady-state levels of Aβ before secretion it is then possible that iAβ could accumulate due to failure in Aβ catabolism.
To date, several Aβ degrading enzymes have been shown to significantly contribute to Aβ homeostasis (17). Numerous studies in vitro and in animal models support the physiological role of insulin-degrading enzyme, neprilysin (NEP), endothelin-converting enzyme-1 (ECE-1) and ECE-2, among others, in Aβ degradation (16). NEP, ECE-1 and ECE-2 are members of the M13 family of metalloproteases, type II membrane-bound zinc metalloproteases sensitive to the inhibitor phosphoramidon (PA).
ECE-1 and ECE-2, coded by different genes, are characterized by the ability to process big endothelin-1 into the potent vasoconstrictor endothelin-1 (18). Similar to NEP, ECE-1 and ECE-2 are expressed in areas relevant to AD (19), and we have previously demonstrated that in ECE-1 and ECE-2 knock-out mice, there is an increase in endogenous levels of Aβ in brain (20).
Although NEP is expressed predominantly on the plasma membrane, the ECE family is more broadly distributed. ECE-1 is composed of four isoforms that are located on the plasma membrane as well as in different intracellular compartments, including the secretory pathway, recycling endosomes, and late endosomes (21). For ECE-2, all four isoforms are strictly intracellular, but their distribution has not yet been properly characterized. Based on the diverse intracellular distribution of the ECE family, we investigated how alterations in ECE activity could lead to iAβ accumulation in SH-SY5Y human neuroblastoma cells overexpressing wild-type APP as an in vitro neuronal model.
EXPERIMENTAL PROCEDURES
Expression Constructs
cDNAs for human wild-type amyloid precursor protein (APP)695 (NM_201414.2), ECE-1a (NM_001113347.1), ECE-1b (NM_001397.2), ECE-1c (NM_001113348.1), ECE-1d (NM_001113349.1), ECE-2 variant 2 (NM_001037324.2), and ECE-2 variant 5 (NM_001100121.1) were subcloned into pcDNA3 (Invitrogen). ECE-2 variants 2 and 5 have shown to be preferentially expressed in brain (22). Variants 2 and 5 were used for transfection of CHO cells. Variant 2 was used for studies in SH-SY5Y cells.
Cell Culture
SH-SY5Y cells stably expressing wild-type human APP695 were maintained in DMEM supplemented with 10% FBS, glutamine, penicillin/streptomycin, and the selective antibiotic geneticin at 400 μg/ml (Invitrogen). Cells were routinely passed by trypsinization. The same medium and maintenance protocol was followed for growing CHO cells. For measurement of Aβ by ELISA, medium and cell extracts were obtained from growing cells at 70–80% confluency in 12-well plates with 500 μl of medium. For transient SH-SY5Y transfection, the “nanojuice” reagent (EMD Chemicals) was used according to the manufacturer's instructions; a booster:DNA ratio of 2:1 was added to DMEM with 10% FBS containing no antibiotics, and cells were incubated for 72 h prior to any assay.
Western Blot
Cells were lysed in cold lysis buffer (PBS with 0.1% Triton X-100 and proteinase inhibitors) and protein homogenates were resolved in Novex® 10–20% Tris-tricine gels and transferred to nitrocellulose membranes. For protein detection, membranes were blocked for 1 h with 10% normal goat serum and incubated with the corresponding primary antibody. Bound antibody was detected with ECL reagents (Millipore) following incubation with the appropriate HRP-conjugated secondary antibody. Signal was digitally recorded with the ImageQuant LAS 4000 (GE Healthcare).
Immunofluorescence
Cells were fixed with cold methanol at −20 °C for 10 min. As blocking reagent, 5% BSA containing 0.1% Tween 20 was used. Following incubation with primary antibodies, Alexa Fluor 488 and 546-conjugated secondary antibodies were used for visualization under a Zeiss Axio Imager Z1 fluorescent microscope.
ELISA Measurements
Levels of Aβ1–40 were measured from conditioned medium and Triton X-100 cell extracts using a highly specific sandwich ELISA. The 33.1.1 antibody against the N-terminal region of the peptide was used for capture and the HRP-conjugated 40-specific antibody, 13.1.1, was used for detection. Aβ ending at position 42 was detected by capturing with anti-Aβ42 antibody 2.1.3 and detecting with 4G8.
Reagents and Antibodies
N-[N-(3,5-Difluorophenacetyl)-l-alanyl]-S-phenylglycine t-butyl ester (DAPT), phosphoramidon, l-thiorphan, chloroquine, and the antibodies for detection of C-terminal APP (A8717), LC3B (L7543), and TGN38 (T9826) were purchased from Sigma-Aldrich. Hoechst was purchased from AnaSpec. CGS35066 was synthesized by the Mayo Clinic Organic Synthesis Core Facility with modifications (23) from the original method (24, 25). Anti-Aβ antibodies 33.1.1 (human Aβ1–16), 13.1.1 (Aβ35–40), and 2.1.3 (Aβ35–42) were from the Mayo Clinic. 4G8-HRP was purchased from Covance. Monoclonal antibodies against rab5 (C8B1) and rab7 (D95F2) were obtained from Cell Signaling. Goat anti-ECE-1 (AF1784) and goat anti-ECE-2 (AF1645) antibodies were obtained from R&D Systems. The secondary fluorescent antibodies Alexa Fluor 488 donkey anti-mouse (A21202), anti-rabbit (A21206), and Alexa Fluor 546 donkey anti-goat (A11056) were purchased from Invitrogen. The synthetic Aβ1–40 peptide (H-1194) was purchased from Bachem.
Data Analysis and Representation
Figures are representative of individual experiments that were repeated at least three times. GraphPad Prism (version 5.01, GraphPad Software, Inc.) was used for all data analysis and graphical representation. For column graphs, column bars represent the mean ± S.E. Student's t test was used for statistical analysis and a significance of p < 0.05 was set for acceptance. Statistical significance was indicated by *, p < 0.05; **, p < 0.01; ***, p < 0.001; and ****, p < 0.0001.
RESULTS
PA Treatment Increases both eAβ and iAβ
To define the contribution of ECE activity to iAβ degradation, SH-SY5Y-APP cells expressing endogenous levels of ECEs were treated with 100 μm PA for 48 h, and levels of Aβ40 and Aβ42 were measured by ELISA in medium and cell extracts. Similar to previous studies (26, 27), we found that PA treatment produced a significant ∼3-fold elevation in the level of eAβ40, and 1.8-fold elevation in eAβ42 (Fig. 1, A and B). In the same manner, in cell extracts, PA treatment also produced significant elevations in iAβ40 and iAβ42 (Fig. 1, C and D).
FIGURE 1.

Effect of PA on extracellular and intracellular Aβ accumulation. 48-h treatment of SH-SY5Y-APP cells with 100 μm PA produced significant elevations in eAβ40 (A) and eAβ42 (B), as measured by ELISA in conditioned medium (p < 0.0001 and p = 0.0003, n = 5). eAβ40 and eAβ42 production were blocked by treatment with the γ-secretase inhibitor DAPT. PA also produced a significant accumulation of iAβ40 (C) and iAβ42 (D) in these cells (p = 0.0004 and p = 0.0225, n = 3). In cell extracts, ELISA measurements remained unchanged upon treatment with DAPT or DAPT+PA, indicating that signal measured in non-treated groups is attributable to background and that the increase in iAβ signal with PA treatment is a true increase in Aβ peptide levels. Similar to results obtained with SH-SY5Y-APP cells, treatment of H4-APP cells with PA produced significant accumulations of eAβ40 (E) (p = 0.0028, n = 3) and iAβ40 (F) (p = 0.0058, n = 3).
Demonstrating iAβ accumulation is particularly challenging due to cross-reactivity of specific Aβ antibodies with APP or cleavage products of APP that contain a portion of, or the full Aβ sequence. To define the specificity of the iAβ measurements by our ELISA system, cells were treated with the γ-secretase inhibitor DAPT to stop Aβ production and increase intracellular accumulation of C-terminal fragments (CTFS) (28). As seen in Fig. 1, A and B, levels of eAβ40 and eAβ42 after a 48-h treatment with 100 μm DAPT or PA + DAPT were nearly undetectable. For iAβ analysis in the absence of PA, Aβ40 and Aβ42 measurements were similar with and without DAPT treatment (Fig. 1, C and D), indicating a significant background signal in the cell extracts that is not due to Aβ or CTFs. Importantly, the significant increases in iAβ40 and iAβ42 measured in cells treated with PA were completely blocked by treatment with DAPT, confirming that PA produces iAβ40 and iAβ42 accumulation.
As additional confirmation of PA-mediated iAβ accumulation, we repeated the experiments with the CNS-derived H4 cell line transfected with wild-type APP (29). Similar to the results obtained with SH-SY5Y-APP cells, treatment of H4-APP cells with 100 μm PA for 48 h produced significant increases in both eAβ and iAβ levels (Fig. 1, E and F). Interestingly, H4-APP cells secrete substantially less Aβ than SH-SY5Y-APP cells and only 4-fold higher than wild-type SH-SY5Y cells (data not shown), and yet H4 cells treated with PA accumulated intracellular Aβ at levels greater than SH-SY5Y-APP cells.
To verify that the Aβ accumulation was due to impaired degradation rather than increased production, we analyzed by Western blot the levels of full-length APP and CTFs, as a measure of secretase activity. Western blot results using a C-terminal APP antibody showed that APP was preferentially processed by α-secretase, as seen by higher levels of CTF-α than CTF-β and that PA did not alter APP processing (Fig. 2B). As expected, in cells treated with DAPT, we detected a significant increase in the levels of CTFs. This increase was not affected by PA treatment. Also, by immunocytochemistry, we observed that PA treatment did not alter APP distribution in the cell (Fig. 2A), a potential cause for altered Aβ levels (15).
FIGURE 2.

Detection of intracellular Aβ accumulation by immunofluorescence and Western blot. Full-length APP and/or CTFs (shown in red) were detected by immunocytochemistry (A) in SH-SY5Y-APP cells using a C-terminal (Cter) APP antibody. PA treatment did not cause an apparent change in amount or localization of APP/CTFs within the cells consistent with Western blot data (B) using the same antibody. Immunostaining with N-terminal Aβ antibody 33.1.1 (shown in green) revealed a punctuated pattern in the cell body of PA-treated cells, whereas in control cells, only faint intracellular staining was detected. Nuclei in these cells were stained in blue (Hoechst) (A). By Western blot (C), a 4-kDa band, co-migrating with synthetic Aβ, was detected in cell lysates of PA-treated cells using both Aβ1–16 antibody 33.1.1 and Aβ17–24 antibody 4G8. This band was not detected in control cells, or in cells treated with both PA and the γ-secretase inhibitor DAPT.
Using N-terminal Aβ antibody 33.1.1 (the capture antibody for our ELISA system), we verified iAβ accumulation by immunocytochemistry. In PA-treated cells, immunoreactivity with a punctuated pattern was detected in the cell body and surrounding the nuclei (Fig. 2A), whereas in control cells, only faint intracellular staining was observed.
Finally, the accumulation of Aβ in cell extracts, detected by sandwich ELISA (Fig. 1), and immunocytochemistry (Fig. 2A) was further confirmed by Western blot using either Aβ1–16 antibody 33.1.1 or Aβ17–24 antibody 4G8. A 4-kDa immunoreactive band was detected in extracts from SH-SY5Y-APP cells treated with PA (Fig. 2C). This band was completely absent in extracts of cells co-treated with PA and DAPT.
NEP Does Not Contribute to Aβ Accumulation in SH-SY5Y Cells
PA inhibits the activity of members of the NEP and ECE families and, less potently, angiotensin-converting enzyme (30). Thus, to differentiate the contribution of the NEP and ECE family members to iAβ removal, we tested the effect of a more selective ECE inhibitor, CGS 35066 (31). As shown in Fig. 3, a 24-h treatment with CGS 35066 increased the levels of both iAβ and eAβ in a dose-dependent manner, with maximal effects similar to that of PA. In contrast, with the potent NEP inhibitor thiorphan, we found no significant increase in either iAβ or eAβ at doses up to 10 μm after 48 h. These results indicate that NEP and other thiorphan/PA-sensitive proteases do not have a major role in Aβ degradation in our cellular model, consistent with previous reports showing that NEP is poorly expressed in SH-SY5Y cells (32). Increased levels of Aβ by PA are likely due to specific inhibition of ECE family members (33), although we cannot completely rule out the presence of an unidentified ECE-like enzyme.
FIGURE 3.

Effect of CGS 35066 and thiorphan on Aβ levels in SH-SY5Y-APP cells. The more selective ECE inhibitor CGS 35066 produced dose-dependent increases in eAβ40 and iAβ40 with maximal effects similar to those of 100 μm PA. In contrast, the potent and more selective NEP inhibitor thiorphan did not alter levels of either eAβ40 or iAβ40.
Accumulated iAβ Does Not Have an Extracellular Origin
There is no clear understanding of the origin and mechanism of iAβ accumulation in neurons. One possibility is an overloading of the mechanism of eAβ removal via endocytosis. Another possibility is that Aβ accumulates due to dysfunction in an undefined step prior to secretion. To study whether PA was inhibiting the degradation of endocytosed Aβ, we treated empty vector-transfected SH-SY5Y cells with Aβ at levels similar to those secreted by SH-SY5Y-APP cells treated with PA. If iAβ originates from internalization of the extracellular pool, incubation with this amount of exogenous Aβ should produce increases in iAβ similar to those observed in SH-SY5Y-APP cells treated with PA.
Fig. 4 shows the results of 48 h of treatment with 5 nm Aβ and 100 μm PA. Levels of eAβ and iAβ were measured soon after Aβ addition and again after 48 h. Aβ measurement after the 48-h incubation period showed a dramatic decrease in the level of exogenous Aβ in the presence or absence of PA, indicating internalization or nearly complete degradation by proteases insensitive to PA. Levels of iAβ were measured to determine whether the peptide had been internalized and accumulated within the cell. There was no increase in iAβ accumulation following treatment with 5 nm Aβ, suggesting that if SH-SY5Y cells endocytose Aβ (34, 35), any internalized peptide is degraded by non-PA sensitive proteases.
FIGURE 4.

Accumulated iAβ does not have an extracellular origin. SH-SY5Y cells expressing endogenous levels of APP were incubated with 5 nm synthetic Aβ1–40 (concentration similar to that accumulated by SH-SY5Y-APP cells treated with PA). Levels of eAβ in Aβ-treated cells were decreased after 48 h in the presence or absence of PA, suggesting that eAβ removal is not dependent on ECE activity. The decrease in eAβ, however, did not reflect an increase in iAβ indicating that ECEs do not degrade internalized eAβ or that all eAβ was degraded in the extracellular space by proteases not sensitive to PA.
Aβ Accumulates within Aβ-producing Compartments
The intracellular location of members of the ECE family coincides with compartments where CTF-β is cleaved by γ-secretase to release Aβ (see “Discussion”). Thus, we tested whether iAβ accumulated in Aβ-producing compartments. Immunostaining with an antibody against the C terminus of APP revealed a punctuated staining pattern that was not seen with an N-terminal APP antibody (not shown) and co-localized with iAβ (Fig. 5). Also, the punctuated iAβ staining localized with rab7-positive compartments (marker for late endosome), suggesting that iAβ is degraded at the site of production within the late endosome.
FIGURE 5.

Aβ accumulates in cellular compartments rich in APP and/or CTFs. In PA-treated SH-SY5Y-APP cells, iAβ immunoreactivity co-localized with APP/CTFs detected with a C-terminal APP antibody and rab7, indicating that iAβ accumulation occurs at a site of production within the late endosomal/lysosomal pathway.
ECE-2 Resides in the Endosomal/Lysosomal Pathway
ECE-2 has not been as thoroughly investigated as ECE-1. Based on its low pH optimum (5–5.5) and substrate affinities similar to those of ECE-1, it has been postulated that ECE-2 resides in the TGN (36). However, this has not been demonstrated convincingly. We confirmed by RT-PCR that SH-SY5Y cells and H4 cells express ECE-2 (data not shown), but endogenous levels of the protein were only weakly detected with commercially available antibodies. Therefore, we evaluated the cellular localization of the enzyme in transfected cells by immunocytochemistry. We observed that in ECE-2 overexpressing SH-SY5Y cells, ECE-2 partially co-localized with rab5 (early endosomal marker), rab7, and LAMP-1 (lysosomal marker) but not with TGN38 (TGN marker) (Fig. 6). Results suggest that ECE-2 Aβ-degrading activity appears restricted to the endosomal/lysosomal pathway and not the secretory pathway in neuronal cells.
FIGURE 6.

Intracellular distribution of ECE-2. SH-SY5Y-APP cells transiently transfected with ECE-2 (variant 2) were co-stained with antibodies against ECE-2 and markers of various intracellular compartments. Co-localization of ECE-2 with rab5, rab7, and lamp-1, markers of early endosome, late endosome, and lysosome, respectively, demonstrated that ECE-2 is mostly located in the endosomal/lysosomal pathway. In contrast, ECE-2 did not co-localize with TGN38, a marker of the trans-Golgi network, suggesting that ECE-2 does not conduct its activity within the secretory pathway.
ECE-1b and ECE-2 Are Active in Aβ-producing Compartments and Prevent iAβ Accumulation
Based on the results above and published evidence showing that ECE-1b is also expressed in late endosomes (21), we next studied whether ECE-2 and ECE-1b directly participate in iAβ degradation. SH-SY5Y-APP cells were transiently transfected with either enzyme prior to incubation with PA. The relative inefficiency of transfection in this cell line permits the comparison of Aβ accumulation within ECE-overexpressing cells and neighboring non-transfected cells. First, we observed that both ECE-1b and ECE-2 partially co-localized with APP and/or APP CTFs (Fig. 7). In addition, in SH-SY5Y-APP cells treated with PA, overexpression of either ECE-2 or ECE-1b dramatically lowered accumulation of iAβ (expressed as the average number of Aβ-positive granules per cell) as compared with surrounding non-ECE transfected cells (Fig. 8, A and B). Even though these cells were treated with PA, we suspect that intracellular ECE activity was not completely inhibited in the overexpressing cells. This hypothesis is supported by the observation that reductions in extracellular Aβ concentration in ECE-1-transfected CHO cells are not reversed 100% by PA treatment at the concentration used in this experiment (see Fig. 10).
FIGURE 7.

Co-localization studies of ECE-2 and ECE-1b with APP. SH-SY5Y-APP cells transiently transfected with either ECE-2 or ECE-1b were stained with the corresponding ECE antibody along with a C-terminal APP antibody. Both ECE-2 and ECE-1b showed partial co-localization with compartments rich in APP and/or CTFs.
FIGURE 8.

Overexpression of ECE-2 or ECE-1b decreases iAβ accumulation. SH-SY5Y-APP cells were transiently transfected with either ECE-1b or ECE-2, treated with 100 μm PA for 48 h, then fixed and co-stained with the corresponding ECE antibody and Aβ antibody 33.1.1. The low efficiency of transfection of these cells permits analysis of iAβ accumulation in neighboring transfected and non-transfected (NT) cells. Cells that overexpressed either ECE-2 or ECE-1b showed a significant reduction in the number of iAβ positive granules per cell (p < 0.0001, n = 41; and p < 0.0001, n = 38, respectively), as compared with non-transfected cells on the same slides (n = 68).
FIGURE 10.
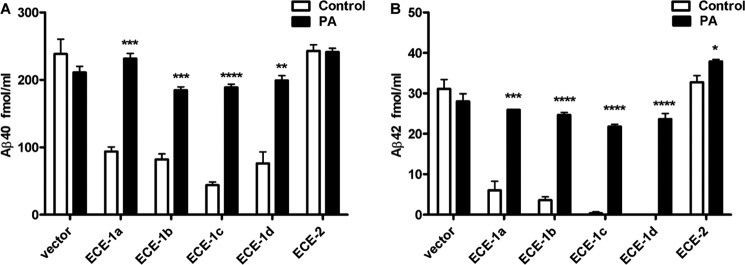
Overexpression of ECE-2 does not influence eAβ levels. A, transfection of CHO cells, which do not express endogenous ECEs, with ECE-1 isoforms a, b, c, or d, resulted in significant decreases in eAβ40 and eAβ42, which were mostly reversed by inhibition of the overexpressed enzyme with PA. In contrast, ECE-2 overexpression had little to no effect on extracellular accumulation of Aβ.
ECE Inhibition Produces Greater iAβ Accumulation under Conditions of Impaired Autophagic/Endocytic Flux
Macroautophagy is a distinct lysosomal degradative pathway specialized in the removal of long-lived proteins, organelles, and other macromolecules by the formation of double membrane vesicles known as autophagosomes (37). Autophagosomes ultimately fuse with lysosomes, resulting in degradation of their contents. Some late endosomes may fuse with autophagosomes upstream of the lysosome, and these and nascent autophagosomes can both be referred to as autophagic vesicles (AVs). There is evidence that Aβ is targeted to degradation by macroautophagy (38), whereas entrapment of membrane domains rich in γ-secretase and CTF-β within autophagosomes provides them with the capacity to produce Aβ (39, 40). Under normal conditions, autophagic-lysosomal fusion is very efficient in neurons, and autophagic vesicles are transient and difficult to detect. In AD, however, dysfunction in this pathway results in accumulation of autophagosomes in the brain (41).
After localizing ECE-1b and ECE-2 to late endosomal compartments, we next tested whether these enzymes also contribute to Aβ degradation within AVs. SH-SY5Y-APP cells were therefore treated with chloroquine (CQ), a lysosomotropic agent that produces AV accumulation by inhibiting AV/lysosomal fusion. Treatment with CQ for 48 h produced a dose-dependent decrease in eAβ levels (Fig. 9A). Although CQ decreased Aβ secretion, CQ at the highest dose (10 μm) produced a significant increase in iAβ. Furthermore, combined PA+CQ treatment produced a significant dose-dependent increase in iAβ, as compared with the treatment with PA or CQ alone.
FIGURE 9.

ECEs participate in clearance of iAβ within autophagosomes. Treatment with CQ for 48 h produced a dose-dependent decrease in eAβ40 levels, even in the presence of PA (A). For iAβ, CQ at the highest dose (10 μm) produced a significant increase in iAβ (p = 0.0075, n = 3), and combined PA+CQ treatment produced a significant dose-dependent increase in iAβ, as compared with the treatment with PA alone (p < 0.0001, n = 3 for 10 μm CQ). By immunocytochemistry (B), we observed a redistribution of the autophagosome marker LC3B (in red) from a cytosolic to a vesicular pattern in CQ-treated cells, consistent with AV accumulation. Co-immunostaining with Aβ antibody 33.1.1 (in green) demonstrated that iAβ was accumulating mainly in LC3B-positive vesicles in CQ+PA-treated cells (B). By Western blot using a C-terminal APP antibody, we observed an accumulation of CTF-α, CTF-β, and CTF-γ in CQ- and CQ+PA-treated groups, confirming the processing of APP C-terminal fragments by γ-secretase within AVs (C). In CQ-treated cells, ECE-2 (in green) co-localized within LC3B-positive vesicles (in red) in ECE-2-overexpressing cells. In contrast, we observed little co-localization of ECE-1b with LC3B in CQ-treated cells (D).
Immunocytochemical analysis of the autophagosome marker LC3B showed that treatment with CQ (5 μm) produced a redistribution from a cytosolic to a vesicular pattern, consistent with AV accumulation (Fig. 9B). Co-immunostaining with Aβ antibody 33.1.1 demonstrated that iAβ was accumulating mainly in LC3B positive vesicles in CQ+PA treated cells. Treatment with PA alone did not alter LC3B distribution (data not shown). By Western blot, we observed a similar accumulation of CTF-α, CTF-β, and CTF-γ in CQ and CQ+PA treated groups, confirming the processing of APP C-terminal fragments by γ-secretase within AVs (Fig. 9C).
ECE-2 Is Present in Autophagic Vesicles
Next, we tested whether ECE-1b and/or ECE-2 were present in AVs. As seen in Fig. 9D, upon CQ treatment, ECE-2 co-localized within LC3B positive vesicles. In contrast, we observed little co-localization of ECE-1b in these compartments. Collectively, our data supports that AVs are a source of iAβ production, but levels are tightly regulated by degradation. In this cellular model, ECE-2, but not ECE-1b, appears to be a major contributor to Aβ degradation within AVs. Our data also suggest that under certain pathological conditions, especially where autophagic/endocytic/lysosomal flux is impaired, iAβ accumulation may occur with no change, or even a decrease, in eAβ.
ECE-2 Does Not Regulate eAβ
To gain further knowledge of how iAβ degradation by ECE-1 and ECE-2 impacts secreted Aβ, we worked with CHO cells, which do not endogenously express members of the ECE family (42). Transfection of CHO cells with all the different isoforms of ECE-1 confirmed previous findings about its role in modulating eAβ (27). Overexpression of ECE-1 isoforms present on the plasma membrane (a and c) as well as intracellular isoforms (b and d) (21) significantly decreased the levels of eAβ40 and eAβ42 (Fig. 10). These effects were reversed by PA treatment, indicating that they were dependent on the activity of the enzymes.
In contrast to the effect of intracellular ECE-1 isoforms on Aβ secretion, overexpression of ECE-2 in CHO cells resulted in little or no effect on eAβ levels (Fig 10). Similar results were obtained with ECE-2 variants 2 (data not shown) and 5, both of which are expressed in brain. To confirm that transfected ECE-2 cells were expressing an active form of the enzyme, ECE activity in membrane fractions was quantified using a big endothelin-1 conversion assay (36) (data not shown). These results indicate that the trafficking and subcellular localization of intracellular ECE-1 and ECE-2 differ in ways impacting their interaction with distinct pools of Aβ. ECE-2 appears not to traffic through the secretory pathway but is active predominantly in late endosomes and AVs where it degrades an intracellular pool of Aβ not destined for secretion.
DISCUSSION
The biogenesis of Aβ is a complex multi-step process involving the traffic of APP through the TGN to the cell surface and clathrin-mediated internalization into endosomes, where the sequential β- and γ-cleavage of APP takes place. Aβ is then secreted via endosomes or the TGN. Alternatively, CTF-β fragment may traffic unprocessed from the endosome to TGN (43) or to the lysosomal pathway (44) where Aβ production may still occur. As evidence suggests, APP sorting and secretase activity are critically important factors modulating Aβ production. The steady-state levels of Aβ, however, also depend on its degradation rate. Because Aβ is believed to be continuously secreted, Aβ catabolism has primarily been considered an extracellular and lysosomal process (after internalization of extracellular Aβ). Although this may hold true for certain Aβ degrading enzymes such as NEP or cathepsins, catabolism within other cellular compartments by enzymes such as the ECEs is less well characterized.
In addition to its strong expression in endothelial cells, ECE-1 is expressed in neurons in different areas of the CNS, including areas relevant to AD, and contributes to the metabolism of different neuropeptides as well as receptor resensitization within the endosomal system (45, 46). The intracellular ECE-1d and ECE-1b isoforms are localized in recycling endosomes with ECE-1b present also in late endosomes. Both intracellular ECE-1 isoforms, however, have also been shown to be transiently expressed at the plasma membrane and secretory pathway (21).
We have previously established that ECE-1a and ECE-1b regulate extracellular levels of Aβ by degrading the peptide primarily before secretion and not in the extracellular space (27). Here, we show that all 4 ECE-1 isoforms are capable of regulating eAβ accumulation. These findings, combined with the cellular distribution of the enzymes, demonstrate that Aβ is actively degraded within the vesicles where it is produced. In addition, the ∼2–3-fold elevation in secreted Aβ upon ECE inhibition highlights ECE activity as a key factor in determining levels of secreted Aβ, perhaps as influential as secretase activity.
In this report, we also demonstrate that ECE dysfunction produces, in addition to increased eAβ, an accumulation of iAβ that can be detected by ELISA, Western blot, and immunocytochemistry. The majority of the accumulated iAβ, however, seems independent from eAβ and is not destined for secretion, judged by the ability of ECE-2 overexpression to prevent its build-up without influencing extracellular levels of the peptide. This result suggests that Aβ accumulating intracellularly is not being produced within early/recycling endosomes and instead originates from an APP pool sorted to other compartments outside of the secretory system. Co-localization of iAβ with rab7 shows the late endosome as a site of iAβ accumulation, consistent with findings in AD brains (14).
Although part of the Aβ produced within recycling endosomes could be routed through the late endosomal/lysosomal pathway, the specific accumulation of CTFs in the late endosome and co-localization with iAβ demonstrates that iAβ is produced within this pathway. Therefore, parallel to ECE-1 cleavage of eAβ prior to secretion, ECE-1b (although we do not exclude other isoforms) and ECE-2 appear to degrade Aβ at the late endosomal site of production. Furthermore, under conditions of impaired lysosomal flux, ECE activity helps to prevent excessive Aβ accumulation within AVs. Co-localization of ECE-2 with LC3B suggests that ECE-2 is at least partially responsible for Aβ clearance within the autophagic pathway. Our results demonstrate that efficient degradation of iAβ occurs upstream of the lysosome.
In conclusion, we demonstrate that a large amount of Aβ is degraded presecretion, challenging the concept that all Aβ is constitutively secreted after production. We also show that inhibition of ECE activity reveals an elusive pool of iAβ that is produced within the endosomal/lysosomal pathway but efficiently degraded by ECEs under normal conditions. The possible physiological relevance of this iAβ pool remains to be established. Considering the results, we propose that intraneuronal Aβ in AD does not originate from internalization of secreted peptide but from impairments in ECE activity. As alterations in the endosomal/lysosomal and autophagic pathways are observed early in the AD course and coincide with iAβ accumulation (6, 14, 47), they could represent an underlying cause of ECE dysfunction and increase in iAβ.
Acknowledgments
We thank Robert Roman, Yunji Kang, Katie Smalley, Colette Whitney, and Daniel Shulkin for technical assistance with this study.
This work was supported by NINDS, National Institutes of Health Grant R01 NS073512 (to E. A. E.).
- AD
- Alzheimer disease
- ECE
- endothelin-converting enzyme
- Aβ
- β-amyloid
- iAβ
- intracellular Aβ
- eAβ
- extracellular Aβ
- NEP
- neprilysin
- PA
- phosphoramidon
- TGN
- trans-Golgi network
- APP
- amyloid precursor protein
- DAPT
- N-[N-(3,5-Difluorophenacetyl)-l-alanyl]-S-phenylglycine t-butyl ester
- CTF
- C-terminal fragment
- AV
- autophagic vesicle
- CQ
- chloroquine.
REFERENCES
- 1. Glenner G. G., Wong C. W., Quaranta V., Eanes E. D. (1984) The amyloid deposits in Alzheimer's disease: their nature and pathogenesis. Appl. Pathol. 2, 357–369 [PubMed] [Google Scholar]
- 2. Hardy J., Selkoe D. J. (2002) The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297, 353–356 [DOI] [PubMed] [Google Scholar]
- 3. Grundke-Iqbal I., Iqbal K., George L., Tung Y. C., Kim K. S., Wisniewski H. M. (1989) Amyloid protein and neurofibrillary tangles coexist in the same neuron in Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 86, 2853–2857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gouras G. K., Tsai J., Naslund J., Vincent B., Edgar M., Checler F., Greenfield J. P., Haroutunian V., Buxbaum J. D., Xu H., Greengard P., Relkin N. R. (2000) Intraneuronal Aβ42 accumulation in human brain. Am. J. Pathol. 156, 15–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gyure K. A., Durham R., Stewart W. F., Smialek J. E., Troncoso J. C. (2001) Intraneuronal aβ-amyloid precedes development of amyloid plaques in Down syndrome. Arch. Pathol. Lab. Med. 125, 489–492 [DOI] [PubMed] [Google Scholar]
- 6. Cataldo A. M., Petanceska S., Terio N. B., Peterhoff C. M., Durham R., Mercken M., Mehta P. D., Buxbaum J., Haroutunian V., Nixon R. A. (2004) Aβ localization in abnormal endosomes: association with earliest Aβ elevations in AD and Down syndrome. Neurobiol. Aging 25, 1263–1272 [DOI] [PubMed] [Google Scholar]
- 7. Skovronsky D. M., Doms R. W., Lee V. M. (1998) Detection of a novel intraneuronal pool of insoluble amyloid β protein that accumulates with time in culture. J. Cell Biol. 141, 1031–1039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lord A., Kalimo H., Eckman C., Zhang X. Q., Lannfelt L., Nilsson L. N. (2006) The Arctic Alzheimer mutation facilitates early intraneuronal Aβ aggregation and senile plaque formation in transgenic mice. Neurobiol. Aging 27, 67–77 [DOI] [PubMed] [Google Scholar]
- 9. Oddo S., Caccamo A., Smith I. F., Green K. N., LaFerla F. M. (2006) A dynamic relationship between intracellular and extracellular pools of Aβ. Am. J. Pathol. 168, 184–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Abramowski D., Rabe S., Upadhaya A. R., Reichwald J., Danner S., Staab D., Capetillo-Zarate E., Yamaguchi H., Saido T. C., Wiederhold K. H., Thal D. R., Staufenbiel M. (2012) Transgenic expression of intraneuronal Aβ42 but not Aβ40 leads to cellular Aβ lesions, degeneration, and functional impairment without typical Alzheimer's disease pathology. J. Neurosci. 32, 1273–1283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Billings L. M., Oddo S., Green K. N., McGaugh J. L., LaFerla F. M. (2005) Intraneuronal Aβ causes the onset of early Alzheimer's disease-related cognitive deficits in transgenic mice. Neuron 45, 675–688 [DOI] [PubMed] [Google Scholar]
- 12. D'Andrea M. R., Nagele R. G., Wang H. Y., Peterson P. A., Lee D. H. (2001) Evidence that neurones accumulating amyloid can undergo lysis to form amyloid plaques in Alzheimer's disease. Histopathology 38, 120–134 [DOI] [PubMed] [Google Scholar]
- 13. Steinerman J. R., Irizarry M., Scarmeas N., Raju S., Brandt J., Albert M., Blacker D., Hyman B., Stern Y. (2008) Distinct pools of β-amyloid in Alzheimer disease-affected brain: a clinicopathologic study. Arch. Neurol. 65, 906–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Takahashi R. H., Milner T. A., Li F., Nam E. E., Edgar M. A., Yamaguchi H., Beal M. F., Xu H., Greengard P., Gouras G. K. (2002) Intraneuronal Alzheimer aβ42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am. J. Pathol. 161, 1869–1879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Small S. A., Gandy S. (2006) Sorting through the cell biology of Alzheimer's disease: intracellular pathways to pathogenesis. Neuron 52, 15–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Eckman E. A., Eckman C. B. (2005) Aβ-degrading enzymes: modulators of Alzheimer's disease pathogenesis and targets for therapeutic intervention. Biochem. Soc. Trans. 33, 1101–1105 [DOI] [PubMed] [Google Scholar]
- 17. Saido T., Leissring M. A. (2012) Proteolytic Degradation of Amyloid β-Protein. Cold Spring Harb. Perspect. Med. 2, a006379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Turner A. J., Murphy L. J. (1996) Molecular pharmacology of endothelin converting enzymes. Biochem. Pharmacol 51, 91–102 [DOI] [PubMed] [Google Scholar]
- 19. Pacheco-Quinto J., Herdt A., Eckman C. B., Eckman E. A. (2012) Endothelin-Converting Enzymes and Related Metalloproteases in Alzheimer's Disease. J. Alzheimers Dis. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Eckman E. A., Watson M., Marlow L., Sambamurti K., Eckman C. B. (2003) Alzheimer's disease β-amyloid peptide is increased in mice deficient in endothelin-converting enzyme. J. Biol. Chem. 278, 2081–2084 [DOI] [PubMed] [Google Scholar]
- 21. Muller L., Barret A., Etienne E., Meidan R., Valdenaire O., Corvol P., Tougard C. (2003) Heterodimerization of endothelin-converting enzyme-1 isoforms regulates the subcellular distribution of this metalloprotease. J. Biol. Chem. 278, 545–555 [DOI] [PubMed] [Google Scholar]
- 22. Ikeda S., Emoto N., Alimsardjono H., Yokoyama M., Matsuo M. (2002) Molecular isolation and characterization of novel four subisoforms of ECE-2. Biochem. Biophys. Res. Commun. 293, 421–426 [DOI] [PubMed] [Google Scholar]
- 23. Fauq A. H., Khan M. A., Eckman C. B. (2004) A concise, high yield synthesis of ECE-inhibitor CGS 35066. Synth. Commun. 34, 775–782 [Google Scholar]
- 24. De Lombaert S., Blanchard L., Stamford L. B., Tan J., Wallace E. M., Satoh Y., Fitt J., Hoyer D., Simonsbergen D., Moliterni J., Marcopoulos N., Savage P., Chou M., Trapani A. J., Jeng A. Y. (2000) Potent and selective non-peptidic inhibitors of endothelin-converting enzyme-1 with sustained duration of action. J. Med. Chem. 43, 488–504 [DOI] [PubMed] [Google Scholar]
- 25. De Lombaert S., Blanchard L., Berry C., Ghai R. D., Trapani A. J. (1995) Non-peptidic inhibitors of neutral endopeptidase 24.11.2 Design and pharmacology of orally active phosphonate prodrugs. Bioorg. Med. Chem. Lett. 5, 151–154 [Google Scholar]
- 26. Fuller S. J., Storey E., Li Q. X., Smith A. I., Beyreuther K., Masters C. L. (1995) Intracellular production of β A4 amyloid of Alzheimer's disease: modulation by phosphoramidon and lack of coupling to the secretion of the amyloid precursor protein. Biochemistry 34, 8091–8098 [DOI] [PubMed] [Google Scholar]
- 27. Eckman E. A., Reed D. K., Eckman C. B. (2001) Degradation of the Alzheimer's amyloid β peptide by endothelin-converting enzyme. J. Biol. Chem. 276, 24540–24548 [DOI] [PubMed] [Google Scholar]
- 28. Dovey H. F., John V., Anderson J. P., Chen L. Z., de Saint Andrieu P., Fang L. Y., Freedman S. B., Folmer B., Goldbach E., Holsztynska E. J., Hu K. L., Johnson-Wood K. L., Kennedy S. L., Kholodenko D., Knops J. E., Latimer L. H., Lee M., Liao Z., Lieberburg I. M., Motter R. N., Mutter L. C., Nietz J., Quinn K. P., Sacchi K. L., Seubert P. A., Shopp G. M., Thorsett E. D., Tung J. S., Wu J., Yang S., Yin C. T., Schenk D. B., May P. C., Altstiel L. D., Bender M. H., Boggs L. N., Britton T. C., Clemens J. C., Czilli D. L., Dieckman-McGinty D. K., Droste J. J., Fuson K. S., Gitter B. D., Hyslop P. A., Johnstone E. M., Li W. Y., Little S. P., Mabry T. E., Miller F. D., Audia J. E. (2001) Functional γ-secretase inhibitors reduce β-amyloid peptide levels in brain. J. Neurochem. 76, 173–181 [DOI] [PubMed] [Google Scholar]
- 29. Haugabook S. J., Yager D. M., Eckman E. A., Golde T. E., Younkin S. G., Eckman C. B. (2001) High throughput screens for the identification of compounds that alter the accumulation of the Alzheimer's amyloid β peptide (Aβ). J. Neurosci. Methods 108, 171–179 [DOI] [PubMed] [Google Scholar]
- 30. Kukkola P. J., Savage P., Sakane Y., Berry J. C., Bilci N. A., Ghai R. D., Jeng A. Y. (1995) Differential structure-activity relationships of phosphoramidon analogues for inhibition of three metalloproteases: endothelin-converting enzyme, neutral endopeptidase, and angiotensin-converting enzyme. J. Cardiovasc. Pharmacol. 26, S65–68 [PubMed] [Google Scholar]
- 31. Jeng A. Y., De Lombaert S., Beil M. E., Bruseo C. W., Savage P., Chou M., Trapani A. J. (2000) Design and synthesis of a potent and selective endothelin-converting enzyme inhibitor, CGS 35066. J. Cardiovasc. Pharmacol. 36, S36–39 [DOI] [PubMed] [Google Scholar]
- 32. Belyaev N. D., Nalivaeva N. N., Makova N. Z., Turner A. J. (2009) Neprilysin gene expression requires binding of the amyloid precursor protein intracellular domain to its promoter: implications for Alzheimer disease. EMBO Rep. 10, 94–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Eckman E. A., Adams S. K., Troendle F. J., Stodola B. A., Kahn M. A., Fauq A. H., Xiao H. D., Bernstein K. E., Eckman C. B. (2006) Regulation of steady-state β-amyloid levels in the brain by neprilysin and endothelin-converting enzyme but not angiotensin-converting enzyme. J. Biol. Chem. 281, 30471–30478 [DOI] [PubMed] [Google Scholar]
- 34. Ida N., Masters C. L., Beyreuther K. (1996) Rapid cellular uptake of Alzheimer amyloid βA4 peptide by cultured human neuroblastoma cells. FEBS Lett. 394, 174–178 [DOI] [PubMed] [Google Scholar]
- 35. Hu X., Crick S. L., Bu G., Frieden C., Pappu R. V., Lee J. M. (2009) Amyloid seeds formed by cellular uptake, concentration, and aggregation of the amyloid-β peptide. Proc. Natl. Acad. Sci. U.S.A. 106, 20324–20329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Emoto N., Yanagisawa M. (1995) Endothelin-converting enzyme-2 is a membrane-bound, phosphoramidon-sensitive metalloprotease with acidic pH optimum. J. Biol. Chem. 270, 15262–15268 [DOI] [PubMed] [Google Scholar]
- 37. Yang Z., Klionsky D. J. (2009) An overview of the molecular mechanism of autophagy. Curr. Top. Microbiol. Immunol. 335, 1–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lünemann J. D., Schmidt J., Schmid D., Barthel K., Wrede A., Dalakas M. C., Münz C. (2007) β-amyloid is a substrate of autophagy in sporadic inclusion body myositis. Ann. Neurol. 61, 476–483 [DOI] [PubMed] [Google Scholar]
- 39. Vingtdeux V., Hamdane M., Loyens A., Gelé P., Drobeck H., Bégard S., Galas M. C., Delacourte A., Beauvillain J. C., Buée L., Sergeant N. (2007) Alkalizing drugs induce accumulation of amyloid precursor protein by-products in luminal vesicles of multivesicular bodies. J. Biol. Chem. 282, 18197–18205 [DOI] [PubMed] [Google Scholar]
- 40. Yu W. H., Cuervo A. M., Kumar A., Peterhoff C. M., Schmidt S. D., Lee J. H., Mohan P. S., Mercken M., Farmery M. R., Tjernberg L. O., Jiang Y., Duff K., Uchiyama Y., Näslund J., Mathews P. M., Cataldo A. M., Nixon R. A. (2005) Macroautophagy–a novel β-amyloid peptide-generating pathway activated in Alzheimer's disease. J. Cell Biol. 171, 87–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nixon R. A., Yang D. S. (2011) Autophagy failure in Alzheimer's disease–locating the primary defect. Neurobiol. Dis. 43, 38–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Xu D., Emoto N., Giaid A., Slaughter C., Kaw S., deWit D., Yanagisawa M. (1994) ECE-1: a membrane-bound metalloprotease that catalyzes the proteolytic activation of big endothelin-1. Cell 78, 473–485 [DOI] [PubMed] [Google Scholar]
- 43. Choy R. W., Cheng Z., Schekman R. (2012) Amyloid precursor protein (APP) traffics from the cell surface via endosomes for amyloid β (Aβ) production in the trans-Golgi network. Proc. Natl. Acad. Sci. U.S.A. 109, E2077–2082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pasternak S. H., Callahan J. W., Mahuran D. J. (2004) The role of the endosomal/lysosomal system in amyloid-β production and the pathophysiology of Alzheimer's disease: reexamining the spatial paradox from a lysosomal perspective. J. Alzheimers. Dis. 6, 53–65 [DOI] [PubMed] [Google Scholar]
- 45. Roosterman D., Cottrell G. S., Padilla B. E., Muller L., Eckman C. B., Bunnett N. W., Steinhoff M. (2007) Endothelin-converting enzyme 1 degrades neuropeptides in endosomes to control receptor recycling. Proc. Natl. Acad. Sci. U.S.A. 104, 11838–11843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cattaruzza F., Cottrell G. S., Vaksman N., Bunnett N. W. (2009) Endothelin-converting enzyme 1 promotes re-sensitization of neurokinin 1 receptor-dependent neurogenic inflammation. Br. J. Pharmacol. 156, 730–739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nixon R. A., Yang D. S. (2012) Autophagy and neuronal cell death in neurological disorders. Cold Spring Harb. Perspect. Biol. 4, a008839. [DOI] [PMC free article] [PubMed] [Google Scholar]