

Background: Rhesus proteins transport NH3 and/or NH4+ in heterologous expression systems.
Results: Heterozygous Rhcg mice develop delayed metabolic acidosis, whereas homozygous KO mice display severe metabolic acidosis. RhCG functions as an NH3 transporter on apical and basolateral membranes.
Conclusion: RhCG is an NH3 but not NH4+ transporter.
Significance: Loss or reduced expression of RhCG may underlie inherited or acquired forms of human acidosis.
Keywords: Acidosis, Ammonia, Animal Models, Kidney Metabolism, Membrane Transport, Collecting Duct
Abstract
Ammonia secretion by the collecting duct (CD) is critical for acid-base homeostasis and, when defective, causes distal renal tubular acidosis (dRTA). The Rhesus protein RhCG mediates NH3 transport as evident from cell-free and cellular models as well as from Rhcg-null mice. Here, we investigated in a Rhcg mouse model the metabolic effects of Rhcg haploinsufficiency, the role of Rhcg in basolateral NH3 transport, and the mechanisms of adaptation to the lack of Rhcg. Both Rhcg+/+ and Rhcg+/− mice were able to handle an acute acid load, whereas Rhcg−/− mice developed severe metabolic acidosis with reduced ammonuria and high mortality. However, chronic acid loading revealed that Rhcg+/− mice did not fully recover, showing lower blood HCO3− concentration and more alkaline urine. Microperfusion studies demonstrated that transepithelial NH3 permeability was reduced by 80 and 40%, respectively, in CDs from Rhcg−/− and Rhcg+/− mice compared with controls. Basolateral membrane permeability to NH3 was reduced in CDs from Rhcg−/− mice consistent with basolateral Rhcg localization. Rhcg−/− responded to acid loading with normal expression of enzymes and transporters involved in proximal tubular ammoniagenesis but reduced abundance of the NKCC2 transporter responsible for medullary accumulation of ammonium. Consequently, tissue ammonium content was decreased. These data demonstrate a role for apical and basolateral Rhcg in transepithelial NH3 transport and uncover an incomplete dRTA phenotype in Rhcg+/− mice. Haploinsufficiency or reduced expression of RhCG may underlie human forms of (in)complete dRTA.
Introduction
Ammonium (NH4+) is the main component of urinary acid excretion. Renal synthesis and excretion of NH4+ rise in response to an acid load, allowing kidneys to regenerate bicarbonate and increase net acid excretion (1, 2). Impaired renal acid excretion characterizes type I distal renal tubular acidosis (dRTA)3 with low urinary ammonium and inappropriately alkaline urinary pH (3).
Ammonium is formed in the proximal tubule from the metabolism of glutamine and added to the luminal fluid. It is reabsorbed into the medullary interstitium in the thick ascending limb creating a cortico-papillary NH3/NH4+ gradient (4, 5). The final step of NH3/NH4+ excretion is achieved by the CD (6). The high tissue concentration of NH3/NH4+ and the pH gradient between interstitium and urine provide the driving forces for NH4+ excretion into urine. NH4+ secretion results from the trapping of NH3 in the tubular lumen after being titrated by H+ ions stemming from active secretion by V-type H+-ATPases (7).
The mechanisms mediating NH3/NH4+ transport across cell membranes into urine have only recently become uncovered. Members of the Rhesus protein family have been identified as pathways for NH3/NH4+ transport in yeast, plants, fish, and mammals (2, 5, 8). In the kidney CD, RhCG and RhBG are expressed (9, 10). Mice lacking Rhbg show either no phenotype or only a very mild reduction in urinary ammonium excretion (11, 12). In contrast, cell-specific or complete Rhcg deficiency causes a massive reduction in urinary ammonium excretion in three different mouse models (13–15). Microperfusion experiments using CDs from Rhcg-deficient mice demonstrated that RhCG is critical for the apical exit of NH3 into urine (13). Thus far, a phenotype of dRTA has only been reported in Rhcg−/− mice, whereas potential changes caused by haploinsufficiency in Rhcg have not been investigated. This issue is relevant because heterozygous abnormalities in RhCG might be more frequent and may affect the renal capacity to cope with an acid load leading to incomplete dRTA. Furthermore, Rhcg+/− mice may also serve as a model to examine the consequences of reduced RhCG expression that may occur during kidney disease. Finally, RhCG localization has been controversial for years. RhCG has been localized by some groups only at the apical side of CD cells, whereas others have found RhCG on both the apical and basolateral membranes (9, 10, 16, 17). The functionality of basolateral RhCG protein remains unknown.
Here, we used a novel Rhcg mouse model to provide the first evidence that haploinsufficiency in Rhcg impairs the handling of a chronic acid load in Rhcg+/− mice, which develop an incomplete metabolic acidosis. Microperfusion studies provide the functional basis of the defect and show that RhCG is absolutely required for apical and partially for basolateral NH3 transport. Moreover, loss of Rhcg is associated with a profound down-regulation of NKCC2 and reduced medullary accumulation of ammonium impairing the gradient necessary for the final excretory step. These data provide new insights into the complex role of RhCG and suggest that congenital or acquired defects in RhCG protein expression may be associated with incomplete dRTA.
EXPERIMENTAL PROCEDURES
Animals
Rhcg+/− mice were purchased from the Texas Institute of Genomic Medicine (Houston TX). Mice were generated by replacing exon 1 by a vector carrying a LacZ/Neo cassette (Fig. 1A). Mice were genotyped by PCR directly on a 3-μl 25 mm NaOH tail digestion product. Genomic DNA was amplified using primer pairs specific for exon 1 forward (AGACCCCACAATGGAAAGCTATAA), wild type reverse (CAACCAGAACTCCCCAGTGTCAGA), and knock-out reverse (ATGGGCTGACCGCTTCCTCGTGCTTTAC). The products were separated by electrophoresis in 1% agarose gel (mutant product, 522 bp; wild type product, 376 bp). Mice were generated by mating Rhcg+/− mice, and mice were bred in the Êntres Vivants Exempts d'Organismes Pathogènes Spécifiques Animal Facility. For acid-loading experiments, mice in metabolic cages were given 0.2 m HCl added to powdered standard food. All experiments were performed according to Swiss Animal Welfare laws and approved by the local veterinary authority (Veterinäramt Zürich).
FIGURE 1.

Rhcg gene targeting and deletion. A, Rhcg gene knock-out was achieved by replacement of exon 1 by a LacZ/neomycin cassette (Texas Institute of Genomic Medicine, Houston, TX). B, RT-quantitative PCR with primers placed in exon 1 of Rhcg demonstrated lower mRNA in kidneys from Rhcg+/− mice and absence of a detectable PCR product in kidneys from Rhcg−/− mice (n = 5 mice). C and D, Rhcg immunodetection in kidney sections from wild type (C) and Rhcg−/− (D) mice. * indicates significantly different p < 0.05.
In Vivo Experiments
All experiments were performed using age- and sex-matched Rhcg wild type (Rhcg+/+), Rhcg knock-out (Rhcg−/−), and Rhcg heterozygote (Rhcg+/−) littermate mice (3–4 month-old) that were housed in metabolic cages (Techniplast, Switzerland). Mice were given deionized water ad libitum and were fed with a standard powdered laboratory chow (Kliba, Augst, Switzerland). Mice were allowed to adapt to metabolic cages for 3 days, and a first retro-orbital blood sample was taken for blood gas analysis under base line. Then two 24-h urine samples were collected under light mineral oil in the urine collector to determine daily urinary parameters. Mice were then allowed to recover for 2 weeks before giving an HCl-containing diet (0.2 m HCl added to powdered standard food) in normal cages. Food, water intake, and urine excretion were monitored following the same procedures as under base-line conditions. Urine collections were performed on the 1st and 2nd day of acid loading and then on the 6th and 7th day. Retro-orbital blood samples were taken on the 2nd and 7th days of HCl diet.
Analytic Procedures
Blood pH, pCO2, and electrolytes were measured with a pH/blood-gas analyzer (ABL 77 Radiometer). Urinary Na+ and K+ concentrations were measured by flame photometry (IL943, Instruments Laboratory); titratable acids were measured using a DL 50 titrator (Mettler Toledo) (18, 19) and creatinine by a modified kinetic Jaffé colorimetric method (20). Urinary pH and bicarbonate were measured with a pH/blood-gas analyzer (ABL 725, Radiometer). Urinary NH4+ was measured with the Berthelot protocol (21).
Immunoblotting
Crude membrane proteins or cytosolic fractions were obtained from kidneys homogenized in 250 mm sucrose, 10 mm Tris-HCl, pH 7.5, and in the presence of protease inhibitors.
Forty micrograms of crude membrane proteins or cytosolic proteins were solubilized in loading buffer containing DTT and separated on 8–10% polyacrylamide gels. For immunoblotting, the proteins were transferred electrophoretically to polyvinylidene fluoride membranes (Immobilon-P, Millipore Corp., Bedford, MA). After blocking with 5% milk powder in Tris-buffered saline, 0.1% Tween 20 for 60 min, the blots were incubated with primary antibodies overnight at 4 °C as follows: phosphate-dependent glutaminase (PDG), which recognizes both the rat (KGA) and human (GAC) kidney-type isoforms of PDG forming the mature PDG protein (66 and 68 kDa; a kind gift from N. Curthoys, Colorado State University; diluted 1:5000) (22); rabbit polyclonal anti-PEPCK (Cayman Chemical, Ann Arbor, MI; diluted 1:5000); rabbit polyclonal anti-NKCC2 (kind gift from Johannes Loffing, Institute of Anatomy, University of Zurich; diluted 1:5000); rabbit polyclonal anti-NHE3 (StressMarq Biosciences Inc., Victoria, British Columbia, Canada); rabbit polyclonal anti-pendrin (Pineda Antibody Service, Berlin, Germany, diluted 1:5000) (23); and mouse monoclonal anti-β-actin antibody (Sigma; diluted 1:5000). After washing and blocking with 5% milk powder for 60 min, membranes were then incubated for 2 h at room temperature with secondary goat anti-rabbit or donkey anti-mouse antibodies (diluted 1:5000) linked to alkaline phosphatase (Promega, Madison, WI). The protein signal was detected with the appropriate substrate (Millipore Corp, Bedford, MA) using the las-4000 image analyzer system (Fujifilm Life Science). All images were analyzed using the software Advanced Image Data Analyzer AIDA (Raytest, Straubenhardt, Germany) to calculate the protein of interest/β-actin ratio.
Immunostaining and Immunogold
Immunohistochemistry and immunogold staining were performed on frozen sections, and specificity was demonstrated on Rhcg−/− tissues. Mouse kidneys were fixed by perfusion retrograde through the aorta with 3% paraformaldehyde in 0.1 m sodium cacodylate buffer, pH 7.2. The tissue was either trimmed into small blocks, further fixed by immersion in 1% paraformaldehyde, infiltrated with 2.3 m sucrose for 30 min, and frozen in liquid nitrogen or prepared for routine paraffin embedding.
For electron microscopy, 70–90-nm cryosections were obtained at −100 °C with an FCS Reichert Ultracut S cryo-ultramicrotome as described previously (24), and 2-μm paraffin sections were obtained with a Leica RM2165 microtome. For LM immunolabeling, the sections were incubated with a rabbit polyclonal antibody against RhCG (a kind gift from Dr. Yves Colin, INSERM, Paris, France (16, 25)) at room temperature for 1 h after preincubation in PBS containing 0.05 m glycine and 1% bovine serum albumin. The sections were subsequently incubated with peroxidase-conjugated secondary antibody (Dako); the peroxidase was visualized with diaminobenzidine, and the sections were counter-stained with Maier's stain for 2 min. The sections were examined in a Leica DMR microscope equipped with a Leica DFC320 camera. Images were transferred by a Leica TFC Twain 6.1.0 program and processed using Adobe Photoshop 8.0. For EM immunolabeling, the sections were incubated with the primary antibody at 4 °C overnight followed by incubation at room temperature for 1 h with 10-nm gold particles coupled to anti-rabbit IgG (BioCell, Cardiff, UK). The cryosections were embedded in methylcellulose containing 0.3% uranyl acetate and studied under a Philips CM100 electron microscope. For controls, sections were incubated with secondary antibodies alone or with nonspecific IgG.
RNA Extraction and Reverse Transcription
Snap-frozen kidneys (five kidneys for each condition) were homogenized in RLT-Buffer (Qiagen, Basel, Switzerland) supplemented with β-mercaptoethanol to a final concentration of 1%. Total RNA was extracted from 200-μl aliquots of each homogenized sample using the RNeasy mini kit (Qiagen, Basel, Switzerland) according to the manufacturer's instructions. Quality and concentration of the isolated RNA preparations were analyzed on the ND-1000 spectrophotometer (Nano-Drop Technologies). Total RNA samples were stored at −80 °C. Each RNA sample was diluted to 100 ng/μl, and 3 μl was used as a template for reverse transcription using the TaqMan reverse transcription kit (Applied Biosystems, Foster City, CA). For reverse transcription, 300 ng of RNA template were diluted in a 40-μl reaction mix that contained (final concentrations) RT buffer (1×), MgCl2 (5.5 mm), random hexamers (2.5 μm), RNase inhibitor (0.4 unites/μl), the multiscribe reverse transcriptase enzyme (1.25 units/μl), dNTP mix (500 μm each), and RNase-free water.
Real Time Quantitative PCR
Quantitative real time quantitative RT-PCR was performed on the ABI PRISM 7700 sequence detection system (Applied Biosystems, Foster City, CA). Primers were chosen using the BLAST tool of Ensemble to result in amplicons no longer than 150 bp spanning intron-exon boundaries to exclude genomic DNA contamination. The specificity of all primers was first tested on mRNA derived from kidney and always resulted in a single product of the expected size (data not shown). Probes were labeled with the reporter dye 6-carboxyfluorescein at the 5′ end and the quencher dye 5-(and 6-)-carboxytetramethylrhodamine at the 3′ end (Microsynth, Balgach, Switzerland). The primers were designed to target the deleted sequence in Rhcg−/− animals (accession number NM_019799) with 5′-ATGCAGGGATGGTTCCATTA-3′ (238–258 bp) as the left primer located within exon 1 and 5′-TGGAAGAAGGTCATAATGAGCAG-3′ (373–392 bp) as the right primer located within exon 2, and 5′-TTACTATCGCTACCCGAGCTTCCAG-3′ as the probe (292–317 bp). Real time PCRs were performed using TaqMan Universal PCR master mix (Applied Biosystems, Foster City, CA). Briefly, 3 μl of cDNA, 0.8 μl of each primer (25 μm), 0.4 μl of labeled probe (5 μm), 5 μl of RNase-free water, and 10 μl of TaqMan Universal PCR master mix reached 20 μl of final reaction volume. Reaction conditions were denaturation at 95 °C for 10 min followed by 40 cycles of denaturation at 95 °C for 15 s and annealing/elongation at 60 °C for 60 s with auto ramp time. All reactions were run in triplicate. For analyzing the data, the threshold was set to 0.2 as this value had been determined to be in the linear range of the amplification curves for all mRNAs in all experimental runs. The expression of the genes of interest was calculated in relation to hypoxanthine-guanine phosphoribosyltransferase (accession number NM_013556, forward primer, 5′-TTATCAGACTGAAGAGCTACTGTAAGATC-3′ (442–471), reverse primer, 5′-TTACCAGTGTCAATTATATCTTCAACAATC-3′ (539–568), and probe, 5′-TGAGAGATCATCTCCACCAATAACTTTTATGTCCC-3′(481–515)). Relative expression ratios were calculated as R = 2(Ct(HPRT) − Ct(test gene), where Ct represents the cycle number at the threshold 0.02; and HPRT is hypoxanthine-guanine phosphoribosyltransferase.
Measurement of Renal Ammonia Content
The renal tissue ammonia content was measured by an enzymatic technique (ammonia assay kit, Sigma) as described previously (26). Mice were anesthetized, and the kidneys were removed and immediately frozen in liquid nitrogen. They were then sliced frozen to yield a column of tissue, which extended from the cortex to the tip of the papilla. Sections were cut along the corticomedullary axis to yield three slices as follows: cortex, outer medulla, and inner medulla. Two kidneys from the same animal were pooled for each sample. Tissue slices were then homogenized in 300 μl of ice-cold 7% trichloroacetic acid, and the solution was centrifuged. The supernatant was drawn off, and the pH of a 250-μl sample was adjusted to near neutral by the addition of 12 μl of 10 mm Na2HPO4 in 9 n NaOH. A 200-μl sample of buffered supernatant was then analyzed for ammonium. The pellet was resuspended in 1 n NaOH, shaken overnight, and analyzed for total protein using the Bio-Rad protein assay.
Microperfusion Studies on Isolated Tubules
Mice were anesthetized with 50 mg/kg pentobarbital sodium or xylazine/ketamine intraperitoneally. Both kidneys were cooled in situ with control bath solution (see below) for 1 min and then removed and cut into thin coronal slices for tubule dissection. CCDs were dissected from the cortex at 10 °C in the control solution. In vitro microperfusion of single CCD segments, intracellular pH, and transepithelial NH3 permeability measurements were performed as described previously (13).
Intracellular pH Measurement
The isolated tubule was transferred to the bath chamber on the stage of an inverted microscope (Axiovert 200, Carl Zeiss, Germany) in the control solution containing (in mm) 138 NaCl, 1.5 CaCl2, 1.2 MgSO4, 2 K2HPO4, 10 HEPES, 5.5 glucose, 5 alanine, pH 7.40, and then was mounted on concentric pipettes and perfused in vitro with Na+-free, ammonium-free solution, where N-methyl-d-glutamine+ (NMDG+) replaced Na+. All solutions were equilibrated with 100% O2 passed through a 3 n KOH CO2 trap. Once the solutions were gassed and the pH checked, they were placed in a reservoir and were continuously bubbled with 100% O2. The average tubule length exposed to bath fluid was limited to 300–350 μm to prevent motion of the tubule. CCDs or OMCDs were loaded with 5 μm of the fluorescent probe 2′,7′-bis(2-carboxyl)-5-(and-6)-carboxyfluorescein (BCECF Invitrogen) for ∼20 min at 37 °C in the control bath solution. The loading solution was then washed out by initiation of bath flow, and the tubule was equilibrated with dye-free control bath solution for 5 min. Bath solution was delivered at a rate of 20 ml/min and warmed to 37 °C by a water jacket immediately upstream to the chamber. After this temperature equilibration in control solution, tubules were first transiently acidified by peritubular Na+ removal (sodium-free, ammonium-free solution) (10 min duration), replaced by NMDG+ to avoid exit of NH4+ by basolateral Na+-coupled transport. This maneuver was done in the luminal absence of Na+. During the fluorescence recording, perfusion solution was delivered to the perfusion pipette via a chamber under an inert gas (N2) pressure (around 1 bar) connected through a manual six-way valve. With this system, opening of the valve instantaneously activates flow of solutions. The majority of the fluid delivery to the pipette exits the rear of the pipette system through a drain port at 4 ml/min. This method results in a smooth and complete exchange of the luminal or the peritubular solution in less than 3–4 s (27). After the fluorescence signal stabilization, luminal medium was instantly (at the rate of 4 ml/min in the draining) replaced by a Na+-free solution containing 20 mm NH4Cl (and 118 mm NMDG-Cl) that elicited a rapid intracellular alkalinization, followed by a sharp acidification. The rate of intracellular alkalinization has been associated with the entry of NH3, whereas the subsequent phase of intracellular acidification in the continued presence of extracellular NH4Cl reflects mostly NH4+ entry (28). Fluorescence monitoring and calibration were performed with a video imaging system (Visitron Systems, Germany) as described previously (11, 13). For peritubular ammonium pulse, peritubular solution was changed by a 6 mm NH4Cl solution, pH 7.40. in the presence of 1 mm furosemide and 2.5 mm ouabain in the bath.
Intrinsic Buffering Capacity Determination
The intrinsic buffering capacity (βi) of CCD cells was determined, as reported previously (29), using a 40 mm NH4Cl basolateral pulse to acidify the cells. To exclude HCO3−/CO2 as a buffering component and block Na+-dependent pHi regulatory mechanisms, Na+-free, HEPES-buffered solutions were used in the perfusate; the bath contained 1 mm amiloride (to inhibit Na+/H+ exchangers), and bath and perfusate also contained 100 μm Sch28080 (to block H+/K+-ATPases) and 200 nm concanamycin A (to block H+-ATPases). Addition of 40 mm NH4Cl to the bath induced an increase following by a decrease in pHi. The pKa of ammonia (9.03) was used to calculate the intracellular NH4+ concentration when cell acidification plateaued. βi was calculated as the ratio of the change in intracellular NH4+ concentration to the change in pHi. Therefore, we determined the correlation between intracellular pH and βi, which was −32.7–22.3·pH in wild types, −2.3–8.2·pH in heterozygotes, and 1,4–5.0· pHi in Rhcg−/− mice. We measured cellular buffering power (βi) in CCDs from all three genotypes of mice.
NH3 Permeability Measurement
The basic approach used to determine NH3 permeability involved construction of a transepithelial gradient of NH3 and measurement of the resulting NH3 flux from the basolateral to the luminal side as described previously (11, 13, 30). The isolated CCD was transferred to the bath chamber on the stage of an inverted microscope (Axiovision A1, Zeiss, Germany) and mounted on concentric glass pipettes for microperfusion. Bath solution was delivered at a rate of 20 ml/min and warmed to 37 °C by a water jacket immediately upstream of the chamber. The perfusion rate was adjusted by hydrostatic pressure to ∼10 nl/min. The tubules were equilibrated for 20–30 min at 37 °C before the beginning of collections. To construct a transepithelial NH3 gradient, the perfusion (lumen) solution contained (in mm) 139 NaCl, 1 NH4Cl, 2.5 K2HPO4, 2 NaHCO3, pH 6.4; the bath solution contained (in mm) 117 NaCl, 1 NH4Cl, 2.5 K2HPO4, 23 NaHCO3, pH 7.4, and in addition, both solutions contained (in mm) 5.5 glucose, 2 CaCl2, 1.2 MgSO4 and 10 HEPES. The osmolarity of the solution was 295 ± 5 mosmol/kg H2O. All solutions were equilibrated with 95% O2, 5% CO2. Once the solutions were gassed and the pH checked, they were placed in a reservoir and continuously bubbled with 95% O2, 5% CO2. The actual pH of the solutions was monitored several times during the experiments, and the pH of solutions was checked at the end of the experiment to ensure that changes did not occur. Carbonic anhydrase (Sigma) was added to the perfusate solution (1 mg/10 ml of solution). The purpose of carbonic anhydrase was to prevent any pH disequilibrium that might arise from proton secretion or NH3 transport. Total ammonium concentration was measured in 10–12-nl samples of peritubular, perfused, and collected fluids using an NH3 diagnostic kit (Sigma) and the flow-through microfluorometer Nanoflow apparatus (World Precision Instruments, UK) (31).
Calculations of Transepithelial NH3 Permeability
Assuming an absence of osmotic or hydrostatic pressure gradients across the epithelium and therefore an absence of net fluid transport, the passive transepithelial transport of total ammonium (Am) may be described by Equation 1,
where PNH3 is diffusive permeability of NH3 (cm/s), AS is tubule luminal surface area (cm2), and ΔCNH3 is the transepithelial concentration difference for NH3 (mm). To calculate the permeability to NH3, Equation 1 is rearranged as Equation 2,
The net rate of transport JAm is calculated as shown in Equation 3,
where [Am]i is the concentration of total ammonium in the perfusate; [Am]o is the concentration of total ammonium in the collected fluid; V is the collection rate (nl/min), as measured in precalibrated constriction pipettes, and L is the perfused tubule length (mm). AS may be calculated as Lπd, where d (mm) is the inner tubule diameter. The total ammonium concentration ([Am]) is equal to the sum of the concentrations of the two species NH3 and NH4+ and is the quantity actually measured by the microfluorimetric assay. The equilibrium between the two species is defined by the Henderson-Hasselbalch Equation 4,
The pKa equals 9.03 at physiological pH and temperature. Knowing the values for pH and [Am], the values for [NH3] and [NH4+] may be determined simultaneously.
Statistics
Data are expressed as means ± S.E. Statistical comparisons were tested by analysis of variance and Student's t test using the Graphpad Prism software (GraphPad). p values < 0.05 were considered as statistically significant.
RESULTS
Deletion of Rhcg in Mice
Mice lacking Rhcg were generated using gene trap technology in a mixed genetic background (129SvEvBrd, C57/BL6) (Fig. 1A) and were purchased from the Texas Institute of Genomic Medicine (32). Rhcg mRNA was undetectable in renal tissue from Rhcg knock-out (Rhcg−/−) mice and reduced by ∼50% in heterozygous mice (Rhcg+/−) mice (Fig. 1B). RhCG protein was completely absent from the kidney of Rhcg−/− mice as confirmed by immunohistochemistry (Fig. 1C).
Heterozygous Mice Develop Metabolic Acidosis While on Long Term Acid Load
We first assessed acid-base status under basal conditions and during an acid load in Rhcg littermates. Throughout the study, food intake was similar in all three genotypes. At base line, no difference in acid-base and electrolyte levels was observed (Tables 1 and 2).
TABLE 1.
Blood values in Rhcg littermates mice under normal diet and during an acid load
| Basal status |
2 days HCl |
7 days HCl |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Rhcg+/+ (n = 10) | Rhcg+/− (n = 16) | Rhcg−/− (n = 9) | Rhcg+/+ (n = 10) | Rhcg+/− (n = 10) | Rhcg−/− (n = 9) | Rhcg+/+ (n = 12) | Rhcg+/− (n = 21) | Rhcg−/− (n = 2) | |
| pH | 7.38 ± 0.02 | 7.38 ± 0.02 | 7.28 ± 0.04 | 7.17 ± 0.02a | 7.17 ± 0.02a | 7.07 ± 0.02a,b | 7.24 ± 0.03a | 7.17 ± 0.02a,b | 6.88 ± 0.07 |
| pCO2 (mm Hg) | 34.0 ± 1.3 | 37.2 ± 0.9 | 38.8 ± 1.4b | 41.2 ± 2.0a | 39.4 ± 0.7 | 37.8 ± 0.9 | 42.0 ± 1.4a | 40.0 ± 0.8 | 44.6 ± 1.6 |
| HCO3 (mm) | 19.0 ± 1.1 | 22.4 ± 1.0 | 18.3 ± 2.1 | 14.3 ± 0.6a | 13.8 ± 0.5a | 11.1 ± 0.6a,b | 18.6 ± 1.3 | 13.9 ± 0.6a,b | 7.9 ± 1.05 |
| pO2 | 74.7 ± 9.7 | 52.3 ± 3.4 | 63.6 ± 6.3 | 60.1 ± 2.3 | 59.4 ± 1.4 | 65.7 ± 4.1 | 57.1 ± 3.0 | 63.1 ± 3.4 | 82.3 ± 4.6 |
| Na+ (mm) | 143.9 ± 1.5 | 145.4 ± 0.9 | 148.0 ± 0.4 | 150.1 ± 1.0 | 145.8 ± 0.8 | 150.4 ± 0.9 | 148.3 ± 0.6 | 149.1 ± 0.5 | 148.5 ± 1.5 |
| Cl− (mm) | 117.5 ± 1.9 | 112.1 ± 1.7 | 111.7 ± 1.5 | 123.7 ± 1.0a | 119.7 ± 1.0a | 126.4 ± 0.9a | 118.9 ± 0.9 | 122.6 ± 0.7a,b | 134.0 ± 0.0 |
| Ionized Ca2+ (mm) | 1.23 ± 0.02 | 1.25 ± 0.01 | 1.29 ± 0.03 | 1.41 ± 0.03a | 1.35 ± 0.01a | 1.42 ± 0.02a | 1.34 ± 0.01a | 1.37 ± 0.01a,b | 1.01 ± 1.9 |
| Glucose (mm) | 10.6 ± 0.7 | 10.9 ± 0.5 | 10.8 ± 0.8 | 7.8 ± 0.4a | 9.0 ± 0.3b | 6.5 ± 0.4a,b | 9.6 ± 0.4 | 8.8 ± 0.3a | NDc |
| Hb (g/dl) | 15.0 ± 1.1 | 15.7 ± 0.3 | 16.3 ± 0.5 | 16.3 ± 0.4 | 15.5 ± 0.3 | 17.5 ± 0.3a,b | 15.2 ± 0.5 | 14.9 ± 0.4 | 17.4 ± 1.0 |
a p < 0.05 versus base-line period in same genotype is shown.
b p < 0.05 versus Rhcg+/+ mice during the same period is shown.
c ND, not determined.
TABLE 2.
Weight, food intake, and urinary values in Rhcg littermate mice under a normal diet and a 2-day HCl load
TA means titratable acid; NA means net acid; UNH4 means urinary ammonium; UCr means urinary creatinine; UTa means urinary titratable acids; UPi means urinary inorganic phosphate; Uurea means urinary urea; UNa means urinary Na+; UCl means urinary Cl−; and UK means urinary K+.
| Basal status |
2 days of HCl |
|||||
|---|---|---|---|---|---|---|
| Rhcg+/+ (n = 14) | Rhcg+/− (n = 13) | Rhcg−/− (n = 13) | Rhcg+/+ (n = 14) | Rhcg+/− (n = 13) | Rhcg−/− (n = 13) | |
| Weight (g) | 26.4 ± 0.6 | 28.7 ± 1.2 | 27.6 ± 1.0 | 24.9 ± 0.5 | 26.6 ± 0.8 | 25.0 ± 1.1 |
| Weight lose in % of body weight under basal status | NDa | ND | ND | 8,3 ± 0.4 | 11.4 ± 1.3 | 15.0 ± 1.0b |
| Food intake (g/24 h/body weight) | 0.31 ± 0.06 | 0.26 ± 0.02 | 0.26 ± 0.02 | 0.24 ± 0.01 | 0.21 ± 0.01 | 0.21 ± 0.02 |
| Urine values | ||||||
| Volume (ml/24 h) | 1.9 ± 0.2 | 2.0 ± 0.1 | 1.9 ± 0.1 | 2.5 ± 0.3 | 2.1 ± 0.2 | 2.1 ± 0.4c |
| Creatinine excretion (μmol/24 h) | 7.6 ± 0.3 | 6.2 ± 0.6 | 7.8 ± 0.4 | 7.5 ± 0.7 | 5.6 ± 1.4 | 8.3 ± 2.3 |
| Urinary pH | 6.13 ± 0.04 | 6.18 ± 0.06 | 6.03 ± 0.06 | 5.47 ± 0.05c | 5.44 ± 0.02c | 5.68 ± 0.06b,c |
| UNH4/UCr (mEq/mmol) | 4.6 ± 1.0 | 5.1 ± 0.7 | 4.1 ± 1.0 | 37.8 ± 4.1c | 43.8 ± 6.0c | 11.1 ± 1.5b,c |
| UTA/UCr (mEq/mmol) | 15.4 ± 1.2 | 14.4 ± 1.0 | 13.6 ± 1.1 | 20.9 ± 2.5 | 15.9 ± 3.2 | 14.5 ± 1.9 |
| UPi/UCr (mEq/mmol) | 16.1 ± 1.7 | 20.3 ± 1.0 | 17.7 ± 1.4 | 9.3 ± 1.5 | 10.7 ± 1.7 | 8.2 ± 1.6 |
| Uurea/UCr (mg/mmol) | 266.3 ± 14.8 | 279.3 ± 14.8 | 280.8 ± 15.0 | 229.2 ± 17.1 | 255.1 ± 24.01 | 146.8 ± 15.7b,c |
| UNA/UCr (mEq/mmol) | 20.0 ± 1.4 | 19.4 ± 0.8 | 17.7 ± 1.3 | 58.7 ± 4.9c | 60.9 ± 9.0c | 25.6 ± 2.9b,c |
| UNa/UCr (mEq/mmol) | 16.8 ± 2.0 | 10.0 ± 2.0 | 15.5 ± 2.0 | 15.4 ± 1.6 | 15.8 ± 3.6 | 9.7 ± 1.7 |
| UCl/UCr (mEq/mmol) | 32.8 ± 2.0 | 30.6 ± 1.7 | 32.6 ± 2.3 | 133.0 ± 14.4c | 132.3 ± 19.8c | 40.5 ± 9.2b,c |
| UK/UCr (mEq/mmol) | 60.1 ± 3.9 | 60.2 ± 4.6 | 63.1 ± 4..9 | 53.9 ± 4.3 | 56.3 ± 7.3 | 27.7 ± 3.2 |
a ND, not determined.
b p < 0.05 versus Rhcg+/+ mice during the same period is shown.
c p < 0.05 versus base-line period in same genotype is shown.
The effects of both acute (2 days) and chronic (7 days) HCl load were tested (Tables 1–3 and Fig. 2). On the 2nd day of the HCl load, blood pH and HCO3− concentration were decreased in all genotypes, as compared with base line (Table 1, and Fig. 2, D and E). Blood pH and HCO3− concentrations were significantly lower in Rhcg−/− mice but similar in Rhcg+/− and Rhcg+/+ mice. Urinary ammonium excretion rate increased significantly on the 1st day of acid loading in Rhcg+/+ and Rhcg+/− mice but much less in Rhcg−/− mice (Tables 2 and 3 and Fig. 2A). Urinary pH decreased in Rhcg+/+ and Rhcg+/− mice as compared with base-line values (Table 2 and Fig. 2B) but to a lesser extent in Rhcg−/− mice. Urinary titratable acid excretion was unaltered in all three genotypes during the acute acid load. Long term HCl loading resulted in the death of most Rhcg−/− mice, which poorly excreted ammonium and exhibited a very severe metabolic acidosis. These animals showed a higher loss of body weight after the acute HCl load presumably due to dehydration (Table 3).
TABLE 3.
Weight, food intake, and urinary values in 3-month-old Rhcg+/+ and Rhcg+/− littermates during 6 or 7 days of HCl load
TA means titratable acid; NA means net acid; UNH4 means urinary ammonium; UCr means urinary creatinine; UTa means urinary titratable acids; UPi means urinary inorganic phosphate; Uurea means urinary urea; UNa means urinary Na+; UCl means urinary Cl−; and UK means urinary K+.
| 6 days of HCl |
7 days of HCl |
||||
|---|---|---|---|---|---|
| Rhcg+/+ (n = 8) | Rhcg+/− (n = 10) | Rhcg−/− (n = 2) | Rhcg+/+ (n = 8) | Rhcg+/− (n = 10) | |
| Weight (g) | NDa | ND | 19.6–22.4 | 25.1 ± 1.2 | 23.3 ± 0.8b |
| Weight lose in % of body weight under basal status | ND | ND | ND | 10.0 ± 1.1 (n = 5) | 14.8 ± 2.1 (n = 5) |
| Food intake (g/24 h/body weight) | ND | ND | 0.31–0.44 | 0.37 ± 0.03 | 0.39 ± 0.02 |
| Urine values | |||||
| Volume (ml/24 h) | 3.8 ± 0.8b | 4.4 ± 0.8b | 2.5–2.6 | 3.1 ± 0.2b | 3.9 ± 0.1b |
| aCreatinine excretion (μmol/24 h) | 8.5 ± 2.0 | 7.2 ± 1.8 | 3.8–4.3 | 6.6 ± 0.3 | 7.9 ± 1.4 |
| aUrinary pH | 5.39 ± 0.13b | 5.59 ± 0.07b | 5.20–5 .52 | 5.57 ± 0.02b | 5.73 ± 0.05b,c |
| aUNH4/UCr (mEq/mmol) | 153.9 ± 31.1b | 173.1 ± 20.6b | 17.3–36.3 | 147.4 ± 26.3b | 171.8 ± 22.3b |
| aUTA/UCr (mEq/mmol) | 25.4 ± 2.6b | 17.9 ± 2.5 | 28.6–36.2 | 26.3 ± 0.6b | 16.6 ± 0.7c |
| aUPi/UCr (mEq/mmol) | ND | ND | ND | 9.8 ± 3.7 | 25.0 ± 8.8 |
| aUNA/UCr (mEq/mmol) | 131.2 ± 6.3b | 128.5 ± 16.4b | 68.9–53.4 | 126.8 ± 9.8 | 118.3 ± 15.0 |
| aUurea/UCr (mg/mmol) | 205.2 ± 11.2b | 220.3 ± 9.9*b | 186.4–230.1 | 208.9 ± 6.3b | 220.6 ± 5.5b |
| aUNa/UCr (mEq/mmol) | 36.9 ± 3.8b | 32.9 ± 2.8b | 67.4–70.9 | 43.0 ± 3.0b | 38.4 ± 4.0b |
| aUCl/UCr (mEq/mmol) | 242.6 ± 11.2b | 253.6 ± 11.4b | 135.1–173.1 | 269.5 ± 12.0b | 263.5 ± 14.9b |
| aUK/UCr (mEq/mmol) | 77.6 ± 4.2 | 79.7 ± 4.1 | 19.6–22.4 | 92.2 ± 2.0 | 85.2 ± 9.9 |
a ND means not determined.
b p < 0.05 versus base-line period in the same genotype.
c p < 0.05 versus Rhcg+/+ mice during the same period.
FIGURE 2.

Incomplete dRTA in Rhcg+/− and Rhcg−/− mice. Mice were observed in metabolic cages under basal conditions and during an acid load for up to 7 days with HCl added to the diet (n = 5–10 per genotype). Rhcg−/− could only be observed for 6 days during the HCl-containing diet because most animals had to be terminated earlier. A, urinary ammonium excretion in 24-h urine collections normalized for urinary creatinine. B, urinary pH in 24-h urine collections. C, urinary net acid excretion (total bars) calculated from urinary ammonium (open bars) plus titratable acid (gray bars). Urinary bicarbonate levels were measured and negligible. D, blood bicarbonate concentrations. E, blood pH values. *, p < 0.05 for Rhcg−/− versus Rhcg+/+. ¥, p < 0.05 for Rhcg+/− versus Rhcg+/+.
In contrast to the Rhcg+/+ mice, which adapted and nearly normalized their blood pH and HCO3− concentration, Rhcg+/− remained acidotic at day 7 of the HCl load, even though both genotypes maintained a high NH4+ excretion. At the end of the chronic acid load, Rhcg+/− mice showed less increase in their titratable acid excretion and had a more alkaline urine pH (Tables 1 and 2 and Fig. 2, B and C). Thus, both Rhcg−/− and Rhcg+/− exhibited renal acid handling defects.
Absence of RhCG Abolishes Medullary Ammonium Accumulation
To assess the cortico-papillary gradient of NH3/NH4+ in kidneys from Rhcg mice, we measured ammonium content in the cortex and outer and inner medulla after 4 days of HCl treatment. There was no difference between Rhcg+/+ and Rhcg+/− mice. However, the inner medulla ammonium content was strongly reduced to 39% in Rhcg−/− mice (Fig. 3). Thus, the absence of Rhcg impairs the ability to concentrate ammonium in the interstitium of the inner medulla.
FIGURE 3.

Medullary ammonium accumulation is altered in Rhcg−/− mice. Kidneys from all three genotypes were dissected after 4 days of HCl load. Cortex (C), outer medulla (OM), and inner medulla (IM) were separated, and tissue ammonium content was measured. In wild type and heterozygous mice, a gradient in renal tissue ammonia content from cortex to inner medulla was observed. In Rhcg−/− mice, the ammonia content increased from cortex to outer medulla, but ammonium content was lower in the inner medulla than in mice from the other two genotypes. (n = 5–8 mice/genotype), * statistically different from Rhcg+/+ mice (p < 0.05). NS, not significant.
RhCG Is Located at the Apical and Basolateral Sides of Cells Along the Distal Nephron
RhCG has been localized to most cells of the CD, including type A intercalated cells as well as principal cells (9, 17). However, the subcellular localization of RhCG has remained controversial because some groups reported both apical and basolateral staining (10, 17), whereas others detected only apical staining for RhCG (9) based on different immunohistochemical methods. We confirmed a strong labeling of both apical and basolateral poles of CD cells in mouse kidneys (Fig. 4 and Table 4). This staining was absent from Rhcg−/− kidneys demonstrating its specificity. In the kidney cortex, RhCG was localized in the distal convoluted tubule, connecting tubule, and CCD (Fig. 4A). In the distal convoluted tubule cells, RhCG was mainly present at the apical side (Fig. 4A). In the connecting tubule, CCD and OMCD, both intercalated and principal cells, were stained. Principal cells and some intercalated cells exhibited RhCG staining at both the apical and basolateral sides (Fig. 4, A–C). However, in some cells RhCG was strictly localized at the apical pole (see arrowheads in Fig. 4, A, B, and D). These cells have been previously identified as non-A/non-B type intercalated cells (33, 34). Finally, in the inner medulla, only strong apical and faint basolateral staining were found in intercalated cells (Fig. 4 and Table 4). Immunogold electron microscopy demonstrated RhCG associated with the apical membrane as well as the basolateral interdigitations of segment specific and intercalated cells (Fig. 4, E and F). No specific immunogold labeling was found in kidneys from Rhcg−/− mice (data not shown). Thus, RhCG protein is expressed on both apical and basolateral membranes in mouse segment-specific principal and intercalated CD cells.
FIGURE 4.

Rhcg immunodetection in mouse kidney and immunogold electron microscopic localization of Rhcg in CCD. A and B, Rhcg protein immunodetection in mouse kidney cortex. Rhc-related staining was detected in the distal convoluted tubule (DCT) starting immediately at the transition from the thick ascending limb (TAL) to the distal convoluted tubule (arrowhead), in the connecting tubule (CNT) and CCD. Most but not all cells are stained. Rhcg staining is observed in most cells at the apical and basolateral sides, but in some cells (arrowheads) only an apical signal is detected. C, Rhcg immunodetection in outer medulla. Rhcg is strictly expressed in collecting ducts at both sides of intercalated and segment-specific cells. D, in the inner medulla, Rhcg is expressed only in intercalated cells. E and F, Rhcg was detected both at the apical membrane (E) and at the invaginations of the basolateral membrane (F) of segment-specific cells in the cortical collecting duct by gold immunoelectron microscopy.
TABLE 4.
Summary of RhCG localization along the mouse nephron
CNT is connecting tubule; DCT is distal convoluted tubule.
| Tubule type | Cell type | Localization | Staining intensity | |
|---|---|---|---|---|
| Cortex | DCT | Apical | High | |
| Basolateral | Weak | |||
| CNT | Principal cells | Plasma membrane | High | |
| Intercalated cells | Plasma membrane/apical | High | ||
| CCD | Principal cells | Plasma membrane | High | |
| Intercalated cells | Plasma membrane/apical | High | ||
| Outer medulla | OMCD | Principal cells | Plasma membrane/apical | High |
| Intercalated cells | ||||
| Inner medulla | IMCD | Principal cells | No staining | |
| Intercalated cells | Plasma membrane/apical | Weak/high |
Total and Apical Membrane Permeabilities for NH3 Are Reduced in CDs from Rhcg+/− Mice
We assessed total transepithelial permeability for NH3 in in vitro microperfused cortical CDs from Rhcg mice after a 2-day HCl diet, a condition causing a strong difference in urinary ammonium excretion between Rhcg+/+ and Rhcg−/− mice. Imposing a bath-to-lumen NH3 gradient in the nominal absence of an NH4+ gradient generated a measurable NH3 secretory flux, which was significantly lower in CCDs from Rhcg+/− and Rhcg−/− mice versus Rhcg+/+ mice. These differences were due to a decrease in transepithelial permeability to NH3 by 54 and 83%, respectively (Table 5 and Fig. 5). Thus, one Rhcg allele was not sufficient to sustain normal transepithelial permeability to NH3 in the mouse collecting duct.
TABLE 5.
CCD in vitro microperfusion data from Rhcg+/+, Rhcg+/−, and Rhcg−/− mice
Values are mean ± S.E.; n means no. of mice.
| Rhcg+/+ (n = 7) | Rhcg+/− (n = 9) | Rhcg−/− (n = 5) | |
|---|---|---|---|
| Tubule length, mm | 0.38 ± 0.04 | 0.36 ± 0.05 | 0.034 ± 0.08 |
| Tubule diameter, μm | 54.98 ± 4.55 | 47.92 ± 4.24 | 44.31 ± 8.88 |
| Collection rate, nl·mm−1·min−1 | 4.61 ± 0.22 | 4.37 ± 0.29 | 3.96 ± 0.42 |
| Perfusate pH | 6.44 ± 0.04 | 6.37 ± 0.04 | 6.50 ± 0.04 |
| Bath pH | 7.42 ± 0.03 | 7.37 ± 0.03 | 7.38 ± 0.03 |
| Total ammonia perfusate, mm | 1.41 ± 0.08 | 1.40 ± 0.08 | 1.42 ± 0.13 |
| [NH3] perfusate, μm | 3.74 ± 0.37 | 3.30 ± 0.32 | 4.16 ± 0.25 |
| Total ammonia bath, mm | 1.41 ± 0.08 | 1.40 ± 0.08 | 1.42 ± 0.13 |
| [NH3] bath, μm | 34.38 ± 3.26 | 32.57 ± 3.72 | 31.39 ± 3.71 |
| Total ammonia collected, mm | 3.88 ± 0.42 | 2.47 ± 0.36 | 1.48 ± 0.35 |
| [NH3] collected, μm | 10.37 ± 1.45 | 5.90 ± 1.24a | 4.25 ± 0.93a |
| Total ammonia flux, pmol·mm−1·min−1 | 32.98 ± 3.32a | 17.84 ± 4.56a | 2.51 ± 3.51a |
| NH3 permeability, mm/s | 0.13 ± 0.02 | 0.08 ± 0.02a | 0.02 ± 0.02a |
a p < 0.05 versus control mice.
FIGURE 5.

Transepithelial NH3 permeability is reduced in the cortical collecting duct from Rhcg+/− and Rhcg−/− mice. Transepithelial permeability to NH3 was assessed in in vitro isolated and microperfused cortical collecting ducts from mice kept for 2 days on an HCl diet by imposing a bath-to-lumen NH3 gradient (see under “Experimental Procedures”) (*, p < 0.05).
Next, we tested whether the apical permeability to NH3 was affected in CDs from Rhcg+/− mice. Therefore, we measured the effects of an inwardly directed gradient on pHi on CCDs isolated from Rhcg+/+ and Rhcg+/− mice (13). Fig. 6A depicts the typical time course of pHi changes when NH3/NH4+ was added to the lumen tubule. The initial rate of cellular alkalinization is proportional to the rate of apical NH3 entry as described previously (13, 35, 36). To directly compare transport rates, we measured intracellular buffering power and calculated the amount of H+ used to titrate NH3 transported across the membrane. Fig. 6B depicts the calculated rate of NH3 transported into CDs from Rhcg+/+ and Rhcg+/− mice, which was drastically reduced in Rhcg+/− tissue.
FIGURE 6.

RhCG is required for apical NH3 permeability of cortical collecting duct cells. Cortical collecting ducts were isolated from kidneys of Rhcg+/+, Rhcg+/−, and Rhcg−/− mice after 2 days of HCl loading, and intracellular pH was monitored with BCECF. 20 mm NH4Cl was applied with the luminal perfusate. A, pHi recording from CCD exposed to a luminal NH4Cl pulse in Rhcg+/+ and Rhcg+/− CCD. Exposure to NH4Cl caused a rapid alkalinization corresponding to NH3 entry into cells. The initial slopes (ΔpHi/Δt) of the alkalinization phase were measured, and the amount of NH3 titrated into NH4+ was finally calculated based on intracellular buffering power. B, bar graph summarizing the amount of titrated NH3 during luminal NH4Cl pulses (n = 8–12 tubules/genotype). *, p < 0.05; NS, not significant.
Basolateral RhCG Accounts for Peritubular Membrane Permeability to NH3
Because RhCG is expressed at both sides of at least a subset of CD cells (Fig. 4 and Table 4), we next tested the effect of Rhcg disruption on NH3 transport across the basolateral membranes of CCD cells. Several transport pathways for NH4+ have been proposed at the basolateral side of CD cells, including the Na+-K+-2Cl− cotransporter NKCC1 and the Na/K-ATPase where NH4+ would substitute for K+ (37, 38). Therefore, we performed experiments in the nominal absence of sodium to block the activity of both transport pathways.
Peritubular NH3/NH4+ prepulses were performed on cortical CDs from Rhcg+/+, Rhcg+/−, and Rhcg−/− mice submitted to HCl loading for 2 days. As summarized in Fig. 7, when NH3/NH4+ (6 mm) was applied to the basolateral side, the calculated rate of NH3 transport into CD cells was unchanged in Rhcg+/− tissue but strongly reduced in Rhcg−/− tissue. Thus, RhCG sustains both apical and basolateral transport of NH3 in CD cells, in agreement with structural data and reconstituted RhCG suggesting that the protein forms a NH3-permeable channel (39, 40).
FIGURE 7.
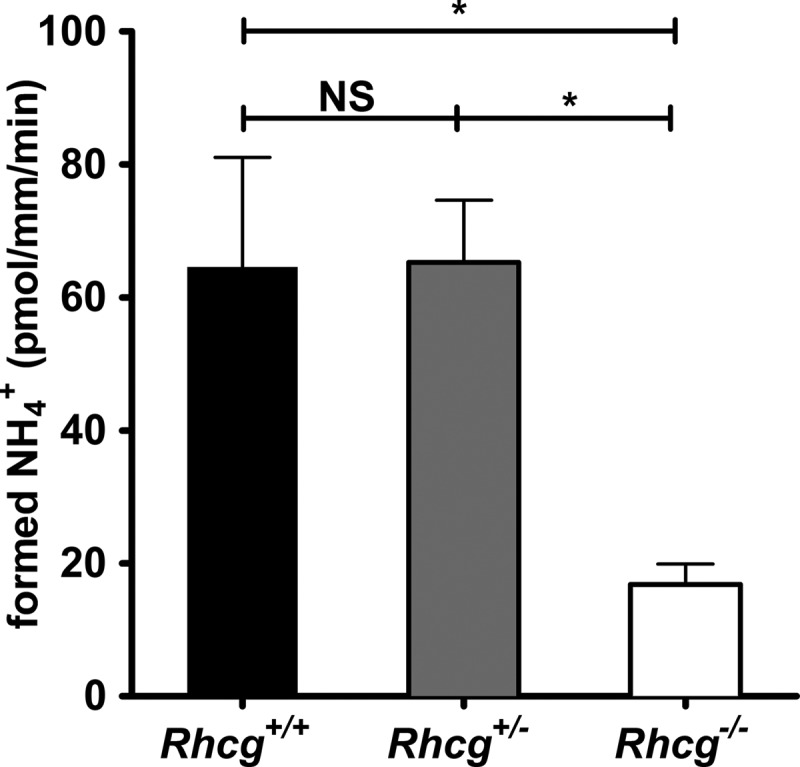
Basolateral NH3 permeability of cortical collecting duct cells. Cortical collecting ducts were isolated from kidneys of Rhcg+/+, Rhcg+/−, and Rhcg−/− mice after 2 days of HCl loading, and intracellular pH was monitored with BCECF. After an equilibrium phase, 6 mm NH4Cl was applied to the bath. Exposure to NH4Cl caused a rapid alkalinization corresponding to NH3 entry into cells. The initial slopes (ΔpHi/Δt) of the alkalinization phase were measured, and the amount of NH3 titrated into NH4+ was finally calculated based on intracellular buffering power. The bar graph summarizes the amount of titrated NH3 (alkalinization phase) during basolateral NH4Cl pulses (n = 5–7 tubules/genotype). *, p < 0.05; NS, not significant.
Compensatory Adaptations to the Loss of Rhcg
We finally examined whether Rhcg deletion affects mechanisms involved in renal ammoniagenesis and ammonium excretion. Surprisingly, immunoblots of whole kidney extracts from mice subjected to a 2-day HCl load revealed no differences in the expression of PEPCK expression (Fig. 8A). In contrast, the expression of the PDG, mediating the initial step of ammoniagenesis was decreased in Rhcg+/− kidneys as compared with both Rhcg+/+ and Rhcg−/− kidneys (Fig. 8B). The abundance of the Na+/H+ exchanger NHE3, involved in NH4+ secretion into the proximal tubule lumen, was unchanged (Fig. 8C). Moreover, the expression of the Na+-K+-2Cl− cotransporter NKCC2, the main apical NH4+ transporter in the thick ascending limb, was highly down-regulated in kidneys from Rhcg+/− and Rhcg−/− mice (Fig. 8D).
FIGURE 8.

Deletion of Rhcg affects the abundance of proteins involved in ammoniagenesis and ammonium transport. Crude membrane and cytosolic fractions were prepared from total kidneys from Rhcg+/+, Rhcg+/,− and Rhcg−/− mice after 2 days of HCl loading, 40 μg loaded onto SDS-PAGE, and their abundance tested. A, PEPCK; B, PDG; C, the Na+-K+-2Cl− cotransporter isoform 2 (NKCC2); and D, Na+/H+ exchanger isoform 3 (NHE3). All membranes were stripped and reprobed for β-actin to control for loading. Bar graphs summarize results from densitometric analysis of proteins of interest normalized against β-actin. n = 5 mice/genotype. * statistically significant, p < 0.05 versus Rhcg+/+ tissue. NS, not significant.
DISCUSSION
Renal ammonium excretion is critical for acid-base homeostasis and is achieved by a complex process involving various nephron segments (2, 5, 6). A critical role for the Rhesus protein RhCG was demonstrated in tissue-specific and complete knock-out mouse models (13–15). However, metabolic effects of Rhcg haploinsufficiency, the role of RhCG in basolateral NH3 transport in CD cells, and the response of other pathways critical for renal ammoniagenesis and ammonium transport have remained unknown. Based on a novel Rhcg mouse model, we show that Rhcg haploinsufficiency causes incomplete metabolic acidosis in mice. Next, RhCG contributes to peritubular NH3 uptake giving functional evidence to the basolateral localization of RhCG in CD cells.
Rhcg−/− Mice Develop a Severe Incomplete Renal Tubular Acidosis
Rhcg−/− develop incomplete dRTA as evident from the inability to respond to an acute oral acid load. They showed severe hyperchloremic metabolic acidosis and did not increase urinary ammonium excretion resulting in low net acid excretion. Moreover, these animals had a drastic reduction in their blood HCO3− concentration and pH. Rhcg−/− mice developed severe dehydration as indicated by high blood hemoglobin and weight loss (Tables 2 and 4). These phenotypes are more pronounced than in mice with partial deletion of Rhcg (14, 15).
Rhcg Haploinsufficiency Leads to Metabolic Acidosis
Similarly, Rhcg+/− mice develop also an incomplete dRTA. However, these mice initially responded to the acid load by increasing urinary ammonium excretion to the same extent as Rhcg+/+ mice. Later, they did not fully adapt to chronic acid loading and remained acidotic after 7 days of HCl loading, being unable to correct blood HCO3− concentration and excreting more alkaline urine, whereas total net acid excretion was comparable with Rhcg+/+ animals. The incomplete chronic metabolic acidosis in Rhcg+/− mice is at least in part due to the inability to maximally acidify urine and to excrete adequate amounts of acid in the form of titratable acidity. The metabolic phenotype of Rhcg+/− mice was further supported by functional studies on microperfused CCDs. Transepithelial permeability to NH3 was reduced in CCDs from Rhcg−/− and Rhcg+/− mice. The reduction in NH3 permeability in CCDs from Rhcg+/− mice was less than in Rhcg−/− mice, suggesting that a 50% reduction in net NH3 permeability in CCDs is still sufficient to support high ammonium excretion but is not enough to correct metabolic acidosis. Of note, medullary accumulation of NH3/NH4+ appeared to be intact. Together, these results represent the first evidence that loss of a single Rhcg allele can lead to chronic hyperchloremic metabolic acidosis.
Renal Adaptation to Rhcg Invalidation
We further examined compensatory mechanisms in Rhcg+/− and Rhcg−/− mice. The generation of the cortico-papillary gradient of ammonium is required for the subsequent uptake of NH3 and NH4+ by intercalated cells and the secretion of NH3 into urine. Surprisingly, Rhcg−/− mice had low tissue ammonium content in the inner medulla after 4 days of acid loading. This result suggests that the absence of RhCG affects the ability of the medulla to generate or maintain a high interstitium ammonium content. Because the key enzymes of proximal tubular ammoniagenesis, PEPCK and PDG, were normally expressed in Rhcg−/− kidney tissue, mice most likely form adequate amounts of ammonium. In Rhcg−/− mice, this ammonium might be either shunted back into systemic circulation or not be absorbed at the level of the thick ascending limb.
Expression of NKCC2 was decreased in both Rhcg+/− and Rhcg−/− kidney tissues. NKCC2 is crucial for thick ascending limb NH4+ absorption and concentration in the medulla. Thus, our results suggest that mechanisms contributing to create a high medullary ammonium concentration are altered in Rhcg-deficient mice. Other mechanisms such as increased removal of ammonium from the interstitium with venous blood might contribute also to the low medullary ammonium content. Metabolic acidosis alters concentrations of various vasoactive substances in the kidney, including higher levels of endothelin and prostaglandins and lower concentrations of NO (41, 42), which may alter medullary blood flow and the ability of the kidneys to maintain the cortico-papillary ammonium gradient.
RhCG Mediates NH3 Transport Also at the Basolateral Side of CD Cells
Subcellular RhCG localization has remained controversial for many years (9, 10, 17). Here, we confirmed a strong labeling of both apical and basolateral poles of CD cells in mouse kidneys. These data were corroborated by our functional study on in vitro microperfused CCDs. We measured a 60% reduction in the NH3 permeability at the basolateral side of Rhcg−/− CCD cells. This is the first evidence that Rhcg is also functional at the basolateral side of CDs cells and participates in basolateral NH3 uptake. Previously, it has been assumed that basolateral uptake occurs mostly if not exclusively in the form of NH4+ and that NH3 plays no major role. Several pathways for NH4+ uptake have been delineated from pharmacological and functional experiments that demonstrated a role for the Na+-K+-2Cl− cotransporter NKCC1, the Na+/K+-ATPase, and possibly potassium channels where NH4+ always would replace potassium (37, 38, 43). We performed our experiments in the absence of sodium to reduce NKCC1 and Na+/K+-ATPase activity, respectively. Thus, our experiments do not allow conclusions regarding the relative importance and contribution of these different pathways to total NH3/NH4+ uptake across the basolateral membrane, but they modify the current model of ammonium transport across the basolateral membrane. However it appears from the data on Rhcg+/− CCD that apical NH3 flux critically depends on full RhCG expression, whereas basolateral NH3 flux is sustained with reduced RhCG expression suggesting that transport of the NH3 form is most important at the apical side. Finally, our findings could also explain the complete lack or only mild phenotype observed in Rhbg knock-out mice (11, 44), where RhCG may have compensated for the lack of RhBG.
In summary, reduced expression or complete loss of expression of RhCG affects the ability of the kidneys to excrete acidic urine and appropriately regulate acid/base homeostasis. We provide the first functional evidence for basolateral RhCG activity and show that RhCG is expressed and functional on apical and basolateral membranes of most cells lining the renal collecting duct. Thus, Rhcg haploinsufficiency or Rhcg deletion may contribute to orphan forms of inherited distal renal tubular acidosis in humans as well as to acquired forms of dRTA.
Acknowledgments
We thank Yves Colin and Isabelle Mourot-Chanteloup for fruitful discussions. We also thank Hanne Siedelmann for skillful technical assistance. The use of the Zurich Integrative Rodent Physiology Core Facility is gratefully acknowledged.
This work was supported in part by Grants 3100A0-122217 and 31003A_138143 from the Swiss National Science Foundation (to C. A. W.), the 7th European Union Framework Project EUNEFRON (to P. H., E. I. C., O. D., and C. A. W.), the Danish Medical Research Council, The Novo Nordisk Foundation (to E. I. C.), and the Swiss National Centre of Competence in Research (to O. D. and C. A. W.).
- dRTA
- distal renal tubular acidosis
- CD
- collecting duct
- CCD
- cortical CD
- BCECF
- 2′,7′-bis(2-carboxyl)-5-(and-6)-carboxyfluorescein
- PEPCK
- phosphoenolpyruvate carboxykinase
- PDG
- phosphate-dependent glutaminase
- NMDG
- N-methyl-d-glutamine
- OMCD
- outer medullary collecting duct
- IMCD
- inner medullary collecting duct.
REFERENCES
- 1. Hamm L. L., Alpern R. J., Preisig P. A. (2008) in Seldin and Giebisch's The Kidney. Physiology and Pathophysiology (Alpern R. J., Hebert S. C., eds) pp. 1539–1585, Academic Press, New York [Google Scholar]
- 2. Weiner I. D., Verlander J. W. (2011) Role of NH3 and NH4+ transporters in renal acid-base transport. Am. J. Physiol. Renal Physiol. 300, F11–F23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Batlle D. C., Kurtzman N. A. (1985) in Renal Tubular Disorders (Gonick H. C., Buckalew V. M., eds) pp. 281–305, Marcel Dekker, Inc., New York [Google Scholar]
- 4. Good D. W. (1994) Ammonium transport by the thick ascending limb of Henle's loop. Annu. Rev. Physiol. 56, 623–647 [DOI] [PubMed] [Google Scholar]
- 5. Wagner C. A., Devuyst O., Belge H., Bourgeois S., Houillier P. (2011) The rhesus protein RhCG. A new perspective in ammonium transport and distal urinary acidification. Kidney Int. 79, 154–161 [DOI] [PubMed] [Google Scholar]
- 6. Wagner C. A., Devuyst O., Bourgeois S., Mohebbi N. (2009) Regulated acid-base transport in the collecting duct. Pflugers Arch. 458, 137–156 [DOI] [PubMed] [Google Scholar]
- 7. Wagner C. A., Finberg K. E., Breton S., Marshansky V., Brown D., Geibel J. P. (2004) Renal vacuolar H+-ATPase. Physiol. Rev. 84, 1263–1314 [DOI] [PubMed] [Google Scholar]
- 8. DuBose T. D., Jr., Good D. W., Hamm L. L., Wall S. M. (1991) Ammonium transport in the kidney: new physiological concepts and their clinical implications. J. Am. Soc. Nephrol. 1, 1193–1203 [DOI] [PubMed] [Google Scholar]
- 9. Quentin F., Eladari D., Cheval L., Lopez C., Goossens D., Colin Y., Cartron J. P., Paillard M., Chambrey R. (2003) RhBG and RhCG, the putative ammonia transporters, are expressed in the same cells in the distal nephron. J. Am. Soc. Nephrol. 14, 545–554 [DOI] [PubMed] [Google Scholar]
- 10. Brown A. C., Hallouane D., Mawby W. J., Karet F. E., Saleem M. A., Howie A. J., Toye A. M. (2009) RhCG is the major putative ammonia transporter expressed in the human kidney, and RhBG is not expressed at detectable levels. Am. J. Physiol. Renal Physiol. 296, F1279–F1290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chambrey R., Goossens D., Bourgeois S., Picard N., Bloch-Faure M., Leviel F., Geoffroy V., Cambillau M., Colin Y., Paillard M., Houillier P., Cartron J. P., Eladari D. (2005) Genetic ablation of Rhbg in the mouse does not impair renal ammonium excretion. Am. J. Physiol. Renal Physiol. 289, F1281–F1290 [DOI] [PubMed] [Google Scholar]
- 12. Bishop J. M., Verlander J. W., Lee H. W., Nelson R. D., Weiner A. J., Handlogten M. E., Weiner I. D. (2010) Am. J. Physiol. Renal Physiol. 299, F1065–F1077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Biver S., Belge H., Bourgeois S., Van Vooren P., Nowik M., Scohy S., Houillier P., Szpirer J., Szpirer C., Wagner C. A., Devuyst O., Marini A. M. (2008) A role for rhesus factor Rhcg in renal ammonium excretion and male fertility. Nature 456, 339–343 [DOI] [PubMed] [Google Scholar]
- 14. Lee H. W., Verlander J. W., Bishop J. M., Igarashi P., Handlogten M. E., Weiner I. D. (2009) Collecting duct-specific Rh C glycoprotein deletion alters basal and acidosis-stimulated renal ammonia excretion. Am. J. Physiol. Renal Physiol. 296, F1364–F1375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Han K. H., Lee S. Y., Kim W. Y., Shin J. A., Kim J., Weiner I. D. (2010) Expression of ammonia transporter family members, Rh B glycoprotein and Rh C glycoprotein, in the developing rat kidney. Am. J. Physiol. Renal Physiol. 299, F187–F198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Eladari D., Cheval L., Quentin F., Bertrand O., Mouro I., Cherif-Zahar B., Cartron J. P., Paillard M., Doucet A., Chambrey R. (2002) Expression of RhCG, a new putative NH3/NH4+ transporter, along the rat nephron. J. Am. Soc. Nephrol. 13, 1999–2008 [DOI] [PubMed] [Google Scholar]
- 17. Kim H. Y., Verlander J. W., Bishop J. M., Cain B. D., Han K. H., Igarashi P., Lee H. W., Handlogten M. E., Weiner I. D. (2009) Basolateral expression of the ammonia transporter family member Rh C glycoprotein in the mouse kidney. Am. J. Physiol. Renal Physiol. 296, F543–F555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jorgensen K. (1957) Titrimetric determination of the net excretion of acid/base in urine. Scand. J. Clin. Lab. Invest. 9, 287–291 [DOI] [PubMed] [Google Scholar]
- 19. Nutbourne D. M. (1961) The effect of dilution on the titratable acid in urine and acidified phosphate buffer solutions, and the correction for this effect in the determination of the rate of elimination of hydrogen ions from the body by the renal tubules. Clin. Sci. 20, 263–278 [PubMed] [Google Scholar]
- 20. Seaton B., Ali A. (1984) Simplified manual high performance clinical chemistry methods for developing countries. Med. Lab. Sci. 41, 327–336 [PubMed] [Google Scholar]
- 21. Berthelot M. (1859) Violet d'aniline. Rep. Chim. App. 1, 284 [Google Scholar]
- 22. Curthoys N. P., Kuhlenschmidt T., Godfrey S. S., Weiss R. F. (1976) Phosphate-dependent glutaminase from rat kidney. Cause of increased activity in response to acidosis and identity with glutaminase from other tissues. Arch. Biochem. Biophys. 172, 162–167 [DOI] [PubMed] [Google Scholar]
- 23. Hafner P., Grimaldi R., Capuano P., Capasso G., Wagner C. A. (2008) Pendrin in the mouse kidney is primarily regulated by Cl− excretion but also by systemic metabolic acidosis. Am. J. Physiol. Cell Physiol. 295, C1658–C1667 [DOI] [PubMed] [Google Scholar]
- 24. Christensen E. I., Nielsen S., Moestrup S. K., Borre C., Maunsbach A. B., de Heer E., Ronco P., Hammond T. G., Verroust P. (1995) Segmental distribution of the endocytosis receptor gp330 in renal proximal tubules. Eur. J. Cell Biol. 66, 349–364 [PubMed] [Google Scholar]
- 25. Marini A. M., Matassi G., Raynal V., André B., Cartron J. P., Chérif-Zahar B. (2000) The human Rhesus-associated RhAG protein and a kidney homologue promote ammonium transport in yeast. Nat. Genet. 26, 341–344 [DOI] [PubMed] [Google Scholar]
- 26. Packer R. K., Desai S. S., Hornbuckle K., Knepper M. A. (1991) Role of countercurrent multiplication in renal ammonium handling. Regulation of medullary ammonium accumulation. J. Am. Soc. Nephrol. 2, 77–83 [DOI] [PubMed] [Google Scholar]
- 27. Watts B. A., 3rd, Good D. W. (1994) Apical membrane Na+/H+ exchange in rat medullary thick ascending limb. pH dependence and inhibition by hyperosmolality. J. Biol. Chem. 269, 20250–20255 [PubMed] [Google Scholar]
- 28. Roos A., Boron W. F. (1981) Intracellular pH. Physiol. Rev. 61, 296–434 [DOI] [PubMed] [Google Scholar]
- 29. Milton A. E., Weiner I. D. (1998) Regulation of B-type intercalated cell apical anion exchange activity by CO2/HCO3−. Am. J. Physiol. 274, F1086–F1094 [DOI] [PubMed] [Google Scholar]
- 30. Flessner M. F., Wall S. M., Knepper M. A. (1992) Ammonium and bicarbonate transport in rat outer medullary collecting ducts. Am. J. Physiol. 262, F1–F7 [DOI] [PubMed] [Google Scholar]
- 31. Zhelyaskov V. R., Liu S., Broderick M. P. (2000) Analysis of nanoliter samples of electrolytes using a flow-through microfluorometer. Kidney Int. 57, 1764–1769 [DOI] [PubMed] [Google Scholar]
- 32. Hansen G. M., Markesich D. C., Burnett M. B., Zhu Q., Dionne K. M., Richter L. J., Finnell R. H., Sands A. T., Zambrowicz B. P., Abuin A. (2008) Large-scale gene trapping in C57BL/6N mouse embryonic stem cells. Genome Res. 18, 1670–1679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Verlander J. W., Miller R. T., Frank A. E., Royaux I. E., Kim Y. H., Weiner I. D. (2003) Localization of the ammonium transporter proteins RhBG and RhCG in mouse kidney. Am. J. Physiol. Renal Physiol. 284, F323–F337 [DOI] [PubMed] [Google Scholar]
- 34. Kim H. Y., Verlander J. W., Bishop J. M., Cain B. D., Han K. H., Igarashi P., Lee H. W., Handlogten M. E., Weiner I. D. (2009) Am. J. Physiol. Renal Physiol. 296, F1364–F1375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kikeri D., Sun A., Zeidel M. L., Hebert S. C. (1989) Cell membranes impermeable to NH3. Nature 339, 478–480 [DOI] [PubMed] [Google Scholar]
- 36. Bleich M., Köttgen M., Schlatter E., Greger R. (1995) Effect of NH4+/NH3 on cytosolic pH and the K+ channels of freshly isolated cells from the thick ascending limb of Henle's loop. Pflugers Arch. 429, 345–354 [DOI] [PubMed] [Google Scholar]
- 37. Wall S. M., Koger L. M. (1994) NH4+ transport mediated by Na+-K+-ATPase in rat inner medullary collecting duct. Am. J. Physiol. 267, F660–F670 [DOI] [PubMed] [Google Scholar]
- 38. Wall S. M., Fischer M. P. (2002) Contribution of the Na+-K+-2Cl− cotransporter (NKCC1) to transepithelial transport of H+, NH4+, K+, and Na+ in rat outer medullary collecting duct. J. Am. Soc. Nephrol. 13, 827–835 [DOI] [PubMed] [Google Scholar]
- 39. Gruswitz F., Chaudhary S., Ho J. D., Schlessinger A., Pezeshki B., Ho C. M., Sali A., Westhoff C. M., Stroud R. M. (2010) Function of human Rh based on structure of RhCG at 2.1 A. Proc. Natl. Acad. Sci. U.S.A. 107, 9638–9643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mouro-Chanteloup I., Cochet S., Chami M., Genetet S., Zidi-Yahiaoui N., Engel A., Colin Y., Bertrand O., Ripoche P. (2010) Functional reconstitution into liposomes of purified human RhCG ammonia channel. PLoS ONE 5, e8921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jones E. R., Beck T. R., Kapoor S., Shay R., Narins R. G. (1984) Prostaglandins inhibit renal ammoniagenesis in the rat. J. Clin. Invest. 74, 992–1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Prabhakar S. S. (2004) Regulatory and functional interaction of vasoactive factors in the kidney and extracellular pH. Kidney Int. 66, 1742–1754 [DOI] [PubMed] [Google Scholar]
- 43. Wall S. M., Fischer M. P., Kim G. H., Nguyen B. M., Hassell K. A. (2002) In rat inner medullary collecting duct, NH uptake by the Na,K-ATPase is increased during hypokalemia. Am. J. Physiol. Renal Physiol. 282, F91–F102 [DOI] [PubMed] [Google Scholar]
- 44. Bishop J. M., Verlander J. W., Lee H. W., Nelson R. D., Weiner A. J., Handlogten M. E., Weiner I. D. (2010) Role of the Rhesus glycoprotein, Rh B glycoprotein, in renal ammonia excretion. Am. J. Physiol. Renal Physiol. 299, F1065–F1077 [DOI] [PMC free article] [PubMed] [Google Scholar]