

Background: hLOXL2 induces metastasis/invasion of breast cancer cells.
Results: The N-glycans and lysine tyrosylquinone (LTQ) cofactor of rhLOXL2 are determined.
Conclusion: N-Glycans are essential for proper folding and secretion of hLOXL2 from S2 cells.
Significance: This is the first determination of 1) LTQ in the hLOX family of proteins and 2) N-glycosylation in the LOX catalytic domain in the LOX family of proteins.
Keywords: Enzyme Catalysis, Glycosylation, Oxidase, Post-translational Modification, Quinones
Abstract
Human lysyl oxidase-like 2 (hLOXL2) is highly up-regulated in metastatic breast cancer cells and tissues and induces epithelial-to-mesenchymal transition, the first step of metastasis/invasion. hloxl2 encodes four N-terminal scavenger receptor cysteine-rich domains and the highly conserved C-terminal lysyl oxidase (LOX) catalytic domain. Here, we assessed the extent of the post-translational modifications of hLOXL2 using truncated recombinant proteins produced in Drosophila S2 cells. The recombinant proteins are soluble, in contrast to LOX, which is consistently reported to require 2–6 m urea for solubilization. The recombinant proteins also show activity in tropoelastin oxidation. After phenylhydrazine derivatization and trypsin digestion, we used mass spectrometry to identify peptides containing the derivatized lysine tyrosylquinone cross-link at Lys-653 and Tyr-689, as well as N-linked glycans at Asn-455 and Asn-644. Disruption of N-glycosylation by site-directed mutagenesis or tunicamycin treatment completely inhibited secretion so that only small quantities of inclusion bodies were detected. The N-glycosylation site at Asn-644 in the LOX catalytic domain is not conserved in human LOX (hLOX), although the LOX catalytic domain of hLOX shares ∼50% identity and ∼70% homology with hLOXL2. The catalytic domain of hLOX was not secreted from S2 cells using the same expression system. These results suggest that the N-glycan at Asn-644 of hLOXL2 enhances the solubility and stability of the LOX catalytic domain.
Introduction
Human lysyl oxidase (hLOX)4 and human lysyl oxidase-like 2 (hLOXL2) represent relatively new therapeutic targets for metastatic/invasive breast cancer (1–3). Recently, the extracellular matrix (ECM) stiffening caused by hLOX and hLOXL2 has been linked to activation of the focal adhesion kinase (FAK)/Src signaling pathway and up-regulation of tissue inhibitor of metalloproteinase-1 (TIMP-1) and matrix metalloproteinase-9 (MMP-9), thereby increasing degradation/remodeling of the ECM and enabling subsequent metastatic dissemination (1, 4, 5). hLOXL2 is generally considered to play an equivalent role to hLOX in the ECM. However, the extent of the post-translational modifications (PTMs) of hLOXL2 and its subcellular localization have not been characterized. Ultimately, the function of hLOXL2 at the molecular level remains unclear.
hLOXL2 belongs to the LOX family of proteins, but differs from hLOX by an additional four N-terminal scavenger receptor cysteine-rich (SRCR) domains, as shown in Fig. 1A (6). LOX is a copper- and lysine tyrosylquinone (LTQ)-dependent amine oxidase that is traditionally known to catalyze the oxidative deamination of the ϵ-amino side chains of lysine residues in collagen and elastin, leading to cross-linking and stabilization of the ECM (7–9). LOX is synthesized as a proprotein (pro-LOX, 50 kDa) that undergoes post-translational modifications such as N-linked and O-linked glycosylation of the propeptide (10), copper- and molecular oxygen-catalyzed biogenesis of the LTQ cofactor (9, 11), and proteolytic cleavage of the propeptide by bone morphogenic protein 1 (BMP-1) in the ECM to yield a 32-kDa nonglycosylated mature protein (12–14). The major obstacle to studying the functions of hLOXL2 and hLOX at the molecular level is the lack of a system for the expression and purification of recombinant proteins suitable for such investigations. This is mostly due to the high cysteine content and highly insoluble nature of the LOX family of proteins, which reportedly requires 2–6 m urea for solubilization. hLOXL2 contains a total of 10 Cys in the LOX catalytic domain and 6 Cys in each SRCR domain (34 Cys total).
FIGURE 1.

A, schematic diagram of full-length hLOXL2 and two truncated recombinant hLOXL2s used in this study. B, SDS-PAGE (lanes 1 and 2) and Western blot (lanes 3 and 4) analyses of affinity-purified Δ1-4SRCR-hLOXL2 (lanes 1 and 3) and Δ1-3SRCR-hLOXL2 (lanes 2 and 4). C, Western blot analyses of PNGase F-treated purified Δ1-3SRCR-hLOXL2 (lanes 1 and 2) and Δ1-4SRCR-hLOXL2 (lanes 3 and 4). D, DIG-labeled lectin analyses of Δ1-3SRCR-hLOXL2 and Δ1-4SRCR-hLOXL2. Protein samples were fractionated by SDS-PAGE and transferred to PVDF membranes. Each membrane strip was incubated sequentially with a DIG-labeled lectin (peanut agglutinin (PNA), Sambucus nigra agglutinin (SNA), Datura stramonium agglutinin (DSA), G. nivalis agglutinin (GNA), or Maackia amurensis agglutinin (MAA)), alkaline phosphatase-conjugated anti-DIG antibody, and nitro blue tetrazolium/5-bromo-4-chloro-3-indolyl phosphate. Lane 1, positive control glycoprotein (0.3 mg) supplied with the kit; lane 2, PNGase F-treated recombinant hLOXL2 (0.1 μg); lane 3, recombinant hLOXL2 (0.1 μg). Arrows indicate detection of high mannose type glycoconjugates. M, protein molecular mass marker.
In this study, we produced two truncated forms of hLOXL2, namely Δ1-3SRCR-hLOXL2 (the first three SRCR domains are truncated, see Fig. 1A) and Δ1-4SRCR-hLOXL2 (all four SRCR domains are truncated, see Fig. 1A), in the culture medium of Drosophila Schneider 2 (S2) cells and analyzed the extent of PTMs. We chose these two truncated hLOXL2s in an attempt to understand how the fourth SRCR domain affects the solubility and catalytic efficacy of the LOX catalytic domain. This is the first study where >95% pure, soluble, and active recombinant hLOXL2s are isolated in quantities sufficient to examine the extent of the PTMs of secreted hLOXL2.
EXPERIMENTAL PROCEDURES
Materials
The cDNA containing the complete open reading frame of hLOXL2 (GenBankTM accession number: NM_002318.2) was purchased from OriGene Technologies (Rockville, MD); SuperFect transfection reagent was from Qiagen (Valencia, CA); DIG glycan differentiation kit was from Roche Applied Science; peptide-N-glycosidase F (PNGase F) was from New England Biolabs (Ipswich, MA); N-acetyl-3,7-dihydroxyphenoxazine (Amplex Red) was from Synchem Laborgemeinschaft (Kassel, Germany); pASK-IBA3 vector, Strep-Tactin affinity column and anti-Strep-tag antibody were from IBA (Göttingen, Germany); Drosophila Schneider 2 cells, pMT/Bip/V5-HisA, and pCoBlast were from Invitrogen; tunicamycin, β-aminopropionitrile (BAPN) fumarate, and phenylhydrazine hydrochloride were from Sigma-Aldrich; and tropoelastin (from chick aorta) was from Elastin Products Co. (Owensville, MO).
Design of Expression Plasmid Constructs and Site-directed Mutagenesis
The Strep-tag II coding sequence was engineered at the 3′ end of our genes of interest (i.e. Δ1-3SRCR-hLOXL2 and Δ1-4SRCR-hLOXL2) by first subcloning the genes into the pASK-IBA3 vector containing Strep-tag II, using the following primer pairs: 5′-GATCACTAGTATGCGCCTGAACGGCGGCCGCAATCCCTACG-3′ (forward) and 5′-CCGCTCGAGTTATTTTTCGAACTGCGGGTGGCTCCAAGC-3′ (reverse); 5′-GATCACTAGTCCTGACCTGGTCCTCAATGCGGAGATG-3′ (forward) and 5′-CCGCTCGATTATTTTTCGAACTGCGGGTG-3′ (reverse), respectively. The hloxl2-StrepII DNAs were then lifted out by PCR and cloned into pMT/Bip/V5-HisA expression vectors at the SpeI and XhoI sites. The asparagine residues at 455 and 644 were mutated to glutamine by QuikChange site-directed mutagenesis (Agilent, Santa Clara, CA) to produce N455Q or N644Q Δ1-3SRCR-hLOXL2 and N644Q Δ1-4SRCR-hLOXL2. The primer pairs used for N455Q and N644Q mutagenesis, respectively, are as follows: 5′-GTGCTGGTGGAGAGACAAGGGTCCCTTGTGTG-3′ (forward) and 5′-CACACAAGGGACCCTTGTCTCTCCACCAGCAC-3′ (reverse); 5′-CTATGACCTGCTGAACCTCCAAGGCACCAAGGTGGCAGAG-3′ (forward) and 5′-CTCTGCCACCTTGGTGCCTTGGAGGTTCAGCAGGTCATAG-3′ (reverse).
Transient and Stable Transfection, and Expression and Purification of Recombinant hLOXL2s
For transient transfection, the expression plasmids for the Δ1-3SRCR-hLOXL2s and Δ1-4SRCR-hLOXL2s were transfected into S2 cells according to the protocol in the Drosophila Expression System manual (Invitrogen). Expression of recombinant proteins was examined by Western blot analysis (using anti-Strep-tag antibody) of medium harvested 4–5 days after induction with 0.7 mm CuSO4.
For stable transfection, the expression plasmids for the Δ1-3SRCR-hLOXL2s and Δ1-4SRCR-hLOXL2 were transfected into S2 cells together with pCoBlast, and the transfected cells were maintained in Drosophila complete medium containing blasticidin, in accordance with the Drosophila Expression System manual. Stable integration of the genes of interest into the genomic DNA of the S2 cells was verified by PCR. Expression of recombinant proteins was examined by Western blot analysis (using anti-Strep-tag antibody) of medium harvested 4–5 days after induction with 0.7 mm CuSO4.
For recombinant protein purification, the stably transfected cells were expanded into serum-free medium, and protein expression was induced by the addition of 0.7 mm CuSO4. The medium was harvested 4–5 days after induction, and the recombinant hLOXL2s were purified by Strep-Tactin affinity chromatography following the kit protocol (IBA). The purity of the isolated proteins was examined by SDS-PAGE and Western blot analysis. Protein concentrations were determined by BCA assay using a Pierce BCA protein assay kit (Thermo Scientific). Typical yields were 0.50–0.75 mg of protein per liter of culture.
Western Blot Analysis
Proteins and biotinylated protein molecular mass marker were electrophoresed together by SDS-PAGE and then electroblotted onto PVDF membranes. The membranes were blocked with 1× TBS containing 0.1% Tween 20 (TBST) and 5% w/v nonfat dry milk, washed three times with 1× TBST, and incubated with a 1:5000 dilution of horseradish peroxidase (HRP)-conjugated anti-Strep-tag antibody and a 1:5000 dilution of HRP-conjugated anti-biotin antibody for detection of recombinant hLOXL2s. The membranes were washed three times with 1× TBST. Membranes were treated with SuperSignal West Pico chemiluminescent substrate (Thermo Scientific) and exposed to x-ray film for the appropriate time to detect the proteins.
Analysis of the Terminal Carbohydrate Structures of Recombinant hLOXL2s
The terminal carbohydrate structures of the recombinant hLOXL2s were analyzed by a DIG glycan differentiation kit according to the kit manual (Roche Applied Science).
Tunicamycin Treatment of Stable S2 Cells and PNGase F Treatment of Recombinant hLOXL2s
Stable S2 cells were expanded into serum-free medium and then grown in the presence of 10 μg/ml tunicamycin 2 h before protein expression was induced by adding 0.7 mm CuSO4. Expression was allowed to proceed for 36 h in the presence of tunicamycin, and then the cells and growth medium were harvested. For deglycosylation by PNGase F, purified recombinant hLOXL2s were incubated with 100 units/ml of PNGase F for 3 h at 37 °C.
Activity Assay
The LOX amine oxidase activity of the recombinant proteins was measured by monitoring the formation of H2O2 using the standard HRP-coupled Amplex Red assay (15) at 37 °C, except that the formation of the oxidized Amplex Red (resorufin) was monitored at 563 nm by a Shimadzu UV-2550 UV-visible spectrophotometer and expressed as milliabsorbance units per minute. The initial rate of substrate oxidation was measured in triplicate from the linear slope of the absorbance change at 563 nm (ΔA563) after tropoelastin was added. The rate of the reaction was converted from milliabsorbance units per minute to the production of pmol of H2O2 formed per minute per μg of recombinant protein (pmol of H2O2/min/μg). The ϵ value for resorufin at 563 nm was determined to be 54,400 ± 600 m−1 cm−1. KaleidaGraph 4.1.1 (Synergy Software, Reading, PA) was used to fit the kinetic data to Equation 1 (i.e. the Michaelis-Menten equation),
 |
by nonlinear regression. kcat was calculated using Equation 2.
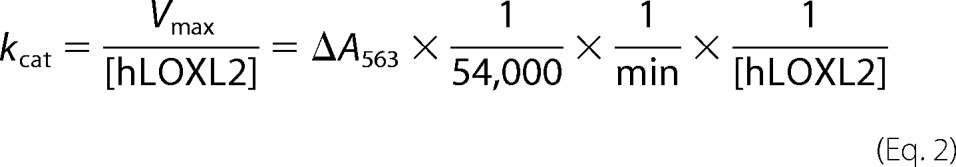 |
Two negative control experiments were also conducted, one without the addition of substrate and the other with 10-fold excess BAPN.
Phenylhydrazine Derivatization, Trypsin Digestion, and Mass Spectrometry
After incubation with 10-fold excess phenylhydrazine and confirmation of no residual activity, Δ1-3SRCR-HLOXL2 (100 μg) was subjected to in-solution trypsin digestion as described elsewhere (16). The protease-digested sample (5 μl, ∼15 μg) was injected onto a C18 column (3-μm particle, 0.3 × 50 mm, CVC Microtech, Fontana, CA) connected to a Waters ACQUITY UPLC system (Milford, MA), which was directly coupled to an electrospray ionization (ESI)-linear ion trap mass spectrometer (ESI-LTQ Velos MS, Thermo Scientific). Aqueous mobile phase consisted of 99.9% water and 0.1% formic acid (solvent A), and organic mobile phase was composed of 99.9% acetonitrile and 0.1% formic acid (solvent B). For reversed phase HPLC, the solvent was held initially at 5% solvent B with a linear increase to 10% solvent B over 5 min, a linear increase to 40% solvent B over 45 min, and a linear increase to 90% solvent B over 10 min, held at 90% solvent B for 10 min before re-equilibrating the column. For mass spectrometry, the electrospray source voltage was 3 kV, and capillary temperature was 250 °C. For MS/MS analysis of peptides by collision-induced dissociation, 30% collision energy was used. LC-MS/MS was set up in data-dependent scan mode where the five most intense ions were chosen for MS/MS analysis with a 3-min dynamic exclusion window. The phenylhydrazine-derivatized LTQ-containing peptide was identified manually using Xcalibur version 2.1 software (Thermo Scientific) by calculating the mass of the cross-linked peptide with and without certain modifications, including the presence and absence of the phenylhydrazine tag, up to two trypsin missed cleavages, and methionine oxidation. The predicted m/z values were searched against the data to identify the MS/MS spectrum containing the appropriate precursor ion mass and b and y ions corresponding to the cross-link.
Peptide Mapping and Glycan Analysis
For analysis of glycopeptides in the MS data, sample preparation and MS data collection were performed as described above, and manual data interpretation was completed by two strategies. The first strategy was to create a prediction table where the mass of a peptide containing an N-linked glycosylation site was added to probable glycoforms specific for Drosophila cells (17), and the predicted m/z values were searched against the MS and MS/MS data. Secondly, the MS/MS data were scanned for characteristic ions indicative of glycopeptide spectra. For example, m/z 366 (a hexose plus an N-acetylhexosamine residue) is often found in the tandem mass spectra of glycopeptides, and it is considered a marker ion for glycopeptides. Upon detection of m/z 366 and other marker ions, the appropriate tandem mass spectra were examined for the appropriate glycan losses for compositional assignment of the glycopeptide.
RESULTS AND DISCUSSION
The LOX family of proteins is traditionally known to require >2 m urea for extraction and/or solubilization. However, our two truncated forms of recombinant hLOXL2, Δ1-3SRCR-hLOXL2 and Δ1-4SRCR-hLOXL2 (Fig. 1A), isolated from the medium of stable S2 cells, are soluble at physiological pHs in buffers without urea or detergent. Both recombinant hLOXL2s exhibited LOX amine oxidase activity toward tropoelastin, an in vitro LOX substrate, in the standard HRP-coupled Amplex Red LOX activity assay (15), and the activity was completely abolished by BAPN, a potent inhibitor of LOX (18). The Km values were determined to be 0.59 ± 0.13 μm (for Δ1-3SRCR-hLOXL2) and 0.62 ± 0.17 μm (for Δ1-4SRCR-hLOXL2), and kcat values were determined to be 2.04 ± 0.17 min−1 and 0.69 ± 0.07 min−1, respectively, at pH 8.0. These values are comparable with the reported values for native LOX from bovine aorta (19, 20). When the fourth SRCR domain was absent, Km was not affected, but kcat was reduced by ∼65%, suggesting that the fourth SRCR domain plays an important role in optimizing the catalytic efficacy of the LOX catalytic domain of hLOXL2. This supports the observation that AB0023, a specific antibody against the fourth SRCR domain, allosterically inhibits hLOXL2 (21, 22).
The molecular masses of Δ1-3SRCR-hLOXL2 and Δ1-4SRCR-hLOXL2 were determined by gel electrophoresis to be ∼45 and ∼30 kDa, respectively (Fig. 1B). The m/z values of Δ1-3SRCR-hLOXL2 and Δ1-4SRCR-hLOXL2 were respectively determined to be 42727.8 ± 2.9 and 29562.0 ± 0.6 by electrospray ionization mass spectrometry on a Q-TOF-2 hybrid mass spectrometer (Micromass Ltd., Manchester, UK) operated in MS mode. The differences between the observed protein masses and those predicted from their primary peptide sequences (40.8 and 28.7 kDa, respectively) are ∼2 and ∼0.8 kDa, respectively, and these recombinant proteins are most likely glycosylated. NetNGlyc 1.0 server predicted three N-linked glycosylation sites (Asn-X-Ser/Thr) in hLOXL2, namely Asn-288 (in the second SRCR domain), Asn-455 (in the fourth SRCR domain) and Asn-644 (in the LOX catalytic domain), as shown in Fig. 1A. Given the differences between the observed m/z and the predicted values, the N-glycan at Asn-644 is expected to be ∼0.8 kDa, and that at Asn-455 is expected to be ∼1.2 kDa.
The sizes of Δ1-3SRCR-hLOXL2 and Δ1-4SRCR-hLOXL2 were reduced by noticeably different amounts after PNGase F treatment (Fig. 1C), supporting the prediction that both Asn-455 and Asn-644 are N-glycosylated in S2 cell-produced recombinant hLOXL2s. In a lectin binding assay, we detected positive stains only with Galanthus nivalis agglutinin (Fig. 1D), suggesting that the N-glycans at Asn-455 and Asn-644 are the high mannose type. The absence of sialic acid is expected as insect cells rarely produce terminal sialic acids (23, 24).
The purified Δ1-3SRCR-hLOXL2 was subjected to in-solution trypsin digestion. The resulting peptide fragments were separated on a C18 column, which was directly coupled to an ESI-LTQ Velos mass spectrometer. Peptide mapping yielded 71% coverage (16). We detected multiple glycopeptides. Among them, peptide 1 with m/z 1255 corresponds to the predominant glycoform of the peptide 455NGSLVWGMVCGQNWGIVEAMVVCR478 (underlined is Asn-455) (Fig. 2A), and peptide 2 at m/z 1153 corresponds to the predominant glycoform of the peptide 628HYHSMEVFTHYDLLNLNGTK647 (underlined is Asn-644) (Fig. 2B). GlycoPep DB (25) and GlycoPep ID (26) were utilized to determine the glycopeptide composition, and all assignments were supported by additional manual interpretation. The subsequent losses of monosaccharides, as labeled on the spectra, can be used to fully deduce the glycopeptide assignment. The N-glycans at Asn-455 and Asn-644 are identified as fucosylated species attached to a trimannose N-linked glycan core (Fig. 2, A and B). This assignment is consistent with our lectin binding assay results (Fig. 1D) and also with the fact that a fucosylated N-linked glycan core is known to be one of the most common glycoforms in insect cells and for proteins expressed in insect cell lines (27, 28).
FIGURE 2.

MS analyses of the PTMs of Δ1-3SRCR-hLOXL2. A, glycopeptide fragment 1 (455–478) detected by mass spectrometry. B, glycopeptide fragment 2 (628–647) detected by mass spectrometry. C, ESI-MS/MS analysis of the peptide fragment containing phenylhydrazine-derivatized LTQ after tryptic digestion. The derivatized LTQ-containing cross-link, shown as an inset as the azo tautomer, is circumscribed by a green oval at the top.
The N-glycosylation site at Asn-455 is unique to hLOXL2, although the site at Asn-644 is conserved in the SRCR domain-containing hLOXLs (hLOXL2, hLOXL3, and hLOXL4) but not in hLOX or in hLOXL1. N-Linked glycosylation has been shown to be essential for proper folding and stability of newly synthesized proteins in the cytosol to prevent self-aggregation and improve solubility (27). To examine whether this is true of the N-linked glycans in hLOXL2, we prepared S2 expression constructs for N455Q and N644Q mutants of Δ1-3SRCR-hLOXL2 and an N644Q mutant of Δ1-4SRCR-hLOXL2. S2 cells were transiently transfected with each of the three expression plasmids, and protein expression was induced by the addition of CuSO4. Four to 5 days after induction, the cells and culture medium were harvested, and the S2 cell extracts were fractionated into soluble and insoluble proteins. We did not detect any of the mutant proteins secreted into the medium. Instead, the N455Q and N644Q mutants of Δ1-3SRCR-hLOXL2 were found in very small quantities, mostly in the insoluble fraction of the cell extracts (Fig. 3A), whereas no protein was detected for the N644Q mutant of Δ1-4SRCR-hLOXL2 (Fig. 3B). We also selected S2 cells stably expressing N455Q and N644Q mutants of Δ1-3SRCR-hLOXL2, with stable integration verified by PCR (Fig. 3C). Neither of these mutants was even detected at all. These results collectively suggest that when either of the glycosylation site asparagines is mutated to prevent N-glycosylation, the proteins fail to fold properly and undergo rapid degradation, most likely through endoplasmic reticulum-associated degradation. It is probable that instances in which we observed expression represent a window during which intermediates in the degradation pathway are present in detectable quantities.
FIGURE 3.

Effect of N-linked glycosylation on the secretion of recombinant hLOXL2s and hLOX from S2 cells. A and B, Western blot analyses of the expression profiles of the glycosylation site mutants of Δ1-3SRCR-hLOXL2 (A)(nonadjacent lanes from the same blot are digitally juxtaposed) and Δ1-4SRCR-hLOXL2 (B) from induced transiently transfected cells. M, cell culture medium; S, soluble cell lysate; I, insoluble cell lysate (cell pellet). C, PCR confirmation of the stable integration of the N455Q and N644Q mutants of Δ1-3SRCR-hLOXL2 into the S2 genome. Stably integrated WT Δ1-3SRCR-hLOXL2 was used as a positive control. D and E, Western blot analyses of the expression profiles of unglycosylated WT Δ1-3SRCR-hLOXL2 (D) and WT Δ1-4SRCR-hLOXL2 (E) from tunicamycin-treated induced stable cells. Panels D and E are γ-adjusted to enhance minimally visible bands. Tunica., tunicamycin. F, Western blot analysis of expression of the LOX catalytic domain of hLOX in stable S2 cells.
To clarify whether the observed inhibition of secretion was caused by a lack of glycosylation or by structural change brought about through site-directed mutagenesis, we treated stable cell lines expressing the wild-type recombinant proteins with tunicamycin, an analog of UDP-N-acetylglucosamine (UDP-GlcNAc) that blocks N-linked glycosylation of newly synthesized proteins by inhibiting GlcNAc C-1-phosphotransferase (29). We discovered that tunicamycin treatment fully inhibited the secretion of the wild-type recombinant proteins and that the nonglycosylated proteins were found only in the insoluble fraction of cell extracts (Fig. 3, D and E). These results demonstrate that it is the N-glycans at Asn-455 and Asn-644 that are essential for proper folding and secretion of hLOXL2 from S2 cells.
The LOX catalytic domains of hLOX and hLOXL1 share 48–49% identity and 66–68% homology with Δ1-4SRCR-hLOXL2. However, the catalytic domains of hLOX and hLOXL1 lack the N-glycosylation site found in Δ1-4SRCR-hLOXL2. We used the same S2 expression system to express the catalytic domain of hLOX, but it was produced only as inclusion bodies (Fig. 3F). These results suggest that the propeptide of LOX is essential for proper folding and secretion of hLOX, supporting the recent study wherein the propeptide of hLOX was shown to be essential for secretion in CHO cells (30). In that study, N-linked glycosylation of the propeptide of hLOX was shown to be neither required for secretion nor required for extracellular proteolytic processing of pro-hLOX in CHO cells; however, N-linked glycosylation was required to achieve optimal enzymatic activity of mature hLOX. This is somewhat puzzling, as the propeptide is not retained in mature hLOX. It is possible that N-glycosylation of the propeptide affects protein folding and biogenesis of the LTQ cofactor. Further study is necessary to evaluate this hypothesis.
The LTQ cofactor was first identified by mass spectrometry of phenylhydrazine-derivatized and protease-digested LOX partially purified from bovine aorta (9). The phenylhydrazine adduct of the LTQ cofactor exists in tautomeric equilibrium, predominantly in the azo form (Fig. 2C, inset) (31, 32). Sequence alignment of hLOXL2 with bovine LOX indicates that Lys-653 and Tyr-689 of hLOXL2 are likely precursor residues for the LTQ cofactor. To confirm this hypothesis, Δ1-3SRCR-hLOXL2 was incubated with 10-fold excess phenylhydrazine and subjected to trypsin digestion after confirming complete loss of catalytic activity. The presence of the LTQ cofactor was determined by electrospray mass spectrometry (Fig. 2C, Table 1). We identified the derivatized LTQ-containing peptide fragment, strongly supporting that Lys-653 and Tyr-689 are precursors for the LTQ cofactor in hLOXL2. The N-glycosylation site at Asn-644 that was determined in this study resides between the predicted Cu2+ binding site (His-626-X-His-628-X-His-630) and Lys-653 of the LTQ cofactor. The accessibility of this site to an endogenous glycotransferase and PNGase F suggests that the active site is solvent-exposed.
TABLE 1.
Theoretical and observed masses of the parent ion containing the phenylhydrazine-derivatized LTQ cross-link
| Mass (theoretical) | Mass (observed) | Parent iona,b,c |
|---|---|---|
| 1739.2 | 1738.7 | (α+β+PH+3H)3+ |
| 1304.7 | 1304.6 | (α+β+PH+4H)4+ |
| 1043.9 | 1044.2 | (α+β+PH+5H)5+ |
a α = peptides 670–690, containing Tyr-689 of the LTQ cofactor, minus 2H+.
b β = peptides 648–669, containing Lys-653 of the LTQ cofactor, minus H+.
c PH = phenyl–N=N–.
CONCLUSION
This study revealed that S2-secreted hLOXL2 is an N-glycosylated protein, where N-linked glycans at Asn-455 and Asn-644 are each essential for protein stability and secretion from S2 cells. Although the LOX catalytic domains of hLOXL2 and hLOX are highly conserved, the latter lacks the N-glycosylation site. Thus, we hypothesize that the difference in glycosylation status of the LOX catalytic domains accounts for the observed disparity in solubility, and furthermore opens the intriguing possibility of functional differences between these proteins. We are currently examining the role of the N-glycan at Asn-288 in the secretion of hLOXL2. We also obtained mass spectrometry data strongly supporting that Lys-653 and Tyr-689 are the precursor residues for the LTQ cofactor of hLOXL2. This is the first reported determination of the presence of the LTQ cofactor in the human LOX family of proteins.
Acknowledgments
We thank Robert Drake for Q-TOF-2 analysis. We also thank Daniel Belz, Marissa Taylor, and Kyla Valaer for technical assistance in this research.
This work was supported, in whole or in part, by National Institutes of Health Grants P20 RR015563 Project 3 (to M. M., Center of Biomedical Research Excellence for Cancer Experimental Therapeutics), GM079446-02 (to M. M.), AHA0765445Z (to M. M.), and RO1RR026061 (to H. D.). This work was also supported by National Science Foundation (NSF) Grants CAREER MCB-0747377 (to M. M.) and CAREER CHE-0645120 (to H. D.).
- hLOX
- human lysyl oxidase
- hLOXL2
- human lysyl oxidase-like 2
- LOX
- lysyl oxidase
- ECM
- extracellular matrix
- SRCR
- scavenger receptor cysteine-rich
- LTQ
- lysine tyrosylquinone
- PTM
- post-translational modification
- PNGase F
- peptide-N-glycosidase F
- DIG
- digoxigenin
- BAPN
- β-aminopropionitrile
- ESI
- electrospray ionization.
REFERENCES
- 1. Barker H. E., Chang J., Cox T. R., Lang G., Bird D., Nicolau M., Evans H. R., Gartland A., Erler J. T. (2011) LOXL2-mediated matrix remodeling in metastasis and mammary gland involution. Cancer Res. 71, 1561–1572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Moreno-Bueno G., Salvador F., Martín A., Floristán A., Cuevas E. P., Santos V., Montes A., Morales S., Castilla M. A., Rojo-Sebastián A., Martínez A., Hardisson D., Csiszar K., Portillo F., Peinado H., Palacios J., Cano A. (2011) Lysyl oxidase-like 2 (LOXL2), a new regulator of cell polarity required for metastatic dissemination of basal-like breast carcinomas. EMBO Mol. Med. 3, 528–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Payne S. L., Hendrix M. J., Kirschmann D. A. (2007) Paradoxical roles for lysyl oxidases in cancer–a prospect. J. Cell. Biochem. 101, 1338–1354 [DOI] [PubMed] [Google Scholar]
- 4. Levental K. R., Yu H., Kass L., Lakins J. N., Egeblad M., Erler J. T., Fong S. F., Csiszar K., Giaccia A., Weninger W., Yamauchi M., Gasser D. L., Weaver V. M. (2009) Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 139, 891–906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Peng L., Ran Y. L., Hu H., Yu L., Liu Q., Zhou Z., Sun Y. M., Sun L. C., Pan J., Sun L. X., Zhao P., Yang Z. H. (2009) Secreted LOXL2 is a novel therapeutic target that promotes gastric cancer metastasis via the Src/FAK pathway. Carcinogenesis 30, 1660–1669 [DOI] [PubMed] [Google Scholar]
- 6. Jourdan-Le Saux C., Tronecker H., Bogic L., Bryant-Greenwood G. D., Boyd C. D., Csiszar K. (1999) The LOXL2 gene encodes a new lysyl oxidase-like protein and is expressed at high levels in reproductive tissues. J. Biol. Chem. 274, 12939–12944 [DOI] [PubMed] [Google Scholar]
- 7. Csiszar K. (2001) Lysyl oxidases: a novel multifunctional amine oxidase family. Prog. Nucleic Acid Res. Mol. Biol. 70, 1–32 [DOI] [PubMed] [Google Scholar]
- 8. Smith-Mungo L. I., Kagan H. M. (1998) Lysyl oxidase: properties, regulation, and multiple functions in biology. Matrix Biol. 16, 387–398 [DOI] [PubMed] [Google Scholar]
- 9. Wang S. X., Mure M., Medzihradszky K. F., Burlingame A. L., Brown D. E., Dooley D. M., Smith A. J., Kagan H. M., Klinman J. P. (1996) A crosslinked cofactor in lysyl oxidase: redox function for amino acid side chains. Science 273, 1078–1084 [DOI] [PubMed] [Google Scholar]
- 10. Vora S. R., Guo Y., Stephens D. N., Salih E., Vu E. D., Kirsch K. H., Sonenshein G. E., Trackman P. C. (2010) Characterization of recombinant lysyl oxidase propeptide. Biochemistry 49, 2962–2972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bollinger J. A., Brown D. E., Dooley D. M. (2005) The formation of lysine tyrosylquinone (LTQ) is a self-processing reaction. Expression and characterization of a Drosophila lysyl oxidase. Biochemistry 44, 11708–11714 [DOI] [PubMed] [Google Scholar]
- 12. Cronshaw A. D., Fothergill-Gilmore L. A., Hulmes D. J. (1995) The proteolytic processing site of the precursor of lysyl oxidase. Biochem. J. 306, 279–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Panchenko M. V., Stetler-Stevenson W. G., Trubetskoy O. V., Gacheru S. N., Kagan H. M. (1996) Metalloproteinase activity secreted by fibrogenic cells in the processing of prolysyl oxidase: potential role of procollagen C-proteinase. J. Biol. Chem. 271, 7113–7119 [DOI] [PubMed] [Google Scholar]
- 14. Uzel M. I., Scott I. C., Babakhanlou-Chase H., Palamakumbura A. H., Pappano W. N., Hong H. H., Greenspan D. S., Trackman P. C. (2001) Multiple bone morphogenetic protein 1-related mammalian metalloproteinases process pro-lysyl oxidase at the correct physiological site and control lysyl oxidase activation in mouse embryo fibroblast cultures. J. Biol. Chem. 276, 22537–22543 [DOI] [PubMed] [Google Scholar]
- 15. Palamakumbura A. H., Trackman P. C. (2002) A fluorometric assay for detection of lysyl oxidase enzyme activity in biological samples. Anal. Biochem. 300, 245–251 [DOI] [PubMed] [Google Scholar]
- 16. Rebecchi K. R., Go E. P., Xu L., Woodin C. L., Mure M., Desaire H. (2011) A general protease digestion procedure for optimal protein sequence coverage and post-translational modifications analysis of recombinant glycoproteins: application to the characterization of human lysyl oxidase-like 2 glycosylation. Anal. Chem. 83, 8484–8491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fabini G., Freilinger A., Altmann F., Wilson I. B. (2001) Identification of core α1,3-fucosylated glycans and cloning of the requisite fucosyltransferase cDNA from Drosophila melanogaster: potential basis of the neural anti-horseradish peroxidase epitope. J. Biol. Chem. 276, 28058–28067 [DOI] [PubMed] [Google Scholar]
- 18. Tang S. S., Trackman P. C., Kagan H. M. (1983) Reaction of aortic lysyl oxidase with β-aminopropionitrile. J. Biol. Chem. 258, 4331–4338 [PubMed] [Google Scholar]
- 19. Bedell-Hogan D., Trackman P., Abrams W., Rosenbloom J., Kagan H. (1993) Oxidation, cross-linking, and insolubilization of recombinant tropoelastin by purified lysyl oxidase. J. Biol. Chem. 268, 10345–10350 [PubMed] [Google Scholar]
- 20. Shah M. A., Scaman C. H., Palcic M. M., Kagan H. M. (1993) Kinetics and stereospecificity of the lysyl oxidase reaction. J. Biol. Chem. 268, 11573–11579 [PubMed] [Google Scholar]
- 21. Barry-Hamilton V., Spangler R., Marshall D., McCauley S., Rodriguez H. M., Oyasu M., Mikels A., Vaysberg M., Ghermazien H., Wai C., Garcia C. A., Velayo A. C., Jorgensen B., Biermann D., Tsai D., Green J., Zaffryar-Eilot S., Holzer A., Ogg S., Thai D., Neufeld G., Van Vlasselaer P., Smith V. (2010) Allosteric inhibition of lysyl oxidase-like-2 impedes the development of a pathologic microenvironment. Nat. Med. 16, 1009–1017 [DOI] [PubMed] [Google Scholar]
- 22. Rodriguez H. M., Vaysberg M., Mikels A., McCauley S., Velayo A. C., Garcia C., Smith V. (2010) Modulation of lysyl oxidase-like 2 enzymatic activity by an allosteric antibody inhibitor. J. Biol. Chem. 285, 20964–20974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Aumiller J. J., Hollister J. R., Jarvis D. L. (2003) A transgenic insect cell line engineered to produce CMP-sialic acid and sialylated glycoproteins. Glycobiology 13, 497–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Marchal I., Jarvis D. L., Cacan R., Verbert A. (2001) Glycoproteins from insect cells: sialylated or not? Biol. Chem. 382, 151–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Go E. P., Rebecchi K. R., Dalpathado D. S., Bandu M. L., Zhang Y., Desaire H. (2007) GlycoPep DB: a tool for glycopeptide analysis using a “Smart Search”. Anal. Chem. 79, 1708–1713 [DOI] [PubMed] [Google Scholar]
- 26. Irungu J., Go E. P., Dalpathado D. S., Desaire H. (2007) Simplification of mass spectral analysis of acidic glycopeptides using GlycoPep ID. Anal. Chem. 79, 3065–3074 [DOI] [PubMed] [Google Scholar]
- 27. Hirsch C., Blom D., Ploegh H. L. (2003) A role for N-glycanase in the cytosolic turnover of glycoproteins. EMBO J. 22, 1036–1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kim Y. K., Shin H. S., Tomiya N., Lee Y. C., Betenbaugh M. J., Cha H. J. (2005) Production and N-glycan analysis of secreted human erythropoietin glycoprotein in stably transfected Drosophila S2 cells. Biotechnol. Bioeng. 92, 452–461 [DOI] [PubMed] [Google Scholar]
- 29. Stanley P., Schachter H., Taniguchi N. (2009) Chapter 8, N-Glycans. in Essentials of Glycobiology (Varki A., Cummings R., Esko J., HH., F., Stanley P., Bertozzi C., Hart G., Etzler M. eds) pp. 101–114, Second Ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 30. Grimsby J. L., Lucero H. A., Trackman P. C., Ravid K., Kagan H. M. (2010) Role of lysyl oxidase propeptide in secretion and enzyme activity. J. Cell. Biochem. 111, 1231–1243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mure M., Wang S. X., Klinman J. P. (2003) Synthesis and characterization of model compounds of the lysine tyrosyl quinone cofactor of lysyl oxidase. J. Am. Chem. Soc. 125, 6113–6125 [DOI] [PubMed] [Google Scholar]
- 32. Wang S. X., Nakamura N., Mure M., Klinman J. P., Sanders-Loehr J. (1997) Characterization of the native lysine tyrosylquinone cofactor in lysyl oxidase by Raman spectroscopy. J. Biol. Chem. 272, 28841–28844 [DOI] [PubMed] [Google Scholar]