

Background: The small GTPase RhoC is an essential mediator of tumor cell invasion and metastasis.
Results: The protein kinase MRK binds to and is activated by both RhoA and RhoC, but phenocopies RhoC in the control of LPA-stimulated tumor cell invasion.
Conclusion: MRK is a novel RhoC effector that mediates tumor cell invasion.
Significance: Dissecting RhoC-mediated signaling is important for understanding tumor metastasis.
Keywords: Cell Invasion, MAP Kinases (MAPKs), Myosin, Rho GTPases, Signal Transduction, RhoC
Abstract
The small GTPase RhoC is overexpressed in many invasive tumors and is essential for metastasis. Despite its high structural homology to RhoA, RhoC appears to perform functions that are different from those controlled by RhoA. The identity of the signaling components that are differentially regulated by these two GTPases is only beginning to emerge. Here, we show that the MAP3K protein MRK directly binds to the GTP-bound forms of both RhoA and RhoC in vitro. However, siRNA-mediated depletion of MRK in cells phenocopies depletion of RhoC, rather than that of RhoA. MRK depletion, like that of RhoC, inhibits LPA-stimulated cell invasion, while depletion of RhoA increases invasion. We also show that active MRK enhances LPA-stimulated invasion, further supporting a role for MRK in the regulation of invasion. Depletion of either RhoC or MRK causes sustained myosin light chain phosphorylation after LPA stimulation. In addition, activation of MRK causes a reduction in myosin light chain phosphorylation. In contrast, as expected, depletion of RhoA inhibits myosin light chain phosphorylation. We also present evidence that both RhoC and MRK are required for LPA-induced stimulation of the p38 and ERK MAP kinases. In conclusion, we have identified MRK as a novel RhoC effector that controls LPA-stimulated cell invasion at least in part by regulating myosin dynamics, ERK and p38.
Introduction
Rho proteins form a subgroup of the Rho family of small GTPases that are involved in tumor progression and metastasis (1, 2). The closely related Rho GTPases RhoA and RhoC have been found to be overexpressed in a variety of tumors (3–6). In some cases their expression correlates with tumor progression (7–9). In addition, RhoC has been shown to play an essential role in metastasis in animal models (10–12), and RhoC expression correlates with increased invasion in several tumor types (5, 13–16).
RhoA and RhoC proteins are 94% identical, and, although they activate the same effectors upon overexpression in cells (17), they appear to have different roles in cell invasion, as RhoC down-regulation inhibits invasion, whereas RhoA depletion increases it, although not in all cell types (9, 18–22). However, the identity of the signaling components involved in RhoC-mediated cell invasion is only beginning to emerge (9, 23, 24).
Lysophosphatidic acid (LPA)7 is a well-established activator of RhoA. LPA stimulates RhoA via Gα13 (25), the α subunit of a heterotrimeric GTPase (26, 27). Both LPA and Gα13 are known to play important roles in cell migration. LPA is an extracellular lipid that mediates hormone and growth factor-like responses in many cell types (28) and stimulates cell migration of both neoplastic and non-neoplastic cells (29). Gα13 is essential for cell migration during development ((30, 31), as well as for migration of cancer cells (32).
Among the known signaling cascades that act downstream of Rho GTPases are the mitogen-activated kinase (MAPK) pathways JNK and p38 (33–37). These cascades relay a wide range of signals downstream of growth factors as well as cellular stress stimuli and cytokines. MRK (MLK-related kinase (38), also known as MLTK (39) and ZAK (40)), is a member of the MAP3K family of protein kinases that activates MAP kinase pathways upon overexpression (38, 39). MRK is activated in response to cellular stresses (38, 39, 41), but its upstream activator/s are not known. In this report, we have used recombinant proteins and RNA interference to show that MRK functions downstream of RhoC and is a novel signaling element in LPA-stimulated tumor cell invasion.
EXPERIMENTAL PROCEDURES
Cell Cultures and Transfections
The osteosarcoma U2-OS cells were obtained from ATCC and cultured as suggested. The ovarian ES-2 cells were kindly provided by Karen Auborn (Feinstein Institute). The M28 cell line was derived from U2-OS Tet-On cells (Clontech) that were engineered to express a trans-activator responsive to doxycyclin, a tetracycline derivative. These cells were transfected with a pTRE plasmid expressing the MRK gene under the control of the Tet-inducible promoter. Individual clones were selected for puromycin resistance and tested for MRK inducibility following doxycyclin treatment. Clone M28 was identified for good inducibility and used in these studies. UMDI and EMDI cells were derived from U2-OS and ES-2 cells, respectively, after stable transfection with plasmid pC4-Fv1E that contains a fusion of the MRK ORF with the binding domain of a FKBP12 drug derivative separated by the following hinge sequence: TCTAGAATTTCCGGTGGTGGTGGTGGAGGTGGTGGTGGTGGAGGAGGAGGTGGTGGTGGAATTACTAGT (the underlined sequences are the restriction sites used in cloning). The pC4-Fv1E plasmid is part of the ARGENT-regulated homodimerization kit, now available from Clontech as the Idimerize System. The resulting construct expresses the MRK protein that is activated upon treatment with the dimerizing drug AP20187, or upon stimulation with physiological stimuli like LPA. Transfections of plasmids or siRNAs were performed using Lipofectamine 2000 (Invitrogen) as previously described (41). Transfections of siRNAs were done using 20 pmol of the siRNA oligos and 2 μl of Lipofectamine 2000 per well of a 6-well plate.
All siRNA duplexes were supplied by Dharmacon (Thermo Fisher Scientific). Specific oligonucleotides for each target gene were as follows: 5′-GAAUGUCUGAGGAGUCUUA-3′ (M1) targeting MRK; 5′-AUGGAAAGCAGGUAGAGUU-3′ (A1) and 5′-GCAGAGAUAUGGCAAACAG-3′ (A2) targeting RhoA;5′-GGAGAGAGCUGGCCAAGAU-3′ (C1) and 5′-GAACUAUAUUGCGGACAUU-3′ (C2) targeting RhoC; and 5′-CGUACGCGGAAUACUUCGA-3′ luciferase siRNA as control.
Western Blot Analysis
Cells were lysed for 30 min in NETN lysis buffer (50 mm Tris/HCl pH 7.5, 150 mm NaCl, 1 mm EDTA, 1% Nonidet P-40, 2 mm DTT, 1 mm Na3OVO4, 1 mm β-glycerophosphate, 5 mm NaFl, supplemented with protease inhibitors (Roche and Benzonase (Novagen). Proteins were resolved on 10% NuPAGE gels (Invitrogen).
Antibodies
The MRK antibodies 4-23 (monoclonal), 40-5 (polyclonal), and phosphospecific antibody were previously described (38, 41). The Gα13 antibody, the monoclonal RhoA antibody (sc-418), the goat RhoC polyclonal antibody (sc-12116) and the ROCKII antibody were from Santa Cruz Biotechnology. The tubulin and Rac antibodies were from Upstate (Millipore). The P-ERK, P-p38, and P-MLC antibodies were from Cell Signaling. The vinculin antibody was from Sigma. The Myc tag antibody was from Roche and the KT3-tag antibody was from Bethyl.
Rho Pull-down Assay
Rho proteins were expressed as GST-fusions in Escherichia coli, purified and loaded with GDP or GTPγS as described in Self and Hall (42). Nucleotide-loaded proteins were incubated with purified MRK or ROCKII, immobilized onto beads as immunoprecipitates from serum-starved cells. Protein complexes were washed and resolved on SDS-PAGE.
Rho Activation Assay
Rho GTPase activity was measured using the EZ-Detect Rho Activation kit from Pierce, using 1 mg of protein lysate.
Cell Motility Assay
Cell motility was measured with the wound-healing assay as previously described in Valster et al. (43).
Matrigel Invasion Assay
Invasion was assayed using Matrigel Invasion Chambers (Becton Dickinson). Cells were transfected with the appropriate siRNAs. Twenty-four hours post-transfection, they were starved from serum overnight. Then, they were trypsinized; trypsin was neutralized by trypsin inhibitor, and 3 × 104 cells were placed in the top chamber in serum-free media supplemented with 0.1% fatty acid-free BSA. LPA (50 μm) in serum-free media containing BSA was added to the bottom chamber. After 24 h of incubation at 37 °C, cells on the top surface of the filter were wiped off with a Q-tip, and the filter was fixed in 4% formaldehyde/PBS. After staining with Crystal Violet, all of the cells on the bottom of the chamber were counted using an IX70 Olympus inverted microscope.
Immunofluorescence
40,000 cells were plated onto coverslips, and 24 h later they were fixed with 4% formaldehyde and processed for immunofluorescence with vinculin antibodies as previously described (44). Phalloidin was used to stain the actin cytoskeleton. Images were collected using an IX70 Olympus inverted microscope equipped with a X60 (1.4 numerical aperture) objective, an Orca II cooled charge coupled device (CCD) camera (Hamatsu) and ESee (Inovision) image analysis software.
RESULTS
Lysophosphatidic Acid Activates MRK
To identify extracellular signals that activate MRK, we tested several growth factors that included epidermal growth factor (EGF), transforming growth factor β (TGFβ), and lysophosphatidic acid (LPA). We determined MRK activity by detecting its autophosphorylation, which we have shown previously to be essential for its activity (38, 41). Because of the low affinity of the MRK phospho-specific antibody, whenever we have examined MRK activity in this study we have used a derivative of the osteosarcoma cells U2-OS (termed UMDI) or a similar derivative of the ovarian cancer cell line ES-2 (EMDI). These cells are stably transfected with a fusion protein of MRK and the drug-binding domain of a FKBP12 derivative (45), which enables forced dimerization in the presence of the homodimerizing drug AP20187. In the absence of the drug, however, the fusion MRK protein is not active, although it can be activated by stimuli like osmotic shock (data not shown). Thus, for this experiment we used the UMDI cells in the absence of the dimerizing drug. We observed that MRK was readily activated by LPA, but not by TGF-β or EGF (Fig. 1A). To confirm that this response was not affected by the fusion with the drug-binding domain, we also examined another derivative of U2-OS cells (termed M28) that expresses recombinant wild type MRK and found that in these cells LPA also activates MRK (Fig. 1B).
FIGURE 1.
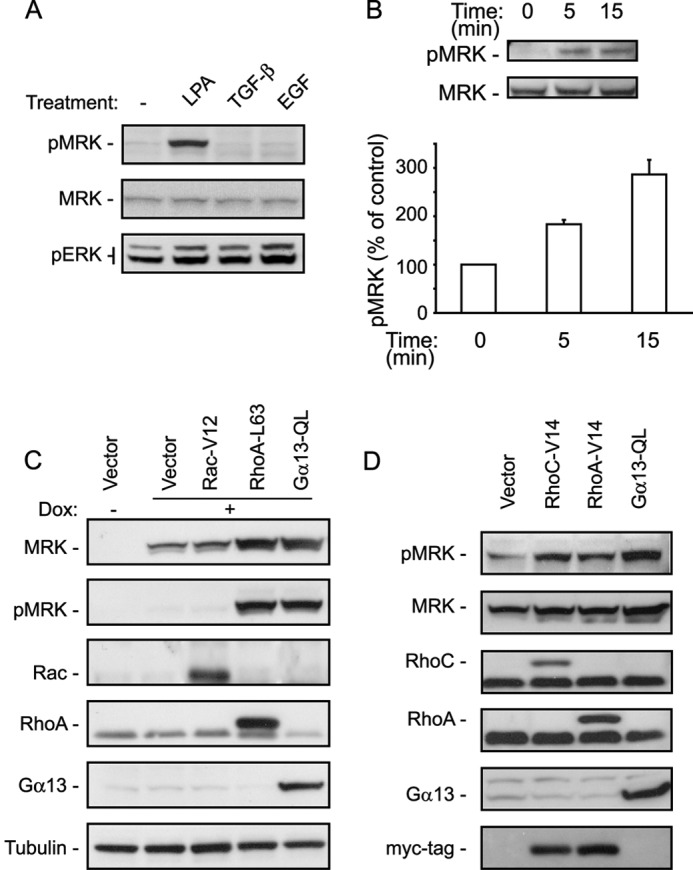
MRK is activated by LPA, Gα13 and Rho proteins. A, UMDI cells, expressing recombinant MRK fused to a drug-binding domain of FKBP12, were treated for 5 min with the following growth factors: 25 μm LPA, 10 ng/ml TGF-β1, and 50 μg/ml EGF, or left untreated (−). Cell lysates were tested by Western blotting for MRK activation using the P-MRK specific antibody. The P-ERK antibody was used to test for cellular stimulation by the growth factors. B, M28 cells that express KT3-tagged MRK after treatment with doxycyclin, were stimulated with LPA for the indicated times and tested for MRK activation as in A. Blots were scanned and the band intensities were quantified using ImageJ. Histograms are the means + S.E. of three independent experiments. C, M28 cells were transfected with vector control, Rac-V12, RhoA-L63, or Gα13-QL constructs. Forty-eight hours later they were treated overnight with doxycyclin (Dox) to induce expression of KT3-MRK and then tested for MRK activity by Western blotting with the P-MRK specific antibody. D, UMDI cells were transfected with the indicated DNA plasmids, 48 h later cells were harvested and tested for MRK activity as in C.
MRK Is Activated by Gα13, RhoA, and RhoC
LPA is known to signal via heterotrimeric G-protein-coupled receptors and Rho GTPases (26, 27). We, therefore, asked whether constitutively active forms of Gα13, Rac1, and RhoA could activate MRK. Constitutive activation of Cdc42 was toxic to the M28 cells and therefore could not be evaluated. M28 cells were transfected with control, Rac1-V12, RhoA-L63, or Gα13-QL plasmids and treated overnight with doxycyclin to induce recombinant MRK expression. Subsequently, cell lysates were analyzed by Western blotting to detect active MRK. Fig. 1C shows that expression of either RhoA-L63 or Gα13-QL induced MRK phosphorylation. Interestingly, activation of MRK was accompanied by an increase in MRK protein levels, suggesting the existence of a positive feedback loop that controls MRK protein levels. We obtained similar results in UMDI cells, where we observed that, in addition to active Gα13, both active RhoA and RhoC can activate MRK (Fig. 1D).
MRK Binds to Both RhoA and RhoC in Vitro
Next, we asked whether MRK acts as an effector of RhoA and RhoC. The definition of a GTPase effector is a protein that selectively binds to the GTP-bound active form of the GTPase, and mediates at least some of the functions of this GTPase. To examine whether MRK directly binds to either RhoA or RhoC, we immunoprecipitated KT3-tagged MRK from serum starved M28 cells and incubated it with purified RhoA or RhoC proteins that had been loaded with either GDP or GTPγS. As a positive control for the quality of the Rho proteins, we used ROCKII, which was reported to bind to RhoC (46). Fig. 2 shows that MRK binds to the GTP-bound forms of both RhoA and RhoC, and to a lesser extent to the GDP-bound form of these GTPases, suggesting a role for MRK as an effector of RhoA and RhoC.
FIGURE 2.

MRK interacts with Rho proteins in vitro. KT3-MRK was immunoprecipitated with KT3 antibodies from M28 cells grown in serum-free medium and incubated in vitro with purified RhoC or RhoA proteins loaded with GDP or GTP-γS. ROCKII protein was used as a positive control for RhoC-binding. Rho proteins were detected using the X-Press antibody that recognizes their tag.
Both RhoA and RhoC Contribute to Gα13-QL as Well as to LPA-stimulated Activation of MRK
To examine whether MRK functions downstream of RhoA or RhoC, we used siRNA-mediated depletion of the Rho proteins in M28 cells transfected with Gα13-QL. Fig. 3 shows that RhoC depletion strongly inhibits MRK activation stimulated by active Gα13, while RhoA depletion partially reduces it. To confirm this result in a more physiological context, we measured MRK activation levels in response to LPA in UMDI cells transfected with RhoA- or RhoC-specific siRNAs.
FIGURE 3.

Rho proteins are necessary for Gα13-QL-induced stimulation of MRK activation. M28 cells were co-transfected with vector control or Gα13-QL plasmids and either RhoA, RhoC, or luciferase (L) siRNAs. After 24 h they were treated with doxycyclin for 18 h to induce MRK expression and then harvested. Whole cell extracts were probed with the indicated antibodies. Two different siRNAs targeting RhoA and RhoC were tested. Histograms are means + S.E. of five independent experiments.
To date, RhoC has not been implicated in LPA-induced signaling. To verify that LPA stimulates RhoC activity, we performed a rhotekin pull-down assay in U2-OS cells. Fig. 4A shows that RhoC is significantly activated by LPA in a time-dependent fashion, albeit to a lesser extent than RhoA. RhoC activation by LPA was also observed in EMDI cells, a derivative of the ES-2 cells that also express the homodimerizing MRK fusion protein (Fig. 4B).
FIGURE 4.

LPA-stimulated MRK activation is inhibited by Rho proteins down-regulation. LPA stimulates activation of RhoA and RhoC proteins. U2OS cells (A) or EMDI cells (B) were treated with 25 μm LPA for the indicated times and then tested for activation of Rho proteins using pull-down assay with GST-Rhotekin-RBD. The last 3 lanes of each blot in A are pull-downs from steady-state lysates of cells transfected with the individual siRNAs that were used to control for band specificity. Each set of experiments was repeated 3–5 times; bands were quantified with the ImageJ program and expressed as means + S.E. in the corresponding histograms. C, UMDI cells, or D, EMDI cells, were transfected with control luciferase, RhoC or RhoA targeting siRNAs and then treated with 25 μm LPA for 5 min. Cell lysates were immunoblotted with the pMRK specific antibody to test for MRK activity, and with the indicated antibodies for controls. The pMRK bands were quantified with ImageJ. Histograms are means + S.E. of three independent experiments. p values representing Student's t test were *, p < 0.05; **, p < 0.01; ***, p < 0.005; ****, p < 0.001.
UMDI cells were starved overnight and subsequently stimulated with LPA for 5 min. MRK activation was determined using the phospho-MRK antibody. Quantification of the phospho-MRK signal indicated that depletion of RhoA did not affect LPA-stimulated MRK activity, although it increased the basal activity of MRK. In contrast, depletion of RhoC strongly inhibited LPA-stimulated MRK activation (Fig. 4C).
We also extended our observations to the EMDI cells, which demonstrated a strong LPA-dependent activation of MRK. However, in these cells, LPA-stimulated MRK activation was dependent on both Rho proteins (Fig. 4D). The EMDI cells also showed an increase in basal MRK activity upon depletion of RhoA.
We note that in some cell types, depletion of RhoA has been shown to cause up-regulation of RhoB (24, 47). Thus, in principle, RhoB could contribute to MRK activity in RhoA-depleted EMDI cells. However, we observed that RhoB levels in these cells are not affected by RhoA down-regulation (data not shown). In conclusion therefore, both RhoA and RhoC contribute to MRK activation.
MRK and RhoC Are Necessary for LPA-stimulated Tumor Cell Invasion
Rho proteins are key regulators of the actin cytoskeleton and cell migration and invasion. To examine the role of MRK in the invasive behavior of tumor cells, we used the ovarian carcinoma cell line ES-2, as the U2-OS cells were poorly invasive in response to LPA. It also is important to note that LPA plays an important role in ovarian cancer, controlling both tumor cell proliferation and invasion (48, 49).
We first tested cell migration using the wound healing assay. Fig. 5A shows that depletion of MRK inhibits cell migration by about 40%. Depletion of MRK also strongly inhibited LPA-stimulated invasion through Matrigel, to an extent that was similar to that achieved by depleting RhoC (Fig. 5B). In contrast, depletion of RhoA resulted in an increase in cell invasion, as previously reported for other cell types (18, 24). Taken together, these results suggest that MRK, even though it can be activated by both RhoA and RhoC, in the context of cell invasion, it functions as an effector of RhoC, rather than RhoA.
FIGURE 5.

MRK and RhoC positively control cell invasion, whereas RhoA negatively regulates it. A, ES-2 ovarian cancer cells were transfected with luciferase (Luc) or MRK siRNAs and then tested for migration in the wound healing assay for 6 and 12 h. B, ES-2 cells, transfected with the indicated siRNAs, were tested for LPA-stimulated cell invasion through Matrigel chambers as described under “Experimental Procedures.” Blots show depletion of the respective proteins by RNA interference. C, enhancement of LPA-stimulated cell invasion by active MRK induced by forced homodimerization. Detection by immunoblotting of MRK activated by the homodimerizing drug AP20187 in the EMDI cells and cell invasion through matrigel in response to LPA (1 μm) and AP20187 (2 μm). Histograms are means + S.E. from 3–6 independent experiments. *, p < 0.005; **, p < 0.001.
To substantiate the role of MRK in tumor cell invasion, we asked whether activating MRK promotes invasion in ES-2 cells. To specifically activate MRK, we reasoned that, like other MLK family members, MRK may homodimerize via its leucine zipper domain, which would lead to its activation (50, 51). To test this possibility, we used the EMDI cells in which MRK was activated by the homodimerizing drug AP20187. We showed that the EMDI cells expressing the activatable MRK construct have a low level of MRK activation in the absence of the AP20187 dimerizing drug, but a high level of activation in response to the drug (Fig. 5C, left panel).
We then examined the invasiveness of these cells in the presence or absence of AP20187. In the absence of LPA, we did not observe any significant increase in cell invasion upon addition of AP20187 (data not shown). However, in the presence of relatively low levels of LPA (1 μm), AP20187 induced a significant increase in cell invasion (Fig. 5C, right panel). No effect of the drug was detected in the parental ES-2 cells (data not shown).
Down-regulation of MRK or RhoC Impairs LPA-stimulated Activation of ERK and p38
We and others have shown that overexpression of MRK leads to activation of ERK and p38 MAP kinases (38, 39). To examine the role of MRK in LPA-stimulated activation of these MAP kinases, we silenced MRK in U2-OS cells and determined the activation levels of ERK and p38 using their respective phospho-specific antibodies. We showed that MRK depletion decreased the activity of these MAP kinases by 50 to 60%, confirming the role of MRK in the regulation of these MAP kinase pathways (Fig. 6, A and B). RhoC depletion also reduced ERK and p38 activity in response to LPA (Fig. 6C). In contrast, depletion of RhoA did not significantly change the activity of these proteins after LPA treatment, although it clearly caused an increase in their basal level (Fig. 6C).
FIGURE 6.

MRK and RhoC down-regulation inhibit LPA-stimulated ERK and p38 activation. UMDI cells were transfected with the MRK siRNA (A and B) for 36 h and with the Rho proteins specific siRNAs (C). Cells were starved overnight and treated with 25 μm LPA for 15 min. Cell lysates were blotted with the indicated antibodies. The pERK and p-p38 bands were quantified using ImageJ. Histograms represent the means + S.E. from three independent experiments. D and E, inhibition of ERK or p38 activity blocks invasion. ES-2 cells were processed for the Matrigel invasion assay as in Fig. 5 in the presence of the ERK inhibitor (D) and the p38 inhibitor (E). Histograms are the means + S.E. from 3–5 independent experiments. *, p < 0.05; **, p < 0.02; ***, p < 0.01; ****, p < 0.001.
The ERK and p38 MAP Kinases Are Necessary for Ovarian Cancer Cell Invasion
Both the ERK and p38 MAP kinase pathways have been implicated in the invasive behavior of a number of tumor cell types, including ovarian cancer cells (52–55). To confirm the role of these pathways in ES-2 cells, we used U0126 and SB203580, inhibitors that specifically block the respective pathways, in the context of LPA-stimulated invasion. Fig. 6, D and E show that the ERK and p38 MAP kinase pathways are both essential for ES-2 cell invasion. These results support the notion that MRK controls cell invasion downstream of RhoC at least in part by stimulating both the ERK and p38 MAP kinase pathways.
MRK and RhoC Are Necessary for Down-regulation of Myosin Activity
A major signaling event downstream of Rho proteins is myosin light chain phosphorylation which controls actomyosin contractility (56). We therefore also investigated the role of MRK in the regulation of myosin light chain phosphorylation in response to LPA. ES-2 cells were transfected with control or MRK-specific siRNAs and the phosphorylation state of myosin light chain (pMLC) was determined after short and long term stimulation with LPA. Fig. 7A shows that, although the pMLC level at 5 min was not affected by MRK depletion, the pMLC level remained relatively high after 60 min, whereas in control cells it was almost back to basal levels. This result suggests that MRK may be important for down-regulating myosin activity.
FIGURE 7.

MRK and RhoC down-regulation cause sustained LPA-induced MLC activation. ES-2 cells were transfected with the indicated siRNAs and 24 h later they were starved overnight. Cells were then stimulated with 25 μm LPA for the indicated time, harvested directly in SDS-containing sample buffer, and processed for Western blot analysis to test for P-MLC. Histograms represent the means + S.E. of 3–4 independent experiments. *, p < 0.05; **, p < 0.02.
Next, we determined the effects of depleting RhoA and RhoC on myosin phosphorylation. As expected, RhoA depletion strongly inhibited myosin phosphorylation (Fig. 7B). Interestingly, however, we found that depleting RhoC had an effect that was very similar to that of depleting MRK: there was no effect on short term phosphorylation of myosin, but it remained elevated at 60 min.
To confirm the role of MRK in down-regulating myosin phosphorylation, we treated EMDI cells, or their vector control counterpart, with LPA and monitored the effects of specific activation of MRK by induced homodimerization on the levels of pMLC. Fig. 8 shows that the pMRK levels stimulated by LPA in the absence of the homodimerizing drug (lane 5) were further increased by the AP drug after 30 min of stimulation. Coincident with this increase in MRK activation, LPA-stimulated activation of ERK and p38 increased, as expected, but LPA-stimulated pMLC levels were reduced, although pMLC levels began to recover at 60 min. These results demonstrate that MRK is necessary for the down-regulation of myosin activity, a function that is shared with RhoC, but not RhoA, further supporting that MRK functions as an effector of RhoC.
FIGURE 8.

Active MRK reduces LPA-stimulated MLC phosphorylation. EMDI cells were starved overnight, they were treated with the homodimerizing drug AP20187 (100 nm) for 1 h and then stimulated with LPA (10 μm) for 10 min. Cells were harvested directly in SDS-containing sample buffer and processed for Western blot with the indicated antibodies. Blot is representative of three independent experiments.
Sustained activation of myosin is expected to result in an increase of focal adhesions and stress fibers (56). To examine this, ES-2 cells were transfected with the respective siRNAs, fixed, and stained with phalloidin to monitor polymerized actin, or vinculin to visualize focal adhesions. Fig. 9 shows that, as previously reported by Vega et al. (24), depletion of RhoA and RhoC have strikingly different effects on cell morphologies: RhoA-depleted cells are elongated, whereas depletion of RhoC causes an increase in cell spreading. Quantification of focal adhesion length and numbers indicated that, whereas RhoA-depleted cells had fewer and shorter focal adhesions compared with controls, they were longer and more numerous in the RhoC- or MRK-depleted cells. Quantification of cell spreading showed about a 20% reduction after depletion of RhoA, in contrast to a ∼50% increase after depletion of either RhoC or MRK. Similar phenotypes were observed in U2-OS cells (data not shown). Thus, taken together, these results are in line with our observations that RhoC and MRK, in contrast with RhoA, promote MLC deactivation.
FIGURE 9.

Down-regulation of MRK or RhoC increases focal adhesion number and length as well as cell area. ES-2 cells were transfected with the indicated siRNAs, 48 h later they were plated on coverslips and 24 h later processed for immunofluorescence. Histograms are means + S.E. of data from 15–30 cells and are representative of two independent experiments. L: luciferase; A: RhoA; C: RhoC; M: MRK. *, p < 0.02; **, p < 0.001.
DISCUSSION
In this work, we provide evidence that MRK functions downstream of RhoC in the control of tumor cell invasion. In addition, our observations indicate that MRK-regulated invasion is mediated at least in part by activation of the ERK and p38 MAP kinase proteins and inactivation of myosin light chain.
Consistent with the paradigm that MAP3Ks can function as effectors of Rho family GTPases (34, 57, 58), this study shows that MRK interacts both with RhoA and RhoC in vitro. In addition, we found that both RhoA and RhoC can contribute to LPA-stimulated activation of MRK. Interestingly, however, our mechanistic analysis of cell invasion shows that depletion of MRK phenocopies depletion of RhoC, in contrast to that of RhoA. Taken together, these findings establish MRK as a novel effector of RhoC in the control of tumor cell invasion.
For a considerable time, the closely related RhoA and RhoC proteins have been assumed to perform similar functions in the context of regulation of the actin cytoskeleton and in the process of cell invasion (59). However, there is now accumulating evidence that RhoA and RhoC play distinct roles in these processes (18, 23, 24, 60). In addition, members of the FMNL family have been shown to specifically bind to RhoC and act as effectors of this GTPase (23, 24). Thus, MRK joins a list of RhoC effectors that can differentially mediate functions that are controlled by RhoC versus RhoA.
A potential mechanism for selective signaling through the RhoC-MRK axis could be the formation of scaffold-mediated signaling complexes that also include Rho GEFs (34, 58, 61). It remains to be seen whether MRK also can mediate biological functions that are specifically regulated by RhoA.
The role of RhoC in the regulation of MLC phosphorylation has not been previously investigated. Rho subfamily proteins are thought to control myosin activity by binding to and activating ROCK kinases, which in turn phosphorylate both myosin light chain and myosin light chain phosphatase, thereby inhibiting its phosphatase activity (56, 62–64). Notably however, recent findings remain equivocal as to whether ROCKs are predominantly controlled by RhoA or RhoC (24, 60). Interestingly, our observations that, unlike depletion of RhoA, depletion of RhoC did not affect initial LPA-stimulated MLC phosphorylation, but retarded its down-regulation, strongly suggest that RhoA and RhoC have opposite roles in the regulation of myosin activity: whereas RhoA stimulates myosin activity, RhoC contributes to myosin dynamics by inhibiting its activity. These findings are in line with observations in prostate cancer cells that RhoA, rather than RhoC, controls ROCK proteins in the regulation of cell morphology and migration (24).
Our observation that depletion of MRK, similar to depletion of RhoC, causes sustained MLC phosphorylation, strongly suggests that MRK mediates the inhibitory effect of RhoC on myosin activation, in line with the notion that MRK acts as an effector of RhoC. How MRK inhibits MLC phosphorylation remains to be elucidated. One possible mechanism is that MRK could activate MYPT1, the myosin phosphatase. This is in line with the finding that depletion of MYPT1, like depletion of RhoC and MRK, causes excess formation of stress fibers and focal adhesions and inhibits cell migration (65). The inhibitory roles of RhoC and MRK on MLC phosphorylation highlight the essential role of dynamic regulation of myosin in the process of cell migration that requires rapid reorganization of the actin cytoskeleton (66).
Rho subfamily proteins have already been implicated in the activation of p38 (36). To our knowledge however, our observations are the first showing a role for RhoC, but not RhoA, in ERK activation. Our results also suggest that RhoC-mediated activation of ERK contributes to the role of RhoC in tumor cell invasion. Whether RhoC-mediated signaling also contributes to additional ERK-regulated functions remains to be determined.
Our study also connects the motogenic and mitogenic extracellular factor LPA to RhoC and MRK. Contrary to findings in other cell types (22), we have shown that LPA significantly stimulates RhoC activity in osteosarcoma and ovarian cancer cells, the two cell lines used in this study. Interestingly, LPA levels have been found to be elevated in plasma from ovarian cancer patients and its receptors are aberrantly expressed in several human cancers (reviewed in Refs. 67). The ecto-enzyme autotaxin that produces LPA is also involved in tumor invasion and metastasis (68). Thus, it is expected that tumors exposed to high levels of LPA may also have high levels of MRK activity.
The requirement of Rho proteins in cancer cell invasion is well established (9, 69). In particular, RhoC has emerged as a key factor in tumor progression in many types of cancer that include invasive breast carcinoma (5,15), gastric cancer (13), non small cell lung carcinoma (14), ovarian carcinoma (8), and esophageal squamous cell carcinoma (70). In addition, RhoC has been shown to be essential for metastasis in animal models (10, 11, 12, 72).
The importance of Rho family GTPases as key signaling hubs in tumorigenesis and metastasis has been recognized since a long time (1, 73). However, despite extensive drug discovery efforts, small GTPases have remained extremely difficult to target (71). Thus, the identification of MRK as a novel effector of RhoC that mediates the role of RhoC in the regulation of tumor cell invasion raises the possibility that small molecule inhibitors of MRK kinase activity may be effective in the treatment of RhoC-dependent metastatic tumors.
Acknowledgment
We thank Eric Sahai for the gift of the RhoC-V14 and the ROCKII plasmids.
This work was supported, in whole or in part, by National Institutes of Health Grant CA100747 (to R. R.), a grant from the Swim Across America Foundation (to M. S.), and an Institutional Grant from the Feinstein Institute for Medical Research (to R. R.).
- LPA
- lysophosphatidic acid
- MRK
- MLK-related kinase
- MLC
- myosin light chain
- GTPγS
- guanosine 5′-O-γ-[thio]triphosphate.
REFERENCES
- 1. Sahai E., Marshall C. J. (2002) RHO-GTPases and cancer. Nat. Rev. Cancer 2, 133–142 [DOI] [PubMed] [Google Scholar]
- 2. Karlsson R., Pedersen E. D., Wang Z., Brakebusch C. (2009) Rho GTPase function in tumorigenesis. Biochim. Biophys. Acta 1796, 91–98 [DOI] [PubMed] [Google Scholar]
- 3. Kamai T., Arai K., Tsujii T., Honda M., Yoshida K. (2001) Overexpression of RhoA mRNA is associated with advanced stage in testicular germ cell tumour. BJU. Int. 87, 227–231 [DOI] [PubMed] [Google Scholar]
- 4. Suwa H., Ohshio G., Imamura T., Watanabe G., Arii S., Imamura M., Narumiya S., Hiai H., Fukumoto M. (1998) Overexpression of the rhoC gene correlates with progression of ductal adenocarcinoma of the pancreas. Br. J. Cancer 77, 147–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. van Golen K. L., Davies S., Wu Z. F., Wang Y., Bucana C. D., Root H., Chandrasekharappa S., Strawderman M., Ethier S. P., Merajver S. D. (1999) A novel putative low-affinity insulin-like growth factor-binding protein, LIBC (lost in inflammatory breast cancer), and RhoC GTPase correlate with the inflammatory breast cancer phenotype. Clin. Cancer Res. 5, 2511–2519 [PubMed] [Google Scholar]
- 6. Fritz G., Just I., Kaina B. (1999) Rho GTPases are overexpressed in human tumors. Int. J. Cancer 81, 682–687 [DOI] [PubMed] [Google Scholar]
- 7. Fritz G., Brachetti C., Bahlmann F., Schmidt M., Kaina B. (2002) Rho GTPases in human breast tumours: expression and mutation analyses and correlation with clinical parameters. Br. J. Cancer 87, 635–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Horiuchi A., Imai T., Wang C., Ohira S., Feng Y., Nikaido T., Konishi I. (2003) Up-regulation of small GTPases, RhoA and RhoC, is associated with tumor progression in ovarian carcinoma. Lab. Invest. 83, 861–870 [DOI] [PubMed] [Google Scholar]
- 9. Vega F. M., Ridley A. J. (2008) Rho GTPases in cancer cell biology. FEBS Lett. 582, 2093–2101 [DOI] [PubMed] [Google Scholar]
- 10. Clark E. A., Golub T. R., Lander E. S., Hynes R. O. (2000) Genomic analysis of metastasis reveals an essential role for RhoC. Nature 406, 532–535 [DOI] [PubMed] [Google Scholar]
- 11. Hakem A., Sanchez-Sweatman O., You-Ten A., Duncan G., Wakeham A., Khokha R., Mak T. W. (2005) RhoC is dispensable for embryogenesis and tumor initiation but essential for metastasis. Genes Dev. 19, 1974–1979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang W., Wu F., Fang F., Tao Y., Yang L. (2008) Inhibition of invasion and metastasis of hepatocellular carcinoma cells via targeting RhoC in vitro and in vivo. Clin. Cancer Res. 14, 6804–6812 [DOI] [PubMed] [Google Scholar]
- 13. Kondo T., Sentani K., Oue N., Yoshida K., Nakayama H., Yasui W. (2004) Expression of RHOC is associated with metastasis of gastric carcinomas. Pathobiology 71, 19–25 [DOI] [PubMed] [Google Scholar]
- 14. Shikada Y., Yoshino I., Okamoto T., Fukuyama S., Kameyama T., Maehara Y. (2003) Higher expression of RhoC is related to invasiveness in non-small cell lung carcinoma. Clin. Cancer Res. 9, 5282–5286 [PubMed] [Google Scholar]
- 15. Kleer C. G., Griffith K. A., Sabel M. S., Gallagher G., van Golen K. L., Wu Z. F., Merajver S. D. (2005) RhoC-GTPase is a novel tissue biomarker associated with biologically aggressive carcinomas of the breast. Breast Cancer Res. Treat. 93, 101–110 [DOI] [PubMed] [Google Scholar]
- 16. Boone B., Van Gele M., Lambert J., Haspeslagh M., Brochez L. (2009) The role of RhoC in growth and metastatic capacity of melanoma. J. Cutan. Pathol. 36, 629–636 [DOI] [PubMed] [Google Scholar]
- 17. Wheeler A. P., Ridley A. J. (2004) Why three Rho proteins? RhoA, RhoB, RhoC, and cell motility. Exp. Cell Res. 301, 43–49 [DOI] [PubMed] [Google Scholar]
- 18. Simpson K. J., Dugan A. S., Mercurio A. M. (2004) Functional analysis of the contribution of RhoA and RhoC GTPases to invasive breast carcinoma. Cancer Res. 64, 8694–8701 [DOI] [PubMed] [Google Scholar]
- 19. Pillé J. Y., Denoyelle C., Varet J., Bertrand J. R., Soria J., Opolon P., Lu H., Pritchard L. L., Vannier J. P., Malvy C., Soria C., Li H. (2005) Anti-RhoA and anti-RhoC siRNAs inhibit the proliferation and invasiveness of MDA-MB-231 breast cancer cells in vitro and in vivo. Mol. Ther. 11, 267–274 [DOI] [PubMed] [Google Scholar]
- 20. Bellovin D. I., Simpson K. J., Danilov T., Maynard E., Rimm D. L., Oettgen P., Mercurio A. M. (2006) Reciprocal regulation of RhoA and RhoC characterizes the EMT and identifies RhoC as a prognostic marker of colon carcinoma. Oncogene 25, 6959–6967 [DOI] [PubMed] [Google Scholar]
- 21. Hutchison N., Hendry B. M., Sharpe C. C. (2009) Rho isoforms have distinct and specific functions in the process of epithelial to mesenchymal transition in renal proximal tubular cells. Cell Signal. 21, 1522–1531 [DOI] [PubMed] [Google Scholar]
- 22. Dietrich K. A., Schwarz R., Liska M., Grass S., Menke A., Meister M., Kierschke G., Längle C., Genze F., Giehl K. (2009) Specific induction of migration and invasion of pancreatic carcinoma cells by RhoC, which differs from RhoA in its localisation and activity. Biol. Chem. 390, 1063–1077 [DOI] [PubMed] [Google Scholar]
- 23. Kitzing T. M., Wang Y., Pertz O., Copeland J. W., Grosse R. (2010) Formin-like 2 drives amoeboid invasive cell motility downstream of RhoC. Oncogene 29, 2441–2448 [DOI] [PubMed] [Google Scholar]
- 24. Vega F. M., Fruhwirth G., Ng T., Ridley A. J. (2011) RhoA and RhoC have distinct roles in migration and invasion by acting through different targets. J. Cell Biol. 193, 655–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bian D., Mahanivong C., Yu J., Frisch S. M., Pan Z. K., Ye R. D., Huang S. (2006) The G12/13-RhoA signaling pathway contributes to efficient lysophosphatidic acid-stimulated cell migration. Oncogene 25, 2234–2244 [DOI] [PubMed] [Google Scholar]
- 26. Buhl A. M., Johnson N. L., Dhanasekaran N., Johnson G. L. (1995) G alpha 12 and Gα13 stimulate Rho-dependent stress fiber formation and focal adhesion assembly. J. Biol. Chem. 270, 24631–24634 [DOI] [PubMed] [Google Scholar]
- 27. Hart M. J., Jiang X., Kozasa T., Roscoe W., Singer W. D., Gilman A. G., Sternweis P. C., Bollag G. (1998) Direct stimulation of the guanine nucleotide exchange activity of p115 RhoGEF by Gα13. Science 280, 2112–2114 [DOI] [PubMed] [Google Scholar]
- 28. Moolenaar W. H., van Meeteren L. A., Giepmans B. N. (2004) The ins and outs of lysophosphatidic acid signaling. Bioessays 26, 870–881 [DOI] [PubMed] [Google Scholar]
- 29. Hama K., Aoki J., Fukaya M., Kishi Y., Sakai T., Suzuki R., Ohta H., Yamori T., Watanabe M., Chun J., Arai H. (2004) Lysophosphatidic acid and autotaxin stimulate cell motility of neoplastic and non-neoplastic cells through LPA1. J. Biol. Chem. 279, 17634–17639 [DOI] [PubMed] [Google Scholar]
- 30. Offermanns S., Mancino V., Revel J. P., Simon M. I. (1997) Vascular system defects and impaired cell chemokinesis as a result of Gα13 deficiency. Science 275, 533–536 [DOI] [PubMed] [Google Scholar]
- 31. Parks S., Wieschaus E. (1991) The Drosophila gastrulation gene concertina encodes a Gα-like protein. Cell 64, 447–458 [DOI] [PubMed] [Google Scholar]
- 32. Radhika V., Onesime D., Ha J. H., Dhanasekaran N. (2004) Gα13 stimulates cell migration through cortactin-interacting protein Hax-1. J. Biol. Chem. 279, 49406–49413 [DOI] [PubMed] [Google Scholar]
- 33. Hill C. S., Wynne J., Treisman R. (1995) The Rho family GTPases RhoA, Rac1, and CDC42Hs regulate transcriptional activation by SRF. Cell 81, 1159–1170 [DOI] [PubMed] [Google Scholar]
- 34. Teramoto H., Crespo P., Coso O. A., Igishi T., Xu N., Gutkind J. S. (1996) The small GTP-binding protein rho activates c-Jun N-terminal kinases/stress-activated protein kinases in human kidney 293T cells. Evidence for a Pak-independent signaling pathway. J. Biol. Chem. 271, 25731–25734 [DOI] [PubMed] [Google Scholar]
- 35. Jia Z., Vadnais J., Lu M. L., Noël J., Nabi I. R. (2006) Rho/ROCK-dependent pseudopodial protrusion and cellular blebbing are regulated by p38 MAPK in tumour cells exhibiting autocrine c-Met activation. Biol. Cell 98, 337–351 [DOI] [PubMed] [Google Scholar]
- 36. Marinissen M. J., Chiariello M., Gutkind J. S. (2001) Regulation of gene expression by the small GTPase Rho through the ERK6 (p38γ) MAP kinase pathway. Genes Dev. 15, 535–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Marinissen M. J., Chiariello M., Tanos T., Bernard O., Narumiya S., Gutkind J. S. (2004) The small GTP-binding protein RhoA regulates c-jun by a ROCK-JNK signaling axis. Mol. Cell 14, 29–41 [DOI] [PubMed] [Google Scholar]
- 38. Gross E. A., Callow M. G., Waldbaum L., Thomas S., Ruggieri R. (2002) MRK, a mixed lineage kinase-related molecule that plays a role in γ-radiation-induced cell cycle arrest. J. Biol. Chem. 277, 13873–13882 [DOI] [PubMed] [Google Scholar]
- 39. Gotoh I., Adachi M., Nishida E. (2001) Identification and characterization of a novel MAP kinase kinase kinase, MLTK. J. Biol. Chem. 276, 4276–4286 [DOI] [PubMed] [Google Scholar]
- 40. Liu T. C., Huang C. J., Chu Y. C., Wei C. C., Chou C. C., Chou M. Y., Chou C. K., Yang J. J. (2000) Cloning and expression of ZAK, a mixed lineage kinase-like protein containing a leucine-zipper and a sterile-α motif. Biochem. Biophys. Res. Commun. 274, 811–816 [DOI] [PubMed] [Google Scholar]
- 41. Tosti E., Waldbaum L., Warshaw G., Gross E. A., Ruggieri R. (2004) The stress kinase MRK contributes to regulation of DNA damage checkpoints through a p38γ-independent pathway. J. Biol. Chem. 279, 47652–47660 [DOI] [PubMed] [Google Scholar]
- 42. Self A. J., Hall A. (1995) Purification of recombinant Rho/Rac/G25K from Escherichia coli. Methods Enzymol. 256, 3–10 [DOI] [PubMed] [Google Scholar]
- 43. Valster A., Tran N. L., Nakada M., Berens M. E., Chan A. Y., Symons M. (2005) Cell migration and invasion assays. Methods 37, 208–215 [DOI] [PubMed] [Google Scholar]
- 44. Chuang Y. Y., Tran N. L., Rusk N., Nakada M., Berens M. E., Symons M. (2004) Role of synaptojanin 2 in glioma cell migration and invasion. Cancer Res. 64, 8271–8275 [DOI] [PubMed] [Google Scholar]
- 45. Crabtree G. R., Schreiber S. L. (1996) Three-part inventions: intracellular signaling and induced proximity. Trends Biochem. Sci. 21, 418–422 [DOI] [PubMed] [Google Scholar]
- 46. Sahai E., Marshall C. J. (2002) ROCK and Dia have opposing effects on adherens junctions downstream of Rho. Nat. Cell Biol. 4, 408–415 [DOI] [PubMed] [Google Scholar]
- 47. Ho T. T., Merajver S. D., Lapière C. M., Nusgens B. V., Deroanne C. F. (2008) RhoA-GDP regulates RhoB protein stability. Potential involvement of RhoGDIα. J. Biol. Chem. 283, 21588–21598 [DOI] [PubMed] [Google Scholar]
- 48. Sengupta S., Wang Z., Tipps R., Xu Y. (2004) Biology of LPA in health and disease. Semin. Cell Dev. Biol. 15, 503–512 [DOI] [PubMed] [Google Scholar]
- 49. Pua T. L., Wang F. Q., Fishman D. A. (2009) Roles of LPA in ovarian cancer development and progression. Future Oncol. 5, 1659–1673 [DOI] [PubMed] [Google Scholar]
- 50. Leung I. W., Lassam N. (1998) Dimerization via tandem leucine zippers is essential for the activation of the mitogen-activated protein kinase kinase kinase, MLK-3. J. Biol. Chem. 273, 32408–32415 [DOI] [PubMed] [Google Scholar]
- 51. Vacratsis P. O., Gallo K. A. (2000) Zipper-mediated oligomerization of the mixed lineage kinase SPRK/MLK-3 is not required for its activation by the GTPase cdc 42 but Is necessary for its activation of the JNK pathway. Monomeric SPRK L410P does not catalyze the activating phosphorylation of Thr258 of murine MITOGEN-ACTIVATED protein kinase kinase 4. J. Biol. Chem. 275, 27893–27900 [DOI] [PubMed] [Google Scholar]
- 52. Chakraborti S., Mandal M., Das S., Mandal A., Chakraborti T. (2003) Regulation of matrix metalloproteinases: an overview. Mol. Cell Biochem. 253, 269–285 [DOI] [PubMed] [Google Scholar]
- 53. Reddy K. B., Nabha S. M., Atanaskova N. (2003) Role of MAP kinase in tumor progression and invasion. Cancer Metastasis Rev. 22, 395–403 [DOI] [PubMed] [Google Scholar]
- 54. Huang C., Jacobson K., Schaller M. D. (2004) MAP kinases and cell migration. J. Cell Sci. 117, 4619–4628 [DOI] [PubMed] [Google Scholar]
- 55. del Barco Barrantes I, Nebreda A. R. (2012) Roles of p38 MAPKs in invasion and metastasis. Biochem. Soc. Trans. 40, 79–84 [DOI] [PubMed] [Google Scholar]
- 56. Katoh K., Kano Y., Noda Y. (2011) Rho-associated kinase-dependent contraction of stress fibres and the organization of focal adhesions. J.R. Soc. Interface 8, 305–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Minden A., Lin A., Claret F. X., Abo A., Karin M. (1995) Selective activation of the JNK signaling cascade and c-Jun transcriptional activity by the small GTPases Rac and Cdc42Hs. Cell 81, 1147–1157 [DOI] [PubMed] [Google Scholar]
- 58. Du Y., Böck B. C., Schachter K. A., Chao M., Gallo K. A. (2005) Cdc42 induces activation loop phosphorylation and membrane targeting of mixed lineage kinase 3. J. Biol. Chem. 280, 42984–42993 [DOI] [PubMed] [Google Scholar]
- 59. Ridley A. J. (2001) Rho family proteins: coordinating cell responses. Trends Cell Biol. 11, 471–477 [DOI] [PubMed] [Google Scholar]
- 60. Bravo-Cordero J. J., Oser M., Chen X., Eddy R., Hodgson L., Condeelis J. (2011) A novel spatiotemporal RhoC activation pathway locally regulates cofilin activity at invadopodia. Curr. Biol. 21, 635–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Pan C. Q., Sudol M., Sheetz M., Low B. C. (2012) Modularity and functional plasticity of scaffold proteins as p (l) acemakers in cell signaling. Cell Signal. 24, 2143–2165 [DOI] [PubMed] [Google Scholar]
- 62. Riento K., Ridley A. J. (2003) Rocks: multifunctional kinases in cell behaviour. Nat. Rev. Mol. Cell Biol. 4, 446–456 [DOI] [PubMed] [Google Scholar]
- 63. Narumiya S., Tanji M., Ishizaki T. (2009) Rho signaling, ROCK and mDia1, in transformation, metastasis and invasion. Cancer Metastasis Rev. 28, 65–76 [DOI] [PubMed] [Google Scholar]
- 64. Street C. A., Bryan B. A. (2011) Rho kinase proteins–pleiotropic modulators of cell survival and apoptosis. Anticancer Res. 31, 3645–3657 [PMC free article] [PubMed] [Google Scholar]
- 65. Xia D., Stull J. T., Kamm K. E. (2005) Myosin phosphatase targeting subunit 1 affects cell migration by regulating myosin phosphorylation and actin assembly. Exp. Cell Res. 304, 506–517 [DOI] [PubMed] [Google Scholar]
- 66. Vicente-Manzanares M., Ma X., Adelstein R. S., Horwitz A. R. (2009) Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat. Rev. Mol. Cell Biol. 10, 778–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Mills G. B., Moolenaar W. H. (2003) The emerging role of lysophosphatidic acid in cancer. Nat. Rev. Cancer 3, 582–591 [DOI] [PubMed] [Google Scholar]
- 68. Stracke M. L., Krutzsch H. C., Unsworth E. J., Arestad A., Cioce V., Schiffmann E., Liotta L. A. (1992) Identification, purification, and partial sequence analysis of autotaxin, a novel motility-stimulating protein. J. Biol. Chem. 267, 2524–2529 [PubMed] [Google Scholar]
- 69. Schmitz A. A., Govek E. E., Böttner B., Van Aelst L. (2000) Rho GTPases: signaling, migration, and invasion. Exp. Cell Res. 261, 1–12 [DOI] [PubMed] [Google Scholar]
- 70. Faried A., Faried L. S., Kimura H., Nakajima M., Sohda M., Miyazaki T., Kato H., Usman N., Kuwano H. (2006) RhoA and RhoC proteins promote both cell proliferation and cell invasion of human oesophageal squamous cell carcinoma cell lines in vitro and in vivo. Eur.J. Cancer 42, 1455–1465 [DOI] [PubMed] [Google Scholar]
- 71. Fritz G., Kaina B. (2006) Rho GTPases: promising cellular targets for novel anticancer drugs. Curr. Cancer Drug Targets 6, 1–14 [PubMed] [Google Scholar]
- 72. Islam M., Lin G., Brenner J. C., Pan Q., Merajver S. D., Hou Y., Kumar P., Teknos T. N. (2009) RhoC expression and head and neck cancer metastasis. Mol. Cancer Res. 7, 1771–1780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Symons M. (1995) The Rac and Rho pathways as a source of drug targets for Ras-mediated malignancies. Curr. Opin. Biotechnol. 6, 668–674 [DOI] [PubMed] [Google Scholar]