

Background: The ER-resident formylglycine-generating enzyme (FGE), essential for post-translational activation of all eukaryotic sulfatase enzymes, is N-terminally processed during secretion.
Results: Processing is mediated by furin at a conserved R/K-YS-R/K↓ cleavage site and leads to FGE inactivation.
Conclusion: Processing serves to regulate the amount of FGE activity following ER exit.
Significance: Processing is never complete, thus suggesting an extra-ER function of FGE.
Keywords: Endoplasmic Reticulum (ER), Intracellular Processing, Post-translational Modification, Protein Processing, Protein Secretion, Formylglycine-generating Enzyme, Furin, Proprotein Convertases, Sulfatases
Abstract
Formylglycine-generating enzyme (FGE) post-translationally converts a specific cysteine in newly synthesized sulfatases to formylglycine (FGly). FGly is the key catalytic residue of the sulfatase family, comprising 17 nonredundant enzymes in human that play essential roles in development and homeostasis. FGE, a resident protein of the endoplasmic reticulum, is also secreted. A major fraction of secreted FGE is N-terminally truncated, lacking residues 34–72. Here we demonstrate that this truncated form is generated intracellularly by limited proteolysis mediated by proprotein convertase(s) (PCs) along the secretory pathway. The cleavage site is represented by the sequence RYSR72↓, a motif that is conserved in higher eukaryotic FGEs, implying important functionality. Residues Arg-69 and Arg-72 are critical because their mutation abolishes FGE processing. Furthermore, residues Tyr-70 and Ser-71 confer an unusual property to the cleavage motif such that endogenous as well as overexpressed FGE is only partially processed. FGE is cleaved by furin, PACE4, and PC5a. Processing is disabled in furin-deficient cells but fully restored upon transient furin expression, indicating that furin is the major protease cleaving FGE. Processing by endogenous furin occurs mostly intracellularly, although also extracellular processing is observed in HEK293 cells. Interestingly, the truncated form of secreted FGE no longer possesses FGly-generating activity, whereas the unprocessed form of secreted FGE is active. As always both forms are secreted, we postulate that furin-mediated processing of FGE during secretion is a physiological means of higher eukaryotic cells to regulate FGE activity upon exit from the endoplasmic reticulum.
Introduction
Sulfatases form a family of enzymes that catalyze the hydrolysis of sulfate esters and sulfamates in a wide variety of substrates like glycosaminoglycans, sulfolipids, and steroid sulfates (1, 2). In pro- and eukaryotic sulfatases, post-translational modification of the crucial cysteine residue in the conserved CXPXR motif to formylglycine (FGly)4 is a hallmark for their activation and sulfatases devoid of this modification are catalytically inactive. Multiple sulfatase deficiency (MSD), a rare but fatal lysosomal storage disorder in humans, is characterized by the production of all 17 human sulfatases with almost no FGly formation in their active sites (3). The formylglycine-generating enzyme (FGE) catalyzes this unique and critical modification in nascent sulfatase polypeptides in the endoplasmic reticulum (ER), and mutations in the FGE encoding gene SUMF1 were discovered as the basis of MSD (4–8). Recently, a role for FGE in regulating cell lineage commitment was reported. FGE, via activation of sulfatases Sulf1 and Sulf2, was shown to control hematopoietic lineage development through FGF and Wnt signaling (9).
In eukaryotes, FGE is localized in the lumen of the ER. The mature 41-kDa protein in humans, lacking the signal peptide (amino acids 1–33), is an N-glycosylated monomer containing 8 cysteines. The core domain containing the active site exhibits a novel fold with remarkably low secondary structural elements that is stabilized by two Ca2+ ions and two intramolecular disulfide bridges (10). The presence of two catalytic cysteines (residues 336 and 341) in the active site, which are involved in binding and oxidation of the cysteine in the sulfatase polypeptide, is highly conserved in FGE homologues from prokaryotes to eukaryotes (11). One of the defining features unique to eukaryotic FGE is the presence of a 55-residue N-terminal extension (amino acid positions 34–88 in the mature form of the protein). We have previously shown that this N-terminal extension of human FGE, for which the structure is unknown, is required for activation of sulfatases in cultured cells. Especially a conserved pair of cysteines (residues 50 and 52) within this extension was shown to be involved, with Cys-52 being critical for this activation (12). Moreover, the N-terminal extension has been shown to confer efficient retention of FGE in the ER by interaction with ERp44, a redox sensor and retention factor for Ero1α and adiponectin (13, 14). FGE that escapes the ER retention machinery is secreted (15). Recently, the reuptake of secreted FGE has also been reported; and interestingly, the endocytosed FGE has been shown to activate sulfatases after it reaches the ER by an unknown mechanism (16).
However, in our previous studies we have shown that a large fraction of secreted FGE is in an N-terminally truncated form starting at glutamate 73 (15). The nature of this proteolytic truncation and the identity of the protease(s) involved have not been defined so far. It is well known that proteolytic processing mediated by proprotein convertases (PCs) along the secretory pathway activates and thereby regulates the function of several secreted proteins (17). In case of FGE, however, the mechanism and relevance of processing for controlling its function as a master regulator in sulfatase biogenesis have not been investigated so far, and truncation of a functionally indispensable N-terminal fragment of FGE during secretion validates a detailed analysis of this process.
In this study, we show that FGE is cleaved off its N-terminal extension during secretion by furin and other furin-like PCs. The N-terminal trimming of secreted FGE is abolished in furin-deficient cells, suggesting that furin is the major protease involved in this processing. The cleavage site is represented by a RYSR72↓ motif, which is conserved among higher eukaryotes and wherein residues Arg-69 and Arg-72 are absolutely required for cleavage of human FGE. Interestingly, residues Tyr-70 and Ser-71 are highly conserved and play a role in lowering the cleavage efficiency. Therefore, FGE is inefficiently cleaved during secretion. Furthermore, the N-terminally truncated FGE is devoid of its formyglycine-generating activity, suggesting that the furin-mediated processing during secretion leads to inactivation of FGE. As always full-length FGE is secreted as well, regulation by processing may in fact address a role of FGE executed outside the ER.
EXPERIMENTAL PROCEDURES
Expression Plasmids
The construction of plasmids used to express FGE-tagged C-terminally with either -RGS-His6 (pSB-FGE-His) or -HA (pBI-FGE-HA), FGE with an appended KDEL motif at the C terminus (pBI-FGE-HA-KDEL), steroid sulfatase (pBI-STS), and myc-ERp44 (pBI-myc-ERp44) was described earlier (12, 15). All variants of the cleavage site motif of FGE used in this study were generated by site-directed mutagenesis PCR with pBI-FGE-HA as template and long-template expand PCR system (Roche Applied Science). The coding sequences of the primers were: FGE-R69A, 5′-TCGGCAGCCGCTCACGCATACTCGCGGGAGGCT-3′; FGE-Y70A, 5′-GCAGCCGCTCACCGAGCCTCGCGGGAGGCTAAC-3′; FGE-S71A, 5′-GCCGCTCACCGATACGCGCGGGAGGCTAACGCT-3′; FGE-R72A, 5′-GCTCACCGATACTCGGCGGAGGCTAACGCTCCG-3′; FGE-R69A/R72A, 5′-GCTCACGCATACTCGGCGGAGGCTAACGCTCCG-3; FGE-S71R, 5′-GCCGCTCACCGATACAGGCGGGAGGCTAACGCT-3′; FGE-Y70A/S71R, 5′-GCCGCTCACCGAGCCAGGCGGGAGGCTAACGCT-3′; FGE-E73P, 5′-CACCGATACTCGCGGCCGGCTAACGCTCCGGGC-3′; FGE-Y70K, 5′-GCAGCCGCTCACCGAAAGTCGCGGGAGGCTAAC-3′; FGE-R69K, 5′-TCGGCAGCCGCTCACAAATACTCGCGGGAGGCT-3′; FGE-R72K, 5′-GCTCACCGATACTCGAAGGAGGCTAACGCTCCG-3′; FGE-Y70F, 5′-GCAGCCGCTCACCGATTCTCGCGGGAGGCTAAC-3′; FGE-Y70S, 5′-GCAGCCGCTCACCGATCCTCGCGGGAGGCTAAC-3′. The resulting constructs were verified by full-length sequencing of the coding region to exclude any PCR prone errors. Plasmids for expression of the PCs encoding human furin (Gene ID 5045), human PACE4 (= PCSK6, Gene ID 5046), murine PC5a (= Pcsk5, Gene ID 18552) and rat PC7 (= Pcsk7, Gene ID 29606) were kindly provided by Abdel-Majid Khatib (18). Note that the PC cDNAs were encoded as pIRES constructs coding also for enhanced green fluorescence protein (EGFP) such that EGFP expression levels serve as a measure of the expression levels of PCs.
Generation of the Phylogenetic Tree and Sequence Logos
The phylogenetic tree (see Fig. 3, left) is based on the Newick format of 13 representative species of a total of 88 SUMF1 sequences from different species. For sequence logo generation through the WebLogo 3.0 program (19, 20) species could be divided into three subgroups, based on the presence of the motif RYSR (group I) or R/K/X-Y-S-R/K/X (group II; X denotes any amino acid) or no common sequence (group III) at position P1-P4 of the cleavage site in most species of the given classifications (Fig. 3). The sequences were centered at P1 (Arg-72) or based on a ClustalW alignment in case of the sequences of group III that do not comprise a R/K/X-Y-S-R/K/X motif. 16 amino acids corresponding to positions 65–80 in human FGE are displayed. All 88 sequences are shown in supplemental Table S1.
FIGURE 3.
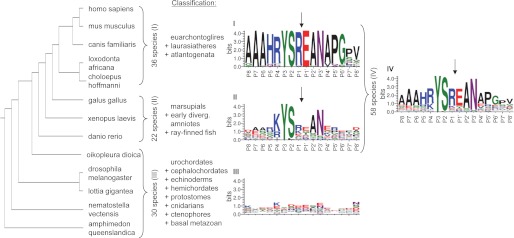
The RYSR motif is conserved in later diverging eukaryotes. The phylogenetic tree (left) of 13 representative species, chosen out of a total of 88 analyzed species (see supplemental Table S1), was generated from Newick format according to modern molecular consensus taxonomy (27) and visualized with the program Phylodendron. The species were divided into three groups based on their taxonomy and the presence of RYSR or YS at the cleavage site position. Group I represents 36 species from euarchontoglires, laurasiatheres, and atlantogenata; group II consists of 22 sequences from marsupials to ray-finned fish; and group III includes 30 species of urochordates to basal metazoan. WebLogo 3.0 was used to create logos (I–III) of the three groups as well as a combined logo (IV) of all 58 later diverging eukaryote sequences (19, 20). The four sequence logos display the degree of conservation of amino acids at positions P8 to P8′ of each group (representing residues 65–80 of human FGE) and were generated as described under “Experimental Procedures.” A high degree of conservation of a single amino acid at a particular position is represented by a large size (in units of bits) of the amino acid letter in the logo. Colors represent chemical properties (polar, basic, acidic, hydrophobic). Cleavage takes place between P1 and P1′, marked by arrows.
Cell Culture and Transfection
HT1080, HeLa, HEK293, and baby hamster kidney cells (American Type Culture Collection) were cultured at 37 °C under 5% CO2 in Dulbecco's modified Eagle's medium (DMEM; Invitrogen) containing 10% fetal calf serum (FCS) (Lonza). CHO-K1 and furin-deficient CHO-FD11 cells (kindly provided by Steven Leppla (21)) were cultured in DMEM supplemented with 40 μg/ml proline (Fisher Scientific). HT1080 Tet-On and MSDi Tet-On cells, for doxycycline-inducible protein expression were maintained as described earlier (12). All transfections were performed with Lipofectamine LTX as recommended by the manufacturer (Invitrogen). A stable CHO-FD11 Tet-On cell line was generated by transfection of CHO-FD11 cells with pUHrT62 (kindly provided by Nadja Jung) encoding the reverse tetracycline controlled transactivator and neomycin resistance vector pSB4.7pA in a 10:1 ratio (15). The stable clones were selected with medium containing 0.8 mg/ml neomycin (Invitrogen) and screened through Western blotting for doxycycline-dependent FGE expression after transient transfection with pBI-FGE-HA plasmid. CHO-FD11 cells stably expressing FGE-RGS-His6 were generated by tranfecting CHO-FD11 cells with pSB-FGE-RGS-His6, and transfectants were selected with 0.8 mg/ml G-418 sulfate (PAA). The stable clone was selected through FGE expression analysis by Western blotting.
Preparation of Cell and Medium Samples for SDS-PAGE and Western Blot Analysis
3 μg of pSB-FGE-RGS-His6 vector was used for single transfections of HT1080, HeLa, HEK293, baby hamster kidney, and CHO cells. Conditioned medium was collected after 24 h and centrifuged (500 × g, 5 min). Cells were washed once with PBS, treated with trypsin (Lonza), and pelleted by 250 × g to remove the medium. Cell pellets were resuspended in PBS (pH 7.4) containing protease inhibitor mixture (Sigma) and lysed by sonication 3 × 10 s on ice. For co-expression of FGE with one of the PCs in CHO-FD11 Tet-On cells 2 μg of pBI-FGE-HA and 2 μg of the plasmids encoding PCs (see above) were used for transient transfection. In case of transient transfections using pBI vector constructs the protein expression was induced by replenishing the medium with medium containing 2 μg/ml doxycycline (BD Biosciences) at 5 h post-transfection. After induction for 24 h, the cells and media were collected and processed as described above for further analysis by SDS-PAGE and Western blotting.
Western blot analyses were carried out using rabbit polyclonal antiserum against FGE and rabbit polyclonal anti-GFP antibody (Living Colors® A.v. Peptide Antibody, Clontech) as primary antibodies and the peroxidase-conjugated goat anti-rabbit secondary antibody (Invitrogen). Quantification of Western blot signals was performed using AIDA 2.1 software package (Raytest), and calculations of FGE amounts were based on 10- or 20-ng FGE standard signals, present on the same blot.
Immunoprecipitation
HT1080 cells were cultured in four 14-cm plates with 5% FCS-containing medium for 48 h. Medium was collected and centrifuged 10 min at 1000 × g, to remove cell debris. Cells of one plate were harvested by trypsinization, pelleted at 1000 × g for 5 min, and lysed by sonication in PBS (pH 7.4) containing 0.1% Triton X-100 and protease inhibitor mixture, followed by centrifugation at 20,000 × g for 15 min. As a negative control fresh medium containing 5% FCS was used, and 0.01% Triton X-100 and protease inhibitor mixture were added to it and the cleared medium. All supernatants were preincubated with rabbit preimmune serum for 30 min at 4 °C, and after addition of protein A-Sepharose CL-4B (Sigma-Aldrich) the bound material was pelleted down by centrifugation at 7000 × g for 10 min. The supernatants of the conditioned medium and the cell lysate were split into two parts, and either rabbit preimmune serum or rabbit FGE antiserum was added. After incubation at 4 °C and addition of protein A-Sepharose CL-4B the bound material was pelleted down by centrifugation at 7000 × g for 10 min. The pellets were washed stepwise as described earlier (22). The pellets were boiled in 1× Laemmli buffer for 5 min at 95 °C and centrifuged at maximum speed for 5 min. 100% of the medium and 30% of the cell lysate supernatants were loaded for 12.5% SDS-PAGE followed by Western blotting and detection with FGE antiserum.
Steroid Sulfatase (STS) Activity Assay and Western Blotting
Activity assays of STS were performed as described earlier (15, 23). For Western blot analysis, polyclonal antisera against FGE and STS were used as primary antibodies. Horseradish peroxidase-conjugated goat anti-rabbit antibody was used as secondary antibodies. Signals of STS are given as relative amounts, i.e. related to signal intensities detected in cells expressing STS only. Relative specific sulfatase activities were calculated, i.e. catalytic activity divided by the Western blot signal (arbitrary units) and referred to that of cells expressing the sulfatase only (relative specific sulfatase activity = 1). Absolute values for this reference are given in the legends.
In Vitro Furin Cleavage Assay and Treatment of Cells with RVKR-Chloromethylketone (CMK) Inhibitor
CHO-FD11 cells stably expressing FGE-His6 or HT1080 cells were grown for 24 h, and cells and medium were harvested. For treatment with furin in vitro cell pellets were resuspended in HEPES (pH 7.5) buffer containing 1 mm CaCl2 and 0.5% Triton X-100 and lysed in the absence of protease inhibitors by sonication on ice. Appropriate amounts of cell lysate and medium were incubated for 3 h at 25 °C with 4 units of furin (NEB) with or without 25 μm decanoyl-RVKR-CMK (Alexis Biochemicals) as indicated in the figures. For in vivo treatment, HT1080 Tet-On cells were transiently transfected with pBI-FGE-HA, and 4 h post-transfection, the medium was replenished with medium containing 20 ng/ml doxycycline and various concentrations of decanoyl-RVKR-CMK or only dimethyl sulfoxide as a carrier control. The cells and medium were collected after 16 h of induction/treatment for further analysis. The samples were boiled in Laemmli buffer and analyzed by SDS-PAGE and Western blotting with FGE antiserum.
Extracellular Processing of FGE-containing Conditioned Medium
CHO-FD cells stably expressing FGE-His6 were cultured on 25-cm plates in 15 ml 5% FCS-containing medium for 24 h. Medium was centrifuged at 500 × g, and supernatants were sterile filtered to remove any floating producer cells. The conditioned medium was added to confluent 10-cm plates of CHO-FD11 (negative control), MSDi, HT1080, HeLa, and HEK293 cells for the indicated incubation times. Medium was collected and centrifuged at 500 × g. Equal amounts of medium were used for Western blot analysis.
In Vitro FGE Activity Assay
FGE activity was assayed using conditioned media that contained either secreted full-length (fl)-FGE (obtained from CHO-FD FGE-His6 cells) or Δ72-FGE (obtained from CHO-FD Tet-On cells after transient co-transfection with pBI-FGE-HA and pIRES-Furin constructs). After expression for 48 h at 1% or 2.5% FCS-containing DMEM, the medium was collected and centrifuged at 500 × g for 5 min. The supernatant was analyzed by SDS-PAGE and Western blotting, and aliquots were used directly for activity testing.
The activity assay was performed in triplicate with three sets of conditioned media at pH 9.3 under standard conditions as described earlier (4) with some modifications; no BSA was added to the reaction mixture, and 2 mm DTT or 5 mm GSH was used as a reducing agent.
RESULTS
FGE Is N-terminally Truncated during Secretion by a Nonsaturable Mechanism in a Post-ER compartment
FGE, an ER-localized enzyme, lacks the canonical (KDEL-like) ER retention signal and is retained in the ER via interactions with ERp44 by a saturable mechanism (13). It is known that recombinantly expressed FGE (41 kDa) in HT1080 cells is released into the medium, and the majority of the secreted protein represents an N-terminally truncated form of 37 kDa in size lacking residues 34–72 (Δ72-FGE) (15). To examine whether this proteolytic processing occurs in a compartment along the secretory route, we expressed FGE and FGE with an appended KDEL sequence at the C terminus (FGE+KDEL) in HT1080 cells (Fig. 1A). Analysis of the cell homogenate and medium clearly shows that the FGE population escaping the ER retention machinery is eventually secreted (Fig. 1A, lane 2) and thereby proteolytically processed, whereas retaining FGE in the ER through an appended KDEL sequence prevents the secretion and in turn the proteolytic processing (Fig. 1A, lanes 3 and 4). These data indicate that the transport of FGE to a post-cis-Golgi compartment during secretion is a prerequisite for cleavage. It should be noted that the unprocessed FGE in the medium (see also Fig. 1C) has a lower electrophoretic mobility in SDS-PAGE compared with the intracellular FGE due to modification of the attached N-glycan in the secretory pathway (see Ref. 15).
FIGURE 1.

FGE is N-terminally truncated in a post-ER compartment. A, FGE is N-terminally processed only upon secretion. HT1080 Tet-On cells were transiently transfected with cDNA encoding FGE-HA or FGE with an appended KDEL signal (FGE-HA-KDEL). 6 h post-transfection, FGE expression was induced with 20 ng/ml doxycycline. After 22 h of induction, cells (C) and medium (M) were analyzed by Western blotting with FGE antiserum. B, FGE truncation is observed in various cell lines. FGE was transiently expressed in the indicated cell lines for 24 h, and cells and medium were analyzed by Western blotting with FGE antiserum. C, N-terminal processing of FGE is independent of its expression level. FGE was transiently expressed in HT1080 Tet-On cells, and expression was induced with the indicated concentrations of doxycycline. After 22 h of induction, cells and medium (in a ratio of 2:1) were analyzed by Western blotting. The amount of FGE in cells and medium was determined by calibration of the Western blot with known amounts of purified FGE (data not shown). D, endogenous FGE is also secreted and proteolytically processed. HT1080 cells were cultured for 48 h, and cell lysates and conditioned media were subjected to immunoprecipitation (IP) with FGE antiserum or preimmune serum (PIS). 100% of IP fractions were analyzed by Western blotting. Nonconditioned (non-c.) cell culture medium served as a control.
To analyze the proteolytic processing in detail and to exclude any cell type-specific effect, we studied the secretion profile of FGE in various cell lines. We transiently expressed FGE in HT1080, HeLa, HEK293, baby hamster kidney, and CHO cells. The analysis of the cell homogenate and medium revealed that FGE is secreted in all cell lines tested (Fig. 1B) and that usually the majority of the secreted FGE is in the truncated form. Of note, the extent of processing was comparable across cell lines except in CHO cells, wherein the truncation was less pronounced. The secretion of the unprocessed fl-form suggests that the processing mechanism could be saturated due to high FGE expression levels. To test this hypothesis, FGE was expressed under the control of a doxycycline-inducible promoter in HT1080 Tet-On cells. FGE expression was induced with increasing concentrations of doxycycline, and quantification of the amount of fl- and Δ72-FGE in the secretions shows that even a 7-fold increased FGE expression does not increase the proportion of the fl-form in the medium (Fig. 1C). Approximately 25–35% of secreted FGE is in the unprocessed form independent of the expression level, indicating that incomplete processing is not due to saturation of the processing machinery. To determine whether endogenous FGE is also secreted and whether it is secreted in a processed form, we immunoprecipitated FGE from untransfected HT1080 and HeLa cell homogenate and medium samples. In both cell lines FGE was traceable in the secretions, as shown in Fig. 1D for HT1080 cells. The majority of secreted FGE was in the truncated form, and surprisingly the unprocessed form was also detected to similar relative levels as observed under overexpression conditions.
The RYSR Motif in the N Terminus of FGE Is Indispensable for Proteolytic Processing of Secreted FGE but Not Required for FGE Activity in Vivo
Because many secreted glycoproteins that are processed along the conventional secretory pathway contain an RXXR-like motif, we speculated that the RYSR motif in FGE (residues 69–72, Fig. 2A) represents a potential PC cleavage site. This is supported by our previous finding that the N-terminally truncated FGE starts at glutamate 73 (15). Surprisingly, an in silico analysis using a widely used PC cleavage site prediction program (24) did not yield any potential cleavage motif in FGE, and only a recently published program PiTou predicts a cleavage at the RYSR motif, albeit with a low score (+0.93) (25).
FIGURE 2.

The RXXR motif is required for proteolytic processing of secreted FGE but not for activity. A, schematic representation of human FGE shows the RYSR motif at the cleavage site (arrow). The cysteine residues are highlighted as black lines, and the calculated molecular masses of fl- and processed Δ72-FGE are indicated. SP, signal peptide. B, HT1080 Tet-On cells were transiently transfected with FGE wild-type (Wt) and the indicated alanine variants of the FGE-RYSR motif. 6 h post-transfection, FGE expression was induced with 20 ng/ml doxycycline. After induction for 20 h, cells (C) and medium (M) at a ratio of 2:1 were analyzed for FGE by Western blotting using FGE antiserum. The amount of FGE in the cells and medium was determined by calibration of the Western blot with known amounts of purified FGE protein. C, MSDi Tet-On cells were transiently transfected with STS and FGE-wt or FGE-RYSR motif variants in the indicated combinations. The amounts of STS and FGE were monitored in the cell extracts by Western blotting. The relative specific activity of STS given below the lanes was calculated from the STS activity (nmol/h per mg of cell protein) divided by the Western blot signal of STS (arbitrary units/mg of cell protein) and referred to that in cells expressing STS only.
To analyze biochemically the role of the RXXR-like motif in the N terminus of FGE, we generated the following alanine variants: FGE-R69A, -R72A, -R69A/R72A, -Y70A, and -S71A. Expression of FGE-R69A, -R72A, or -R69A/R72A led to a clear resistance to truncation, and all secreted FGE was in the unprocessed fl-form (Fig. 2B, lanes 4, 6, 8) indicating that both arginine residues (Arg-69 and Arg-72) are equally essential for proteolytic cleavage. Expression of FGE-S71A led to a nearly complete processing of secreted FGE (Fig. 2B, lane 12), whereas the Y70A mutation conferred partial resistance to cleavage, with 1.5-fold more FGE secreted in the fl-form compared with wild-type (wt) FGE (Fig. 2B, compare lanes 2 and 10). These data indicate that the RYSR motif is a bona fide cleavage motif and that residues Tyr-70 and Ser-71 determine cleavage efficiency.
Expression of sulfatases in immortalized MSD cells that lack endogenous FGE activity leads to the production of inactive sulfatases whereas co-expression of a sulfatase with FGE in these cells yields active sulfatases, thus providing a reliable system to investigate FGE-mediated FGly generation in vivo (12). Using this method we examined whether the RYSR motif is required for the activation of sulfatases. We transfected cDNAs encoding STS alone or co-transfected along with cDNAs encoding RYSR motif alanine variants of FGE in immortalized MSD Tet-On cells (Fig. 2C). Upon doxycycline induction, STS was barely active when expressed alone, whereas co-expression with FGE-wt increased the activity of STS approximately 50-fold, as observed previously (12). Co-expressing the alanine variants of the RYSR motif led to an increase in STS activity to similar levels observed for FGE-wt, indicating that the integrity of the RYSR motif is not required for the activity of FGE in vivo. In conclusion, the RYSR motif in the N terminus of FGE serves as an authentic cleavage motif rather than a role in the activity of cellular FGE.
The RYSR Motif Is Highly Conserved among Higher Eukaryotes
Because the RYSR motif was only recognized as a weak potential cleavage site by the prediction program, we analyzed the conservation of the cleavage site flanking regions across species in the animal kingdom. The whole N terminus including the cleavage site as well as the important Cys-Gly-Cys-motif that has been studied earlier (12, 13) is encoded by exon 1 of the SUMF1 gene. Notably, this region has no counterpart in homologous prokaryotic genes (26) and in fact arose as an extension in early eukaryotes and persisted to their present day descendants.
A phylogenetic tree with 13 representative species (Fig. 3, left) provides an overview for our set of 88 available eukaryotic sequences, which were arranged according to modern molecular taxonomy (27; see supplemental Table S1 for the full set of sequences). Within this taxonomy-based list of species we could identify three groups. Group III consisted of the 30 earlier diverging eukaryote species from sponge (basal metazoan) to sea pineapple (urochordates), among which no significant sequence conservation was detected in the relevant region (Fig. 3, WebLogo III). By contrast, in the 58 later diverging eukaryote species from northern pike (ray-finned fish) to human the sequences are significantly conserved forming the cleavage site motif K/R-YS-R/K (Fig. 3, WebLogo IV). The 22 species from northern pike to short tailed opossum (marsupials) mainly carry either a Lys or an Arg residue at positions P4 and P1 (group II), whereas the 36 sequences representing the full range of placental mammal species form a highly conserved RYSR core motif (group I). Of note, some of the earlier diverging eukaryote species (group III) also bear a similar motif like the later diverging eukaryotes, e.g. KYKR in the mountain pine beetle (Dendroctonus ponderosae) (supplemental Table S1).
Interestingly, especially the residues Tyr-70 and Ser-71 within this motif are almost 100% conserved among the 58 later diverging eukaryotes; and, additionally, Glu-73, Ala-74, and Asn-75, representing the neo-N terminus of processed FGE, are as highly conserved as the R/K-YS-R/K itself (Fig. 3, WebLogo IV). The high degree of conservation of these seven consecutive residues in later diverging eukaryotes is best explainable by a strong selective pressure implying important functionality of this sequence.
Notably, all 88 species from human to sponge contain the essential Cys-Gly-Cys sequence mentioned above, which is required for sulfatase activation (12). Therefore, the cleavage site motif arose later in the molecular evolution of the N-terminus and gained a function that may serve as a tool to regulate FGE activity in later diverging eukaryotes.
RYSR72↓E Represents a Unique Cleavage Motif That Imparts Suboptimal Cleavage Efficiency
Our observation that under any condition tested approximately 20–30% of secreted FGE-wt is in the unprocessed form (Figs. 1 and 2) suggested that the processing of the N-terminal extension of secreted FGE is suboptimal. The cleavage efficiency of mammalian PCs has been shown to be directly dependent on ∼20 amino acid residues surrounding the cleavage site, and especially the positions P4 to P1′ (RYSR72↓E in FGE) are important. To determine biochemically the functional advantage of the conserved residues in the RYSR72↓E motif, we transiently expressed motif variants in HT1080 cells and quantified the extent of processing in the secretions by SDS-PAGE and Western blotting. Expression of the motif variants KYSR72↓E and RYSK72↓E, representing motifs found in group II species that contain Lys at position P4 or P1, respectively, led to truncation of the secreted protein, but the cleavage efficiency was lowered, as indicated by a doubling of the fl-FGE/Δ72-FGE ratio compared with that of FGE-wt (Fig. 4A). The occurrence of proline at position P1′ is known to compromise the cleavage efficiency of the PCs, and as expected, substitution of Glu-73 with proline (RYSR72↓P) also led to a reduction in processing, indicating that Glu-73 is conducive to cleavage.
FIGURE 4.

Analysis of the conserved RYSR↓E cleavage motif by alanine scanning mutagenesis. HT1080 Tet-On cells were transiently transfected with pBI plasmids encoding FGE-wt (with RYSR↓E motif) or mutants thereof (with the mutated residues in the RYSR↓E motif indicated in bold). FGE expression was induced with 20 ng/ml doxycycline, 6 h after transfection. After 20 h of induction, cells (C) and medium (M) (at a ratio of 2:1) were analyzed by Western blotting with FGE antiserum (upper panels). The cleavage efficiency was quantified from these blots and expressed as signal ratio of fl-FGE/Δ72-FGE in the medium, as indicated below each medium lane in a bar graph. These data are representative of two independent experiments.
Mutating the highly conserved Tyr-70 at position P3 to Phe (RFSR↓E) or Lys (RKSR↓E) led to a clear increase in cleavage efficiency, whereas substitution with Ser (RSSR↓E) only had a minor effect (Fig. 4B). A dramatic increase in the cleavage efficiency was observed when Ser-71 at position P2 was mutated to a positively charged arginine residue (RYRR↓E or RARR↓E) to a level where virtually all of the secreted FGE was processed. From these data we conclude that RYSR↓E is a unique cleavage motif with the highly conserved Tyr (at position P3) and Ser (P2) conferring an inefficient cleavage property to FGE. Whether these residues, apart from probably being unfavorable for PC recognition, make contact to the FGE core domain (or to other proteins), which may restrict access of the cleavage site, remains to be determined.
Furin Mediates the Proteolytic Processing of Secreted FGE
To verify that the N terminus of FGE containing the RYSR motif is subject to processing by furin or furin-like convertases, we studied the secretion profile of FGE in cells in the presence of decanoyl-RVKR-CMK, an inhibitor for PCs (Fig. 5A). HT1080 Tet-On cells expressing FGE under the control of doxycycline were treated with only dimethyl sulfoxide (as a carrier control) or increasing concentrations of the inhibitor for 16 h in culture. Indeed, the amount of the truncated form of FGE in the secretions was decreased by the inhibitor in a concentration-dependent manner (Fig. 5A). These data clearly indicate that the N-terminal processing during secretion of FGE is mediated by PCs.
FIGURE 5.

Proteolytic processing of FGE is mediated by furin. A, processing of secreted FGE is inhibited by the PC inhibitor RVKR-CMK. HT1080 cells transiently expressing FGE were treated with the indicated concentrations of the inhibitor. After 16 h of treatment, cells (C) and medium (M) were analyzed by Western blotting. B, processing of secreted FGE is impaired in furin-deficient LoVo cells. Cells and medium from HT1080 and LoVo cells stably expressing FGE were analyzed by Western blotting. C, FGE processing is abolished in furin-deficient CHO cells (CHO-FD11) but efficiently restored by co-expression of furin. Wild-type CHO (CHO-K1) and CHO-FD11 cells transiently expressing FGE or CHO-FD11 cells transiently co-expressing furin and FGE were cultured for 24 h. Cells and media were analyzed by Western blotting. D, cellular and secreted FGE are processed by rFurin in vitro. Cell lysate and medium of CHO-FD11 cells stably expressing FGE were incubated at 25 °C for 3 h either in the presence or absence of rFurin. E, endogenous cellular FGE from HT1080 cells is cleaved by rFurin in vitro, and this processing is inhibited by the RVKR-CMK inhibitor. Equal amounts of HT1080 cell lysate were incubated with rFurin and 25 μm CMK inhibitor as indicated. A–E, all Western blots were probed with FGE antiserum.
In mammals, the proprotein convertase family comprises nine enzymes that differ in their substrate specificity and tissue-specific expression and/or subcellular localization (17). Furin is the best characterized mammalian PC with a ubiquitous tissue distribution, and it is the major convertase in the secretory pathway. To analyze the role of furin in the processing of FGE we expressed FGE in cells that are deficient for furin. When expressed in LoVo cells, the major fraction of secreted FGE in the medium was found in the unprocessed form, which contrasts with the observations in other cells; however, a significant fraction was still in the truncated form (Fig. 5B). Strikingly, expression of FGE in furin-deficient CHO cells (CHO-FD11) led to secretion of FGE in the fl-form exclusively (Fig. 5C, lane 4), whereas expressing FGE in CHO-K1 cells, which contain endogenous furin, led to processed FGE in the secretion medium (Fig. 5C, lane 2). This clearly indicates that furin is involved in processing the N terminus of FGE. Remarkably, when furin was replenished in CHO-FD11 cells by exogenous expression, all secreted FGE was in the truncated form (Fig. 5C, lane 6), providing unequivocal evidence that FGE is processed by furin along the secretory pathway. Further, FGE was found to be processed by furin in vitro. When cell homogenate and medium from CHO-FD11 cells stably expressing FGE-His were treated with commercially available recombinant furin (rFurin), almost half the amount of FGE was processed, as observed for both cells and medium (Fig. 5D). Using this in vitro furin cleavage assay, we also examined whether endogenous intracellular FGE is susceptible to furin processing. Indeed, rFurin readily converted almost 90% of endogenous fl-FGE from untransfected HT1080 cells to the truncated form (Fig. 5E). Also, here the RVKR-CMK inhibitor completely abrogated the furin-mediated processing of FGE. In summary, these data unambiguously show that furin mediates the N-terminal truncation of FGE.
Other Members of the PC Family Are Able to Process FGE during Secretion and in Some Cells Processing Additionally Occurs Extracellularly
The observation of a low but significant truncation of secreted FGE in LoVo cells (Fig. 5B) led us to speculate that FGE could also serve as a substrate to other furin-related PCs. It is reported that PACE4, PC5a, and PC7 have sequence specificity similar to that of furin (28, 29). To verify this, pIRES vector constructs coding for EGFP and furin, PACE4, PC5a, or PC7 were transiently transfected along with pBI-FGE in CHO-FD11 Tet-On cells; thereafter, cell and medium samples were subjected to Western blot analysis for FGE processing (Fig. 6A). Successful detection of EGFP signals in transfected cell lysates indirectly verified expression of untagged PCs (Fig. 6A, lower panel). Co-expression of furin led to nearly 100% Δ72-FGE in secretions, whereas PACE4 and PC5a co-expression led to processing of FGE, albeit with low efficiency. Of note, a physiological role for PC7 on the processing of FGE is unclear due to its lower expression level compared with other PCs. However, this experiment indicates that FGE is a better substrate for furin than for other PCs.
FIGURE 6.

Proteolytic processing of FGE by other furin-like proteases and extracellular processing of secreted FGE. A, cells (C) and medium (M) of CHO-FD11 Tet-On cells transiently expressing FGE alone or co-expressing the PCs furin, PACE4, PC5a, or PC7 for 24 h were analyzed by Western blotting using FGE antiserum (upper panel). The PC expression level was indirectly determined by analysis of the cell lysates for expression of EGFP, driven from the downstream IRES element (see “Experimental Procedures”), using an anti-GFP antibody (lower panel). B, conditioned medium from CHO-FD11 cells stably expressing FGE was added to MSDi, HeLa, HEK293, CHO-FD11, or HT1080 cells, or left untreated (control) and incubated for 20 h before being analyzed by Western blotting using FGE antiserum. C, conditioned medium from CHO-FD11 cells was incubated with HEK293 cells for the indicated time points and analyzed as above.
PCs are localized mainly in the trans-Golgi network but also cycle between the endosomal compartments and the cell surface (30). In addition, a soluble form of furin can be generated by sheddases (31). To investigate the possibility of post-secretion processing, we incubated fl-FGE containing conditioned medium obtained from CHO-FD11 cells stably expressing FGE with different cell lines (Fig. 6B). In fact, FGE was processed after incubation with HEK293 cells but not with HeLa, HT1080, or immortalized MSD patient cells. In parallel, conditioned medium was incubated with CHO-FD11 cells to show that truncation of FGE does not occur due to the cultivation conditions used.
Further, increased incubation times with HEK293 cells led to increased level of Δ72-FGE in the conditioned medium (Fig. 6C). These data clearly show that FGE can be processed extracellularly by surface-exposed proteases and this cleavage is cell type-specific and time-dependent. The cell type specificity may represent the furin expression level of the cell line or, at least, the amount of cell surface-exposed and/or soluble furin.
Processing of the N Terminus Leads to Inactivation of FGE
We have recently shown that the N-terminal part of FGE (residues 34–68) is essential to activate sulfatases in vivo (12). By contrast, the N-terminal extension was not required for in vitro FGly-generating activity of purified secreted Δ72-FGE. However, this in vitro activity is dependent on the presence of the reductant DTT (26, 32). To assess the physiological consequence of the N-terminal truncation by furin on the function of FGE, we analyzed the FGly-generating activity of the processed and the unprocessed forms of secreted FGE in the presence of GSH, a physiological reductant, or DTT serving as a control. The in vitro FGE activity assay (based on mass spectrometry; see “Experimental Procedures”) was performed with conditioned media obtained from CHO-FD11 cells expressing either FGE alone or co-expressing FGE plus furin leading to secretion of unprocessed FGE or processed Δ72-FGE, respectively (Fig. 7B, Western blot panels). Reaction conditions (amount of FGE and incubation time) in presence of DTT were set to 50% turnover of the cysteine-containing substrate peptide (2526.3 m/z) to the FGly-containing product peptide (2508.3 m/z). The unprocessed FGE is active in the presence of both DTT and GSH as shown by the appearance of the product (2508.3 m/z) in the representative spectra (Fig. 7A, panels a and b), whereas processed FGE (Δ72-FGE) showed activity only in the presence of DTT but not when GSH was used as reductant (Fig. 7A, panels c and d). Quantitative analysis of the FGly-generating activity (percentage substrate turnover) of FGE and Δ72-FGE in the presence of GSH normalized to that of DTT (100%) revealed that the unprocessed form is as active with GSH as with DTT, but Δ72-FGE is barely active in the presence of GSH. These data clearly indicate that the FGly-generating activity of FGE with a physiological reductant is exclusively dependent on the presence of the N-terminal extension, and furin-mediated processing functionally inactivates FGE during secretion. This agrees with the earlier observation that the forced expression of truncated FGE in MSD patient cells does not lead to intracellular sulfatase activation in vivo (12).
FIGURE 7.

Furin-mediated processing of FGE leads to inactivation. FGly-generating activity was measured in vitro using conditioned media from CHO-FD11 cells containing fl-FGE, or from CHO-FD11 cells containing Δ72-FGE due to co-expression of FGE and furin. The activity assay was performed in triplicate with three sets of conditioned media as described under “Experimental Procedures.” A, representative spectra of MALDI-TOF mass spectrometry analysis of the substrate peptide after incubation with FGE (for 20 min; a and b) or Δ72-FGE (for 30 min; c and d) containing conditioned medium using either 2 mm DTT or 5 mm GSH as reducing agents, as indicated. The cysteine substrate peptide is detected showing a monoisotopic mass at 2526.3 m/z (a–d), whereas the corresponding signal of the FGly-containing product peptide appears at 2508.3 m/z (a–c), as indicated. B, bar graph displaying relative substrate peptide turnover with GSH or DTT as reductant for fl-FGE (a and b) and Δ72-FGE (c and d); substrate turnover in the presence of GSH is normalized to that of the corresponding DTT sample (100%). Mean values of triplicates of one representative experiment are shown (top panel). Analysis of the assayed conditioned media by Western blotting (WB) using FGE antiserum is shown in the bottom panel. Error bars, S.D.
Accessibility of the N Terminus of FGE for Furin-mediated Cleavage Is Abolished When FGE Is in Complex with ERp44
Although FGE is an ER resident protein, it lacks a canonical ER retention motif. We recently showed that FGE is retained in the ER by ERp44 via a thiol-independent mechanism but nevertheless forms a disulfide-linked covalent complex with ERp44 through its N-terminal Cys-50 and Cys-52 (13). Interestingly, when co-expressed together with ERp44 lacking the RDEL sequence (ERp44ΔRDEL), a larger fraction of FGE was secreted, mainly in the fl-form. This indicates that FGE when secreted along with ERp44ΔRDEL as a complex escapes N-terminal trimming. In this study, using the in vitro furin cleavage assay, we assessed furin-mediated processing of the FGE-ERp44 covalent complex (Fig. 8). Homogenates, pretreated with N-ethylmaleimide (to prevent post-lysis disulfide shuffling), from HT1080 cells expressing either FGE alone or FGE plus myc-ERp44 together from a bi-directional promoter were incubated with rFurin and analyzed by SDS-PAGE under nonreducing conditions. Upon overexpression, intracellular FGE via its N-terminal cysteines (Cys-50 and Cys-52) forms covalently cross-bridged homodimers (Fig. 8, lanes 1 and 2) and, additionally, FGE-ERp44 heterodimers when co-expressed with ERp44 (Fig. 8, lanes 4 and 6) as previously observed (13). Removal of the N terminus by furin cleavage should lead to a disappearance of the homo-/heterodimeric FGE forms, which serves as an indicator for accessibility of the furin cleavage motif in FGE. Upon treatment with rFurin, the signals for monomeric as well as the homodimeric fraction of FGE decrease with a concomitant increase in the signal corresponding to the N-terminally truncated form (Δ72-FGE), indicating that in both monomer and homodimer the furin-cleavage site is accessible to furin (Fig. 8, compare lanes 2 and 3; lanes 4 and 5). However, rFurin treatment does not abolish the covalent interaction between FGE and ERp44 as evidenced by the presence of an intact FGE-ERp44 heterodimer observed in untreated samples (Fig. 8, compare lanes 4 and 5; 6 and 7) and a quantitative increase of the unprocessed FGE form due to heterodimer formation when analyzed under reducing (+SH) conditions (Fig. 8, lanes 3 and 5). These data show that the cleavage motif in the N terminus of FGE is inaccessible when in complex with ERp44 and is thereby protected from furin cleavage.
FIGURE 8.

FGE in complex with ERp44 resists furin cleavage. Equal amounts of NEM-treated HT1080 Tet-On cell lysates (lysed without protease inhibitor) expressing either FGE alone or co-expressing FGE and myc-ERp44 were subjected to in vitro furin cleavage (see “Experimental Procedures”). Samples were boiled in SDS-PAGE sample buffer (with or without β-mercaptoethanol), subjected to SDS-PAGE under nonreducing (−SH, upper panel) or reducing (+SH, lower panel) conditions and analyzed by Western blotting using either anti-FGE or anti-myc antibodies or both, as indicated. Note that FGE processing by endogenous furin (as indicated by the appearance of band * in lane 2) is abolished in the presence of protease inhibitor (lane 1).
DISCUSSION
FGE as a Noncanonical Substrate for Furin and Other PCs
In this study we could show that furin-like PCs cleave the N terminus of secreted FGE, with furin itself being the most effective and primary protease that performs this N-terminal processing. Several lines of evidence support our conclusion. FGE, like other proteins that are processed by PCs, bears a conserved cleavage site sequence containing the minimal consensus motif [R/K]Xn[R/K]↓. Our phylogenetic analysis revealed that the RYSR motif of FGE is conserved in later diverging eukaryote species (Fig. 3); experimentally we show that this RYSR sequence represents an authentic processing motif, as alanine variants of the conserved arginines conferred resistance to cleavage. Moreover, the processing of FGE was abolished either when treated with RVKR-CMK, a peptide based inhibitor of PCs, or when expressed in CHO-FD11 cells that are deficient for furin; replenishing furin in these cells by transient expression led to a complete processing of secreted FGE. Expression of PACE4 or PC5a also led to FGE processing, albeit to a lesser extent compared with furin. Further, both intracellular and secreted FGE was processed by recombinant furin under in vitro conditions. All of these data unambiguously lead to the conclusion that furin is the primary PC that mediates the processing of FGE during secretion.
The observation that this N-terminal processing occurs for endogenous secreted FGE signifies the physiological relevance of this processing event. Interestingly, a fraction (∼20–30%) of secreted FGE, independent of its expression level, is found in the unprocessed form. The escape from complete processing even at the very low amounts of endogenous FGE exiting the ER may reveal a kinetic limitation during the processing step and, at the same time, the importance of secreted fl-FGE. Because we observed an intracellular accumulation of FGE upon treatment with brefeldin A indicating that an intact Golgi is necessary for secretion of FGE,5 it is very unlikely that the fraction of the unprocessed form represents a population of FGE secreted via unconventional trafficking routes. Rather, we find that the processing is inefficient due to properties of the cleavage motif. A classical furin cleavage motif is defined by the presence of arginines at P1 and P4, but residues in close vicinity of the cleavage site play an important role in the recognition and cleavage efficiency of PCs (33). The presence of a positively charged amino acid at P2 (mostly lysine or arginine) has been shown to improve the processing efficiency of furin, whereas no clear consensus has been found for residues at position P3. Interestingly, the presence of Ser at position P2 in some substrates has been suggested to be unfavorable for cleavage by mammalian furin and other PCs (29). Our mutational analysis of Tyr-70 and Ser-71 residues indicates that this holds true also for FGE. Modifying the residues at positions P2 and P3 to positively charged residues, thus representing a more favorable motif, led to a strongly increased efficiency of processing with a maximum effect observed by mutating Ser-71. Thus, the presence of Ser at position P2 is the major cause for inefficient processing of FGE. Interestingly, Tyr-70 and Ser-71 are absolutely conserved throughout evolution to a degree even higher than the essential basic (arginine) residues, supporting our conclusion that the RYSR motif in FGE is a noncanonical cleavage motif that evolved to confer suboptimal cleavage efficiency.
Why Should FGE Be Processed at All?
Both phylogenetic comparison and experimental data clearly demonstrate that the FGE protein during evolution gained a function by addition of the N-terminal extension harboring the Cys-Gly-Cys sequence. This motif is critical for the biological activity of eukaryotic FGE (12) and fully conserved from human to sponge. The N-terminal extension is found only in eukaryotes (encoded by exon 1), and the fact that eukaryotic FGly generation is a co-translational event occurring in a specialized compartment, the ER, suggests that the N-terminal extension serves to adapt FGE to ER-based functioning in eukaryotes. One of the gained properties is ER retention through interaction of ERp44 with the N terminus (13); another seems to be related to inter- or intramolecular activation of the catalytic domain of FGE by the Cys-Gly-Cys motif (12). Further ER-specific aspects, such as competence to act on nascent sulfatase polypeptides emerging at ER import sites, might be associated with the N-terminal extension.
Interestingly, prokaryotic FGE from Streptomyces coelicolor, which lacks the N-terminal extension, when expressed in the cytoplasm of eukaryotic cells was shown to possess FGly-generating activity acting on engineered cytosolic model substrates containing the FGly modification signature (34, 35). On the other hand, we have shown that N-terminally truncated human FGE, when expressed in the ER, did not possess FGly-generating activity, which agrees with the hypothesis that N-terminal processing irreversibly abrogates ER-based FGE functioning. On the basis of these two observations one can speculate that furin-mediated removal of the N terminus during secretion could serve a possible mechanism in later diverging eukaryotes to generate FGE that is active under extracellular conditions. However, an extracellular formylglycine generation has not been reported so far.
Processing by PCs as a Means of Enzyme Inactivation
Our data indicate that a direct consequence of N-terminal processing of secreted human FGE is its inactivation. The N-terminal extension was found to be essential for in vitro activity in the presence of glutathione, a physiological reductant, which does not sustain activity of N-terminally truncated FGE. This corroborates our previous observation that the N terminus is essential for biological activity in cultured cells. Because removal of this part renders secreted FGE inactive, we propose that the observed furin-mediated processing during secretion is a physiological means for regulation of FGE function/activity. The need for inactivation of the enzyme is not clear, but it might be a mechanism necessary to avoid generation of toxic aldehydes in extracellular or cell surface proteins in case that FGE evades its indirect ERp44-mediated ER retention.
Proteolytic processing mediated by PCs along the secretory pathway is a widely used necessary step in the activation or maturation of many proteins that are involved in various cellular processes. In contrast, inactivation of a protein function by furin-mediated processing only recently has been recognized as a novel mode of regulation, as shown for PCSK9 (36). The function of PCSK9, in mediating LDL receptor internalization and degradation, was shown to be inactivated by furin-catalyzed cleavage of PCSK9. FGE could represent another protein that is inactivated by proteolytic processing mediated by PCs. Nevertheless, one cannot exclude the possibility that the secreted N-terminally truncated FGE could perform as yet unknown extracellular functions other than FGly generation.
Incomplete Processing as a Means to Provide Active FGE to the Extracellular Space?
On the other hand, it is tempting to assume that the inefficient processing could represent a functional tool for regulated release of FGE in the unprocessed active form. The low but significant secretion of fl-FGE even under endogenous expression levels might indicate such a regulated release of active FGE. In fact, it has been shown that secreted FGE can enter other cells by mannose receptor-mediated internalization and activate sulfatases in a paracrine manner after reaching the ER of the recipient cells (16). To exert this function, FGE should escape furin-mediated inactivation during secretion. As a proof of principle our data show that FGE, when in complex with ERp44, is barred from furin processing. This indicates that masking the cleavage site by an interacting protein during secretion could significantly hinder recognition by furin and could lead to the secretion of FGE in the unprocessed form. Alternatively, a population of FGE destined to be secreted in the active form could have an altered conformation wherein the cleavage site is masked due to intramolecular interactions of the N terminus to the core of the protein, thus preventing truncation. In combination with the suboptimal cleavage motif, an impaired processing by furin due to an inaccessible cleavage site (either by inter- or intramolecular interactions) could further increase the fraction of unprocessed secreted FGE. The secretion of FGE (14, 15) and possible reuptake by other cells (16) has been shown to be a multistep-regulated process, and our findings extend this complex regulation of FGE function to an additional level.
Perspective
The finding that FGE is secreted in the unprocessed form by furin-deficient CHO cells should pave the way for production and detailed in vitro analysis of fl-FGE, which hopefully will lead to a more complete understanding of the structure-function relationship of FGE. It should further allow studying the cell biology of FGE in more detail to address the questions and concepts put forward in this study. One of the concepts to be tested involves uptake of recombinant fl-FGE by recipient cells, which if true, might be developed into strategies for enzyme-replacement therapy in MSD patients.
Supplementary Material
Acknowledgments
We thank Steven Leppla for the furin-deficient CHO-FD11 cells; Karsten Niehaus and Manuela Meyer for help with mass spectrometry measurements; Kerstin Böker and Nicole Eiselt for excellent technical assistance; Lars Schlotawa, Marc-André Frese, Md Sarfaraz Alam, and Andrea Nolting for valuable discussions; and Peter Rehling for continuous support.
This work was supported by Deutsche Forschungsgemeinschaft Grants DI 575/7, SCHM 830/2, and SFB 860.

This article contains supplemental Table S1.
E. C. Ennemann, K. Radhakrishnan, M. Mariappan, M. Wachs, T. H. Pringle, B. Schmidt, and T. Dierks, unpublished results.
- FGly
- formylglycine
- CHO
- Chinese hamster ovary
- CMK
- chloromethylketone
- EGFP
- enhanced GFP
- ER
- endoplasmic reticulum
- FD
- furin-deficient
- FGE
- formylglycine-generating enzyme
- fl
- full-length
- MSD
- multiple sulfatase deficiency
- PC
- proprotein convertase
- rFurin
- recombinant furin
- STS
- steroid sulfatase.
REFERENCES
- 1. von Figura K., Schmidt B., Selmer T., Dierks T. (1998) A novel protein modification generating an aldehyde group in sulfatases: its role in catalysis and disease. BioEssays 20, 505–510 [DOI] [PubMed] [Google Scholar]
- 2. Diez-Roux G., Ballabio A. (2005) Sulfatases and human disease. Annu. Rev. Genomics Hum. Genet. 6, 355–379 [DOI] [PubMed] [Google Scholar]
- 3. Schmidt B., Selmer T., Ingendoh A., von Figura K. (1995) A novel amino acid modification in sulfatases that is defective in multiple sulfatase deficiency. Cell 82, 271–278 [DOI] [PubMed] [Google Scholar]
- 4. Dierks T., Schmidt B., Borissenko L. V., Peng J., Preusser A., Mariappan M., von Figura K. (2003) Multiple sulfatase deficiency is caused by mutations in the gene encoding the human C(α)-formylglycine-generating enzyme. Cell 113, 435–444 [DOI] [PubMed] [Google Scholar]
- 5. Cosma M. P., Pepe S., Annunziata I., Newbold R. F., Grompe M., Parenti G., Ballabio A. (2003) The multiple sulfatase deficiency gene encodes an essential and limiting factor for the activity of sulfatases. Cell 113, 445–456 [DOI] [PubMed] [Google Scholar]
- 6. Cosma M. P., Pepe S., Parenti G., Settembre C., Annunziata I., Wade-Martins R., Di Domenico C., Di Natale P., Mankad A., Cox B., Uziel G., Mancini G. M., Zammarchi E., Donati M. A., Kleijer W. J., Filocamo M., Carrozzo R., Carella M., Ballabio A. (2004) Molecular and functional analysis of SUMF1 mutations in multiple sulfatase deficiency. Hum. Mutat. 23, 576–581 [DOI] [PubMed] [Google Scholar]
- 7. Annunziata I., Bouche V., Lombardi A., Settembre C., Ballabio A. (2007) Multiple sulfatase deficiency is due to hypomorphic mutations of the SUMF1 gene. Hum. Mutat. 28, 928. [DOI] [PubMed] [Google Scholar]
- 8. Schlotawa L., Steinfeld R., von Figura K., Dierks T., Gärtner J. (2008) Molecular analysis of SUMF1 mutations: stability and residual activity of mutant formylglycine-generating enzyme determine disease severity in multiple sulfatase deficiency. Hum. Mutat. 29, 205. [DOI] [PubMed] [Google Scholar]
- 9. Buono M., Visigalli I., Bergamasco R., Biffi A., Cosma M. P. (2010) Sulfatase modifying factor 1-mediated fibroblast growth factor signaling primes hematopoietic multilineage development. J. Exp. Med. 207, 1647–1660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dierks T., Dickmanns A., Preusser-Kunze A., Schmidt B., Mariappan M., von Figura K., Ficner R., Rudolph M. G. (2005) Molecular basis for multiple sulfatase deficiency and mechanism for formylglycine generation of the human formylglycine-generating enzyme. Cell 121, 541–552 [DOI] [PubMed] [Google Scholar]
- 11. Landgrebe J., Dierks T., Schmidt B., von Figura K. (2003) The human SUMF1 gene, required for posttranslational sulfatase modification, defines a new gene family which is conserved from pro- to eukaryotes. Gene 316, 47–56 [DOI] [PubMed] [Google Scholar]
- 12. Mariappan M., Gande S. L., Radhakrishnan K., Schmidt B., Dierks T., von Figura K. (2008) The non-catalytic N-terminal extension of formylglycine-generating enzyme is required for its biological activity and retention in the endoplasmic reticulum. J. Biol. Chem. 283, 11556–11564 [DOI] [PubMed] [Google Scholar]
- 13. Mariappan M., Radhakrishnan K., Dierks T., Schmidt B., von Figura K. (2008) ERp44 mediates a thiol-independent retention of formylglycine-generating enzyme in the endoplasmic reticulum. J. Biol. Chem. 283, 6375–6383 [DOI] [PubMed] [Google Scholar]
- 14. Fraldi A., Zito E., Annunziata F., Lombardi A., Cozzolino M., Monti M., Spampanato C., Ballabio A., Pucci P., Sitia R., Cosma M. P. (2008) Multistep, sequential control of the trafficking and function of the multiple sulfatase deficiency gene product, SUMF1, by PDI, ERGIC-53 and ERp44. Hum. Mol. Genet. 17, 2610–2621 [DOI] [PubMed] [Google Scholar]
- 15. Preusser-Kunze A., Mariappan M., Schmidt B., Gande S. L., Mutenda K., Wenzel D., von Figura K., Dierks T. (2005) Molecular characterization of the human Cα-formylglycine-generating enzyme. J. Biol. Chem. 280, 14900–14910 [DOI] [PubMed] [Google Scholar]
- 16. Zito E., Buono M., Pepe S., Settembre C., Annunziata I., Surace E. M., Dierks T., Monti M., Cozzolino M., Pucci P., Ballabio A., Cosma M. P. (2007) Sulfatase modifying factor 1 trafficking through the cells: from endoplasmic reticulum to the endoplasmic reticulum. EMBO J. 26, 2443–2453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Seidah N. G. (2011) What lies ahead for the proprotein convertases?. Ann. N.Y. Acad. Sci. 1220, 149–161 [DOI] [PubMed] [Google Scholar]
- 18. Khatib A. M., Siegfried G., Prat A., Luis J., Chrétien M., Metrakos P., Seidah N. G. (2001) Inhibition of proprotein convertases is associated with loss of growth and tumorigenicity of HT-29 human colon carcinoma cells: importance of insulin-like growth factor-1 (IGF-1) receptor processing in IGF-1-mediated functions. J. Biol. Chem. 276, 30686–30693 [DOI] [PubMed] [Google Scholar]
- 19. Schneider T. D., Stephens R. M. (1990) Sequence logos: a new way to display consensus sequences. Nucleic Acids Res. 18, 6097–6100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Crooks G. E., Hon G., Chandonia J. M., Brenner S. E. (2004) WebLogo: A sequence logo generator. Genome Res. 14, 1188–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gordon V. M., Klimpel K. R., Arora N., Henderson M. A., Leppla S. H. (1995) Proteolytic activation of bacterial toxins by eukaryotic cells is performed by furin and by additional cellular proteases. Infect. Immun. 63, 82–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gieselmann V., Schmidt B., von Figura K. (1992) In vitro mutagenesis of potential N-glycosylation sites of arylsulfatase A: effects on glycosylation, phosphorylation, and intracellular sorting. J. Biol. Chem. 267, 13262–13266 [PubMed] [Google Scholar]
- 23. Mariappan M., Preusser-Kunze A., Balleininger M., Eiselt N., Schmidt B., Gande S. L., Wenzel D., Dierks T., von Figura K. (2005) Expression, localization, structural, and functional characterization of pFGE, the paralog of the Cα-formylglycine-generating enzyme. J. Biol. Chem. 280, 15173–15179 [DOI] [PubMed] [Google Scholar]
- 24. Duckert P., Brunak S., Blom N. (2004) Prediction of proprotein convertase cleavage sites. Protein Eng. Des. Sel. 17, 107–112 [DOI] [PubMed] [Google Scholar]
- 25. Tian S., Huajun W., Wu J. (2012) Computational prediction of furin cleavage sites by a hybrid method and understanding mechanism underlying diseases. Sci. Rep. 2, 261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dierks T., Schlotawa L., Frese M. A., Radhakrishnan K., von Figura K., Schmidt B. (2009) Molecular basis of multiple sulfatase deficiency, mucolipidosis II/III and Niemann-Pick C1 disease: lysosomal storage disorders caused by defects of non-lysosomal proteins. Biochim. Biophys. Acta 1793, 710–725 [DOI] [PubMed] [Google Scholar]
- 27. Erwin D. H., Laflamme M., Tweedt S. M., Sperling E. A., Pisani D., Peterson K. J. (2011) The Cambrian conundrum: early divergence and later ecological success in the early history of animals. Science 334, 1091–1097 [DOI] [PubMed] [Google Scholar]
- 28. Hosaka M., Nagahama M., Kim W. S., Watanabe T., Hatsuzawa K., Ikemizu J., Murakami K., Nakayama K. (1991) Arg-X-Lys/Arg-Arg motif as a signal for precursor cleavage catalyzed by furin within the constitutive secretory pathway. J. Biol. Chem. 266, 12127–12130 [PubMed] [Google Scholar]
- 29. Remacle A. G., Shiryaev S. A., Oh E. S., Cieplak P., Srinivasan A., Wei G., Liddington R. C., Ratnikov B. I., Parent A., Desjardins R., Day R., Smith J. W., Lebl M., Strongin A. Y. (2008) Substrate cleavage analysis of furin and related proprotein convertases A comparative study. J. Biol. Chem. 283, 20897–20906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Molloy S. S., Bresnahan P. A., Leppla S. H., Klimpel K. R., Thomas G. (1992) Human furin is a calcium-dependent serine endoprotease that recognizes the sequence Arg-X-X-Arg and efficiently cleaves anthrax toxin protective antigen. J. Biol. Chem. 267, 16396–16402 [PubMed] [Google Scholar]
- 31. Plaimauer B., Mohr G., Wernhart W., Himmelspach M., Dorner F., Schlokat U. (2001) “Shed” furin: mapping of the cleavage determinants and identification of its C terminus. Biochem. J. 354, 689–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fey J., Balleininger M., Borissenko L. V., Schmidt B., von Figura K., Dierks T. (2001) Characterization of posttranslational formylglycine formation by luminal components of the endoplasmic reticulum. J. Biol. Chem. 276, 47021–47028 [DOI] [PubMed] [Google Scholar]
- 33. Tian S., Jianhua W. (2010) Comparative study of the binding pockets of mammalian proprotein convertases and its implications for the design of specific small molecule inhibitors. Int. J. Biol. Sci. 6, 89–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Carlson B. L., Ballister E. R., Skordalakes E., King D. S., Breidenbach M. A., Gilmore S. A., Berger J. M., Bertozzi C. R. (2008) Function and structure of a prokaryotic formylglycine-generating enzyme. J. Biol. Chem. 283, 20117–20125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wu P., Shui W., Carlson B. L., Hu N., Rabuka D., Lee J., Bertozzi C. R. (2009) Site-specific chemical modification of recombinant proteins produced in mammalian cells by using the genetically encoded aldehyde tag. Proc. Natl. Acad. Sci. U.S.A. 106, 3000–3005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Essalmani R., Susan-Resiga D., Chamberland A., Abifadel M., Creemers J. W., Boileau C., Seidah N. G., Prat A. (2011) In vivo evidence that furin from hepatocytes inactivates PCSK9. J. Biol. Chem. 286, 4257–4263 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.