

Abstract
The c-abl proto-oncogene encodes a unique protein-tyrosine kinase (Abl) distinct from c-Src, c-Fes, and other cytoplasmic tyrosine kinases. In normal cells, Abl plays prominent roles in cellular responses to genotoxic stress as well as in the regulation of the actin cytoskeleton. Abl is also well known in the context of Bcr-Abl, the oncogenic fusion protein characteristic of chronic myelogenous leukemia. Selective inhibitors of Bcr-Abl, of which imatinib is the prototype, have had a tremendous impact on clinical outcomes in chronic myelogenous leukemia and revolutionized the field of targeted cancer therapy. In this minireview, we focus on the structural organization and dynamics of Abl kinases and how these features influence inhibitor sensitivity.
Keywords: Protein/Drug Interactions, Protein Dynamics, SH2 Domains, SH3 Domains, Tyrosine Protein Kinase (Tyrosine Kinase), Chronic Myelogenous Leukemia, Hydrogen Exchange Mass Spectrometry, Imatinib
Structural Overview of the c-Abl Kinase Core
The kinase core of the c-Abl protein has a domain organization similar to that of the Src family kinases, with sequential Src homology (SH)3 3 and SH2 domains, an SH2/kinase linker, and a bilobed kinase domain (Fig. 1). This core is flanked by an N-terminal “cap” (N-cap) region with a signal sequence for myristoylation, which serves dual roles in regulation of kinase activity and in membrane localization. C-terminal to the kinase domain is a long region of >600 amino acids encoded by a single exon, which controls interaction of Abl with other SH3-containing proteins and the actin cytoskeleton. This region also regulates nuclear-cytoplasmic shuttling of the kinase (1–4). These key structural and regulatory features are discussed in detail below.
FIGURE 1.

Domain organization of Abl kinases and crystal structure of the down-regulated c-Abl core. Upper, the c-Abl-1b protein kinase consists of a myristoylated (Myr) N-cap region, followed by SH3 and SH2 domains, the SH2/kinase linker, the tyrosine kinase domain, and a long last exon region with a C-terminal actin-binding domain (ABD). In Bcr-Abl, N-terminal fusion to Bcr sequences removes most of the N-cap, leaving the remainder of Abl intact. The Bcr-derived portion includes an N-terminal coiled-coil (CC) oligomerization domain as well as Dbl and pleckstrin homology domains (DH/PH). Lower, the x-ray crystal structure of the c-Abl-1b core protein in the down-regulated state (PDB code 2FO0) is rendered as a ribbon (lower left) and with the surface added (right) to emphasize the tight molecular packing of the regulatory regions (N-cap/SH3/SH2/linker) against the kinase domain. The unstructured portion of the myristoylated N-cap that engages the C-lobe of the kinase domain is shown as a dotted line. Domains in the structure are color-coded and correspond to the schematic shown at the top. In the kinase domain, the positions of helix αC and the activation loop (Act. Loop) are rendered in cyan and magenta, respectively. The side chain of the activation loop autophosphorylation site (Tyr412) is also shown.
The Myristoylated N-cap Is Critical for Down-regulation of Abl
The N-cap is ∼80 amino acids in length and is myristoylated in the 1b splice variant of Abl (5). The first crystal structure of the Abl core (residues 1–531) revealed that this N-terminal myristic acid group binds a deep hydrophobic pocket in the C-terminal lobe (C-lobe) of the kinase domain (Fig. 1) (6). Binding of the myristoyl group into this pocket induces a bend in C-lobe helix αI, allowing the SH2 domain to dock onto the C-lobe of the kinase domain (Fig. 2). Interaction of the myristoylated N-cap with the C-lobe is critical to maintenance of the autoinhibited state, as mutation of the myristoylation signal sequence results in a highly active kinase (7). Interestingly, small molecules that bind to this site also modulate kinase activity, supporting an allosteric connection between this regulatory pocket and the kinase active site (8–11).
FIGURE 2.

Reorientation of the SH2 domain as a function of Abl kinase activation. A, left, the position of the SH2 domain (blue) in the down-regulated structure of the c-Abl core is shown (PDB code 2FO0). Right, the interface of the SH2 domain with the back of the kinase domain is highlighted. The distal end of helix αI of the kinase domain C-lobe (cyan) is rotated away from the SH2 domain. Tyrosine residues from the SH2 (Tyr158) and kinase (Tyr361) domains form a pi-stacking interaction that contributes to the stabilization of the down-regulated conformation. B, the SH2 domain interacts with the N-lobe of the kinase domain to stabilize an active kinase domain conformation (PDB code 1OPL). Left, overview of this active top-hat conformation; right, close-up view of the SH2/N-lobe interface. Ile164 (cyan) from the SH2 domain plays a critical role in this interaction.
In addition to binding the C-lobe of the kinase domain, the N-cap also influences kinase regulation via the SH3 and SH2 domains. Although the N-cap region was disordered in the first crystal structure of the c-Abl core, a more recent structure with a modified N-cap revealed that Ser69 (numbered according to Protein Data Bank (PDB) code 2FO0)4 is phosphorylated and contacts the short connector joining the SH3 and SH2 domains. Mutation of Ser69 increased Abl activity, identifying this site as a potential input for regulatory kinases (12). Additional contacts were observed between N-cap residues 65–80 and the SH2 domain, supporting the idea that the N-cap helps to clamp the SH3 and SH2 domains to the back of the kinase domain.
The Abl SH3 and SH2 Domains Work Together to Suppress Kinase Activation
The Abl SH3 domain is composed of two short antiparallel β-sheets packed against each other to form a barrel-shaped structure. Like other SH3 domains, the Abl SH3 domain binds to proline-rich peptides that adopt a polyproline type II (PPII) helical conformation (13). In the down-regulated Abl core, the SH3 domain binds to the linker that connects the SH2 and kinase domains and forms a PPII helix (Fig. 3). Deletion or mutation of the SH3 domain, as well as alanine substitution of linker prolines, causes up-regulation of Abl kinase activity, supporting a negative regulatory role for this interaction in kinase regulation (14, 15).
FIGURE 3.

The Abl SH3 domain interacts with the SH2/kinase linker in down-regulated Abl. The positions of the SH3 domain (red) and the linker (orange) within the down-regulated c-Abl core structure (PDB code 2FO0) are shown (upper) and are also enlarged (lower). Note that the linker adopts a PPII helix that engages the SH3 domain. The side chains of three regulatory tyrosine phosphorylation sites are also shown. Two are located on the binding surface of the SH3 domain that faces the linker (Tyr89 and Tyr134), whereas the third is on the linker (Tyr245) and faces the N-lobe of the kinase domain.
Although analogous SH3/linker interactions play major roles in the regulation of Abl and Src family kinases, there are interesting differences in the sequences and structures of the linkers between these kinase families. The Abl linker adopts a unique conformation that results in part from the insertion of three additional residues relative to Src family kinases. As described in more detail below, hydrogen exchange mass spectrometry (HX-MS) supports the persistence of Abl SH3/linker interaction in the absence of the kinase domain and shows that these additional residues stabilize the linker PPII helical conformation required for SH3 binding. This is in striking contrast to the Src family kinase Hck, where SH3/linker interaction does not occur in the absence of the kinase domain. Thus, SH3/linker interaction may have a more prominent role in regulating c-Abl kinase activity relative to Src family kinases (16–18).
The Abl SH2 domain mediates sequence-specific recognition of phosphotyrosine-containing sequences. This phosphotyrosine-binding function contributes to recognition and processive phosphorylation of some Abl substrate proteins (19). Structurally, the Abl SH2 domain consists of a central antiparallel β-sheet flanked by α-helices on each side. The central β-sheet divides the domain into two functionally distinct pockets, one of which binds the phosphotyrosine side chain in the target protein. The second pocket interacts with the side chain of the third amino acid C-terminal to the phosphotyrosine residue to confer sequence specificity. In Src family kinases, the SH2 domain engages a conserved phosphotyrosine residue in the C-terminal tail, and this intramolecular interaction is critical to down-regulation of kinase activity (20). In contrast, the phosphotyrosine-binding function of the Abl SH2 domain is not known to contribute to kinase down-regulation. Instead, the Abl SH2 domain packs against the C-lobe of the kinase domain through a network of hydrogen bonds and a unique pi-stacking interaction involving the side chains of SH2 Tyr158 and C-lobe Tyr361 (PDB code 2FO0) (Fig. 2A). Binding of the myristoylated N-cap to the C-lobe, as described above, reorients the kinase domain C-lobe helix αI to allow SH2 docking (6, 7, 12). SH2/C-lobe interface mutations (particularly Tyr158) increase kinase activity, demonstrating the importance of this interaction to Abl down-regulation (7).
More recent studies show that the SH2 domain may undergo significant reorientation during kinase activation. In the most highly activated state, evidence suggests that the SH2 domain moves from its negative regulatory position from the back of the C-lobe to a new position on “top” of the kinase domain (Fig. 2B). Sometimes referred to as the “top-hat” conformation (10, 21), this orientation allows the SH2 domain to interact with the N-terminal lobe (N-lobe) to stabilize the active form of the kinase domain. Mutagenesis of amino acids involved in this SH2/N-lobe interface, particularly SH2 Ile164, impairs the kinase activity of both Abl and Bcr-Abl, supporting a positive regulatory role for this interaction in kinase function (22, 23). A similar SH2/N-lobe interaction has also been reported to stabilize the active conformation of the c-Fes protein-tyrosine kinase (22).
Abl Kinase Domain
The Abl kinase domain catalyzes the transfer of γ-phosphate from ATP onto tyrosine residues in substrate proteins and peptides. The relative orientations of the N- and C-lobes, as well as conserved residues in the active site, coordinate the dynamic interconversion of the active and inactive conformations of the kinase domain with catalytic function (24). Structural analyses of the isolated Abl kinase domain and the larger core proteins described above have revealed key regulatory features (25–28). The N-lobe of c-Abl is composed of a five-stranded antiparallel β-sheet and a single α-helix called helix αC. In contrast, the C-lobe is mainly helical and encompasses the peptide substrate-binding site. Both lobes contribute important conserved residues to the active site, which is located between them. Amino acids connecting strands β1 and β2 in the N-lobe form the phosphate-binding loop, which is critical for coordination of the ATP-Mg2+ co-substrate complex. In the crystal structure of the down-regulated kinase domain with imatinib bound, a conserved glutamate residue from helix αC (Glu286) forms an ion pair with Lys271 in the N-lobe (26). This pairing is important in coordinating the phosphate group of ATP and is conserved in virtually all protein kinase structures.
The relative orientation of N-lobe helix αC plays a major role in regulating the interconversion of the active versus inactive conformations of many kinase domains. In the structures of the inactive forms of c-Src and cyclin-dependent kinases, helix αC is rotated away from the active site, and the conserved Glu-Lys ion pair is disrupted (20). Interestingly, the orientation of helix αC is essentially unchanged, and this ion pair is maintained in the Abl kinase domain structure when bound to imatinib (PDB code 1IEP) (Fig. 4A). In addition, the Abl kinase domain can access a c-Src/Cdk-like inactive conformation when complexed with an ATP-peptide conjugate instead of imatinib (PDB code 2G1T) (29).
FIGURE 4.

Structural features of the Abl kinase domain important for activity and inhibitor binding. A, close-up view of the Abl kinase domain bound to imatinib (PDB code 1IEP), which stabilizes a down-regulated conformation of the active site. Key structural elements of this off-state include the inward rotation of N-lobe helix αC (cyan), which positions Glu286 for ion pairing with Lys271. The DFG motif (Asp381, Phe382, and Gly383), which is located at the N-terminal end of the activation loop (Act Loop; green), is rotated away from the active site to accommodate imatinib binding (DFGout). The activation loop tyrosine (numbered as Tyr393 in this structure) points into the active site and makes a hydrogen bond with the catalytic aspartate (Asp363; purple). Also shown is the side chain of the gatekeeper residue (Thr315; orange), which forms a critical hydrogen bond with imatinib. B, close-up view of the Abl catalytic site in an active conformation with dasatinib bound (PDB code 2GQG; with ligand carbon atoms in yellow). Note that helix αC and the Glu286 ion pair with Lys271 are positioned as per the inactive state in A, but the activation loop is completely reoriented. The phosphorylated activation loop tyrosine (phospho-Tyr393) is now paired with a nearby arginine residue (Arg386), releasing the catalytic aspartate and stabilizing the active conformation. The DFG motif is now flipped inward (DFGin); this conformation is not compatible with imatinib binding due to steric clash. The gatekeeper threonine also makes a hydrogen bond with dasatinib.
The C-lobe contributes to the active site through the activation loop, another dynamic structural feature common to many kinases. Phosphorylation of a single tyrosine site in the Abl activation loop (Tyr393 in PDB code 2GQG) drives electrostatic interaction with a neighboring arginine residue (Fig. 4B), stabilizing an “open” conformation of the active site and allowing access to the peptide substrate (30). In the inactive imatinib-bound conformation, the activation loop tyrosine is not phosphorylated and instead forms a hydrogen bond with the catalytic aspartate (Asp363 in PDB code 1IEP). This interaction causes the activation loop to occlude the active site by mimicking the binding mode of substrates in a manner reminiscent of pseudo-substrate inhibition (Fig. 4A).
N-terminal to the activation loop is an aspartate-phenylalanine-glycine (DFG) motif common to many eukaryotic protein kinases. In the active state, the aspartate residue of the DFG motif is oriented toward the active site (“DFG-in” conformation) (Fig. 4B), where it coordinates a catalytically important magnesium ion. In the inactive conformation, the aspartate moves away from the active site while the phenylalanine moves inward. This so-called “DFG-out” or “DFG-flipped” conformation (Fig. 4A) is incompatible with Mg2+ binding and catalysis. Molecular dynamics simulations have established that the protonation state of the DFG aspartate controls the DFG flip and hence the conformational changes associated with kinase activation and imatinib binding (31).
The DFG-out conformation is essential for imatinib binding (Fig. 4A) and was originally proposed to account for imatinib specificity for Abl (25, 26). However, this view changed when c-Src, a very weak binder of imatinib, was also observed to access the DFG-out conformation in the crystalline state (27, 28). These studies went on to show that a “kinked” conformation unique to the Abl kinase domain phosphate-binding loop accounts for imatinib specificity (c-Src cannot kink this loop).
Regulation of Abl Kinase Activity by Phosphorylation and Interaction with Other Proteins
The kinase activity of Abl is tightly regulated in cells, and in the absence of activating stimuli, neither endogenous nor overexpressed Abl is phosphorylated on tyrosine (32, 33). As described above, the crystal structures of down-regulated Abl reveal that multiple intramolecular interactions among the non-catalytic domains work together to suppress kinase activity. Disruption of these inhibitory interactions by site-directed mutagenesis increases Abl phosphotyrosine content, which is positively correlated with activity (34). Phosphorylation of tyrosine sites in the activation loop, the SH3 domain, the linker, and elsewhere contributes to kinase activation, presumably by affecting intramolecular regulatory interactions and protein dynamics. One example is Tyr245 in the SH2/kinase linker. Mutagenesis of this site to prevent phosphorylation reduces maximal activation of Abl by ∼50% in vitro, indicating an important role in kinase activation that may result from disruption of SH3/linker or SH3/N-lobe interaction (32). Other phosphorylation sites implicated in Abl regulation include Tyr89 and Tyr134 in the SH3 domain, which impact binding to the SH2/kinase linker (see Fig. 3). These tyrosine sites can be phosphorylated by other kinases, such as Src family members, providing critical points for regulatory inputs in Abl signaling networks and in Bcr-Abl (35–37).
Abl kinase activity is also regulated by interacting partners, and the interaction mechanisms are often complex (1). Abl interacts with other proteins through its SH2 or SH3 domain, whereas the SH2 or SH3 domain on the partner proteins may associate with respective phosphotyrosine sites and PPII helices on Abl. For example, the Abl interactor proteins ABI1 and ABI2 bind to Abl through reciprocal SH3/PPII helical interactions. ABI1 binding facilitates Abl oligomerization and autophosphorylation, leading to enhanced phosphorylation of Abl substrates (38–40). Interaction with ABI1 may link Abl kinases to remodeling of the actin cytoskeleton (41–43). Another example is the Ras effector protein RIN1, which engages both the Abl SH2 and SH3 domains (44), leading to kinase activation through a domain displacement mechanism. In contrast to the ABI proteins and RIN1, other Abl-interacting partners have been implicated in the negative regulation of kinase activity, including PAG, FUS1, and the NR2D subunit of the NMDA receptor (45–47). The structural mechanism of Abl kinase suppression by interaction with these proteins is less clear but may involve stabilization (as opposed to disruption) of intramolecular interactions.
Exploring the Conformational Dynamics of Abl Proteins via HX-MS
Abl function and regulation cannot be fully understood based on the static structural views provided by x-ray crystallography alone. In this regard, HX-MS has proven very useful for exploring the dynamic changes that accompany kinase activation (48). Below, we review the HX-MS analyses of Abl that help to explain its dynamic regulation. Application of HX-MS to the more general study of protein dynamics is reviewed elsewhere (49–52).
The fundamental principle of HX-MS is based on the exchange of hydrogen atoms in proteins with deuterium atoms upon exposure to D2O solvent (53). Deuteration causes the protein to gain mass; the rate of hydrogen exchange can therefore be monitored over time (typically seconds to hours) by MS (54). In folded proteins, hydrogens in highly dynamic regions exposed to solvent undergo rapid deuteration, whereas regions less exposed to solvent or involved in hydrogen bonding display slower exchange kinetics. Spatial resolution of deuterium incorporation requires pepsin digestion of the protein after quenching the exchange reaction but before chromatography and MS.
HX-MS Reveals Cooperative Unfolding in the Abl SH3 Domain
HX-MS is capable of detecting the interconversion of folded and unfolded protein states over time, as governed by the following relationship (Reaction 1) (53, 55),
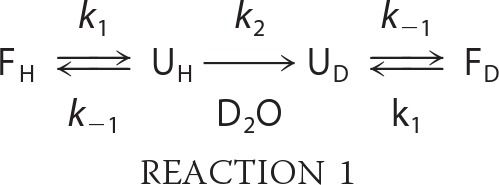 |
where F and U represent the folded versus unfolded states, respectively, with subscripted H and D designating the starting protein (all hydrogen) versus deuterated forms. When the refolding rate of a protein is much greater than the deuterium exchange rate (i.e. k−1 ≫ k2), the resulting mass spectra exhibit a single peak that uniformly increases in mass over the labeling time course (EX2 kinetics). A rarer event in protein dynamics (EX1 kinetics) occurs when the rate of protein refolding is much slower than the deuterium exchange rate (k−1 ≪ k2). Proteins undergoing EX1 exchange exhibit a unique bimodal isotopic distribution in the mass spectra due to the presence of unfolded species that undergo cooperative deuterium uptake and thus display a higher mass relative to the folded species.
Using HX-MS, EX1 kinetics was discovered in the SH3 domain of Abl (56). The Abl SH3 domain undergoes a slow cooperative unfolding-refolding event with a half-life of ∼10 min (56). Cooperative unfolding was greatly slowed when the SH3 domain was bound to a peptide ligand, providing a useful measure of SH3 domain occupancy (16). These observations allowed us to establish HX-MS as a biophysical approach to better understand dynamic changes in the Abl SH3 domain as a function of ligand binding and linker interaction.
Characterization of Abl SH3/Linker Interaction Dynamics by HX-MS
Association of the Abl SH3 domain with the SH2/kinase linker was first studied with HX-MS by monitoring changes in cooperative unfolding of the Abl SH3 domain in a series of recombinant Abl SH3/SH2/linker proteins that lacked the kinase domain (16). These studies showed that the presence of the linker slowed down the unfolding rate, providing the first direct evidence for Abl SH3/linker interaction in solution. HX-MS also showed a stabilizing influence of the N-cap on intramolecular SH3/linker interaction (57) even in the absence of the kinase domain and myristoylation. This observation suggested that the N-cap may compensate for the absence of the negative regulatory C-terminal tail, which in Src family kinases engages the SH2 domain in the down-regulated conformation (58).
Src family kinases phosphorylate the Abl SH3 domain at Tyr89, a phosphorylation event required for the full transforming function of Bcr-Abl (36). In the crystal structure of the down-regulated Abl core, Tyr89 is located at the interface of the SH3 domain and the SH2/kinase linker (Fig. 3), suggesting that phosphorylation may disturb this key regulatory interaction. HX-MS studies provide direct support for this idea, showing that phosphorylation of Tyr89 by the Src family kinase Hck causes SH3 release from the linker (59). This result supports a model in which phosphorylation by Src family kinases may directly contribute to Bcr-Abl oncogenic potential and drug resistance.
Characterization of the Abl Kinase Core by HX-MS
HX-MS was later used to investigate conformational changes in larger c-Abl kinase core proteins consisting of the N-cap, SH3 and SH2 domains, the linker, and the kinase domain. One study compared changes in deuterium uptake resulting from imatinib resistance mutations. The most recalcitrant imatinib-resistant mutant of Bcr-Abl observed clinically occurs in the kinase domain at the “gatekeeper” position (Thr315 in PDB code 1IEP) (Fig. 4A). Substitution of the Bcr-Abl gatekeeper position with isoleucine (T315I) results in the loss of a direct hydrogen bond with the drug and also creates steric clash (60–62). The Abl T315I protein showed increased deuterium incorporation at the site of the mutation relative to the wild-type protein, reflecting local conformational changes. Remarkably, the T315I mutant also showed changes in deuterium uptake in the SH3 domain, providing evidence for the long-range conformational impact of this mutation that helps to explain its activating effect on the kinase (62, 63).
HX-MS has also been a very useful approach to understand the stabilizing influence of inhibitor binding to the Abl kinase domain through both the active site (e.g. imatinib and nilotinib) and the myristic acid-binding pocket (e.g. GNF-2/GNF-5). Binding of GNF-5 to the myristic acid-binding pocket resulted in a local reduction in the rate of deuterium uptake relative to both the unbound protein and the ATP-binding site >30 Å away (9, 10). This observation provides important evidence that binding of myristic acid and small molecules to this C-lobe pocket has a stabilizing inhibitory influence on the active site. Furthermore, simultaneous binding of GNF-5 and the ATP-competitive inhibitor dasatinib to the Abl T315I mutant induced conformational changes similar to those observed with wild-type Abl in the presence of dasatinib alone (9, 10). These observations support the idea that binding of small molecule antagonists to the myristate-binding pocket helps to stabilize the down-regulated conformation of the active site and/or remodel the active site such that ATP-competitive inhibitors can interact with Abl mutants.
What Do Structural Studies of the Abl Core Tell Us about Bcr-Abl Structure and Regulation?
Bcr-Abl is the chimeric oncoprotein that drives chronic myelogenous leukemia (CML) pathogenesis. As a result of the Philadelphia chromosome translocation associated with CML, Bcr fusion removes most of the N-cap from Abl (Fig. 1). As a consequence, the negative regulatory influence of the myristic acid group and N-cap interactions with the SH3 and SH2 domains are lost. In addition, fusion to Bcr adds a coiled-coil domain to the N terminus of Bcr-Abl (64, 65). This Bcr domain causes oligomerization of Bcr-Abl and is believed to result in juxtaposition of multiple Abl kinase domains and subsequent transphosphorylation of the activation loop and other sites that contribute to kinase activation (66). Short peptides that disrupt Bcr coiled-coil interactions reduce kinase and transforming activities, supporting this activation model (67).
One important question that remains unanswered concerns the regulatory influence of the SH3 and SH2 domains on the kinase domain in the context of Bcr-Abl. Although there are no structural or HX-MS data currently available for full-length active Bcr-Abl, other studies suggest that the SH3 and SH2 domains retain their influence over the kinase domain. One line of evidence comes from studies of imatinib-resistant mutants of Bcr-Abl. As described above, imatinib binds to the DFG-out conformation of the Abl kinase domain (Fig. 4A). Although many imatinib-resistant mutants of Bcr-Abl involve kinase domain residues that are directly involved in drug binding (e.g. T315I), others occur at a distance from the binding site and map to the SH2 and SH3 domains and the linker. Imatinib resistance also results from overexpression of the Src family kinase Hck in the absence of Bcr-Abl mutations (35, 68) and may be due to Hck-mediated phosphorylation of the Bcr-Abl SH3 domain (35). These observations support a mechanism in which either mutation or phosphorylation of the SH3/SH2 regulatory region disturbs its interaction with the kinase domain, causing it to adopt a conformation incompatible with drug binding.
Recent work from our laboratories approached this question more directly by systematically increasing the proline content of the Abl SH2/kinase linker to enhance intramolecular SH3 engagement (69). Remarkably, myeloid cells transformed with Bcr-Abl proteins carrying high affinity linkers showed enhanced sensitivity to apoptosis induced by both imatinib and GNF-2. These effects correlated with inhibition of Bcr-Abl kinase activity and were observed with both wild-type and imatinib-resistant forms of Bcr-Abl. HX-MS of the corresponding recombinant Abl core proteins revealed that enhanced SH3/linker interaction correlated with long-range dynamic stabilization of both the kinase domain active site and the C-lobe myristate-binding pocket. These results provide strong evidence that regulatory SH3/linker interaction is maintained in the context of Bcr-Abl and suggest that chemical stabilization of the SH3/linker interface may sensitize Bcr-Abl to existing CML drugs.
Other studies have explored the structural positioning of the SH2 domain relative to the kinase domain in the context of Bcr-Abl (23). As described earlier, the SH2 domain interacts with the back of the C-lobe in the down-regulated conformation of the Abl core but can undergo a dramatic shift to the top-hat conformation upon kinase activation (Fig. 2B) (22, 23). In this conformation, SH2 interacts with the kinase domain N-lobe to stabilize the active state. Bcr-Abl proteins with mutations designed to disrupt SH2/N-lobe interaction show reduced kinase activity and transforming function, strongly supporting the existence of this active conformation in the context of Bcr-Abl.
At first glance, the observation that the Abl SH2/SH3 unit can influence inhibitor sensitivity and kinase domain conformation seems at odds with a requirement for the top-hat conformation in Bcr-Abl activation. However, these seemingly disparate observations can be reconciled when all possible active conformations of Bcr-Abl are considered. Closer examination of the activity data from the study of Grebien et al. (23) suggests that some but not all Bcr-Abl molecules in a given population adopt the top-hat conformation. In most of the experimental paradigms employed in this study, mutations predicted to disrupt the SH2/kinase interface reduced but did not eliminate Bcr-Abl kinase activity and function. Interestingly, experiments with a “monobody” designed to target the SH2/kinase interface reduced Bcr-Abl kinase activity by ∼50%, at which point the concentration-response curve reached a plateau. These observations support the existence of other active states of Bcr-Abl that are not sensitive to this reagent and therefore may not require the SH2/kinase interface to function. Such alternative active states may arise from the unique mechanism by which the Abl core is activated in the context of Bcr-Abl. In this case, fusion to the Bcr coiled coils induces clustering of the Abl kinase core, resulting in transphosphorylation of the activation loop as described above. Such a mechanism may not require regulatory domain displacement from the back of the kinase domain or a shift to the top-hat conformation. The broader concept that multidomain kinases (including Abl) may occupy multiple conformational states and take more than one path to activation is supported by previous small angle x-ray scattering studies of the Src family kinase Hck (70).
In summary, both Abl and Bcr-Abl are regulated by conserved kinase domain features as well as unique structural elements of the N-cap, SH3 and SH2 domains, and the SH2/kinase linker. Future drug discovery efforts targeting allosteric mechanisms unique to this kinase system may provide a path to exceptional inhibitor selectivity.
This work was supported, in whole or in part, by National Institutes of Health Grants CA169962 and CA101828 (to T. E. S.) and GM086507 and GM101135 (to J. R. E.) and by a research collaboration with the Waters Corporation (to J. R. E.).
Three PDB files were used to create the structural models of Abl used in this minireview. One uses the numbering convention of the Abl-1b splice variant (code 2FO0), in which the activation loop tyrosine is numbered as Tyr412. The other structures use Abl-1a numbering (codes 1IEP and 2GQG); this splice variant is not myristoylated and is 19 amino acids shorter than Abl-1b. The activation loop tyrosine is therefore located at amino acid 393 in these structures. In referring to specific amino acid positions in text, we indicate the PDB code for the residue positions in question.
- SH
- Src homology
- N-cap
- N-terminal cap
- C-lobe
- C-terminal lobe
- N-lobe
- N-terminal lobe
- PDB
- Protein Data Bank
- PPII
- polyproline type II
- HX-MS
- hydrogen exchange mass spectrometry
- CML
- chronic myelogenous leukemia.
REFERENCES
- 1. Colicelli J. (2010) ABL tyrosine kinases: evolution of function, regulation, and specificity. Sci. Signal. 3, re6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hantschel O., Superti-Furga G. (2004) Regulation of the c-Abl and Bcr-Abl tyrosine kinases. Nat. Rev. Mol. Cell Biol. 5, 33–44 [DOI] [PubMed] [Google Scholar]
- 3. Pendergast A. M. (2002) The Abl family kinases: mechanisms of regulation and signaling. Adv. Cancer Res. 85, 51–100 [DOI] [PubMed] [Google Scholar]
- 4. Van Etten R. A. (1999) Cycling, stressed-out and nervous: cellular functions of c-Abl. Trends Cell Biol. 9, 179–186 [DOI] [PubMed] [Google Scholar]
- 5. Pluk H., Dorey K., Superti-Furga G. (2002) Autoinhibition of c-Abl. Cell 108, 247–259 [DOI] [PubMed] [Google Scholar]
- 6. Nagar B., Hantschel O., Young M. A., Scheffzek K., Veach D., Bornmann W., Clarkson B., Superti-Furga G., Kuriyan J. (2003) Structural basis for the autoinhibition of c-Abl tyrosine kinase. Cell 112, 859–871 [DOI] [PubMed] [Google Scholar]
- 7. Hantschel O., Nagar B., Guettler S., Kretzschmar J., Dorey K., Kuriyan J., Superti-Furga G. (2003) A myristoyl/phosphotyrosine switch regulates c-Abl. Cell 112, 845–857 [DOI] [PubMed] [Google Scholar]
- 8. Adrián F. J., Ding Q., Sim T., Velentza A., Sloan C., Liu Y., Zhang G., Hur W., Ding S., Manley P., Mestan J., Fabbro D., Gray N. S. (2006) Allosteric inhibitors of Bcr-Abl-dependent cell proliferation. Nat. Chem. Biol. 2, 95–102 [DOI] [PubMed] [Google Scholar]
- 9. Zhang J., Adrián F. J., Jahnke W., Cowan-Jacob S. W., Li A. G., Iacob R. E., Sim T., Powers J., Dierks C., Sun F., Guo G. R., Ding Q., Okram B., Choi Y., Wojciechowski A., Deng X., Liu G., Fendrich G., Strauss A., Vajpai N., Grzesiek S., Tuntland T., Liu Y., Bursulaya B., Azam M., Manley P. W., Engen J. R., Daley G. Q., Warmuth M., Gray N. S. (2010) Targeting Bcr-Abl by combining allosteric with ATP-binding-site inhibitors. Nature 463, 501–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Iacob R. E., Zhang J., Gray N. S., Engen J. R. (2011) Allosteric interactions between the myristate- and ATP-site of the Abl kinase. PLoS ONE 6, e15929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yang J., Campobasso N., Biju M. P., Fisher K., Pan X. Q., Cottom J., Galbraith S., Ho T., Zhang H., Hong X., Ward P., Hofmann G., Siegfried B., Zappacosta F., Washio Y., Cao P., Qu J., Bertrand S., Wang D. Y., Head M. S., Li H., Moores S., Lai Z., Johanson K., Burton G., Erickson-Miller C., Simpson G., Tummino P., Copeland R. A., Oliff A. (2011) Discovery and characterization of a cell-permeable, small-molecule c-Abl kinase activator that binds to the myristoyl binding site. Chem. Biol. 18, 177–186 [DOI] [PubMed] [Google Scholar]
- 12. Nagar B., Hantschel O., Seeliger M., Davies J. M., Weis W. I., Superti-Furga G., Kuriyan J. (2006) Organization of the SH3-SH2 unit in active and inactive forms of the c-Abl tyrosine kinase. Mol. Cell 21, 787–798 [DOI] [PubMed] [Google Scholar]
- 13. Pisabarro M. T., Serrano L., Wilmanns M. (1998) Crystal structure of the Abl-SH3 domain complexed with a designed high-affinity peptide ligand: implications for SH3-ligand interactions. J. Mol. Biol. 281, 513–521 [DOI] [PubMed] [Google Scholar]
- 14. Franz W. M., Berger P., Wang J. Y. (1989) Deletion of an N-terminal regulatory domain of the c-Abl tyrosine kinase activates its oncogenic potential. EMBO J. 8, 137–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mayer B. J., Baltimore D. (1994) Mutagenic analysis of the roles of SH2 and SH3 domains in regulation of the Abl tyrosine kinase. Mol. Cell. Biol. 14, 2883–2894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen S., Brier S., Smithgall T. E., Engen J. R. (2007) The Abl SH2-kinase linker naturally adopts a conformation competent for SH3 domain binding. Protein Sci. 16, 572–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hochrein J. M., Lerner E. C., Schiavone A. P., Smithgall T. E., Engen J. R. (2006) An examination of dynamics crosstalk between SH2 and SH3 domains by hydrogen/deuterium exchange and mass spectrometry. Protein Sci. 15, 65–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lerner E. C., Trible R. P., Schiavone A. P., Hochrein J. M., Engen J. R., Smithgall T. E. (2005) Activation of the Src family kinase Hck without SH3-linker release. J. Biol. Chem. 280, 40832–40837 [DOI] [PubMed] [Google Scholar]
- 19. Mayer B. J., Hirai H., Sakai R. (1995) Evidence that SH2 domains promote processive phosphorylation by protein-tyrosine kinases. Curr. Biol. 5, 296–305 [DOI] [PubMed] [Google Scholar]
- 20. Huse M., Kuriyan J. (2002) The conformational plasticity of protein kinases. Cell 109, 275–282 [DOI] [PubMed] [Google Scholar]
- 21. Dixit A., Verkhivker G. M. (2009) Hierarchical modeling of activation mechanisms in the ABL and EGFR kinase domains: thermodynamic and mechanistic catalysts of kinase activation by cancer mutations. PLoS Comput. Biol. 5, e1000487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Filippakopoulos P., Kofler M., Hantschel O., Gish G. D., Grebien F., Salah E., Neudecker P., Kay L. E., Turk B. E., Superti-Furga G., Pawson T., Knapp S. (2008) Structural coupling of SH2-kinase domains links Fes and Abl substrate recognition and kinase activation. Cell 134, 793–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Grebien F., Hantschel O., Wojcik J., Kaupe I., Kovacic B., Wyrzucki A. M., Gish G. D., Cerny-Reiterer S., Koide A., Beug H., Pawson T., Valent P., Koide S., Superti-Furga G. (2011) Targeting the SH2-kinase interface in Bcr-Abl inhibits leukemogenesis. Cell 147, 306–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cox S., Radzio-Andzelm E., Taylor S. S. (1994) Domain movements in protein kinases. Curr. Opin. Struct. Biol. 4, 893–901 [DOI] [PubMed] [Google Scholar]
- 25. Schindler T., Bornmann W., Pellicena P., Miller W. T., Clarkson B., Kuriyan J. (2000) Structural mechanism for STI-571 inhibition of Abelson tyrosine kinase. Science 289, 1938–1942 [DOI] [PubMed] [Google Scholar]
- 26. Nagar B., Bornmann W. G., Pellicena P., Schindler T., Veach D. R., Miller W. T., Clarkson B., Kuriyan J. (2002) Crystal structures of the kinase domain of c-Abl in complex with the small molecule inhibitors PD173955 and imatinib (STI-571). Cancer Res. 62, 4236–4243 [PubMed] [Google Scholar]
- 27. Seeliger M. A., Nagar B., Frank F., Cao X., Henderson M. N., Kuriyan J. (2007) c-Src binds to the cancer drug imatinib with an inactive Abl/c-Kit conformation and a distributed thermodynamic penalty. Structure 15, 299–311 [DOI] [PubMed] [Google Scholar]
- 28. Seeliger M. A., Ranjitkar P., Kasap C., Shan Y., Shaw D. E., Shah N. P., Kuriyan J., Maly D. J. (2009) Equally potent inhibition of c-Src and Abl by compounds that recognize inactive kinase conformations. Cancer Res. 69, 2384–2392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Levinson N. M., Kuchment O., Shen K., Young M. A., Koldobskiy M., Karplus M., Cole P. A., Kuriyan J. (2006) A Src-like inactive conformation in the Abl tyrosine kinase domain. PLoS Biol. 4, e144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tokarski J. S., Newitt J. A., Chang C. Y., Cheng J. D., Wittekind M., Kiefer S. E., Kish K., Lee F. Y., Borzillerri R., Lombardo L. J., Xie D., Zhang Y., Klei H. E. (2006) The structure of dasatinib (BMS-354825) bound to activated ABL kinase domain elucidates its inhibitory activity against imatinib-resistant ABL mutants. Cancer Res. 66, 5790–5797 [DOI] [PubMed] [Google Scholar]
- 31. Shan Y., Seeliger M. A., Eastwood M. P., Frank F., Xu H., Jensen M. Ø., Dror R. O., Kuriyan J., Shaw D. E. (2009) A conserved protonation-dependent switch controls drug binding in the Abl kinase. Proc. Natl. Acad. Sci. U.S.A. 106, 139–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Brasher B. B., Van Etten R. A. (2000) c-Abl has high intrinsic tyrosine kinase activity that is stimulated by mutation of the Src homology 3 domain and by autophosphorylation at two distinct regulatory tyrosines. J. Biol. Chem. 275, 35631–35637 [DOI] [PubMed] [Google Scholar]
- 33. Dorey K., Engen J. R., Kretzschmar J., Wilm M., Neubauer G., Schindler T., Superti-Furga G. (2001) Phosphorylation and structure-based functional studies reveal a positive and a negative role for the activation loop of the c-Abl tyrosine kinase. Oncogene 20, 8075–8084 [DOI] [PubMed] [Google Scholar]
- 34. Barilá D., Superti-Furga G. (1998) An intramolecular SH3-domain interaction regulates c-Abl activity. Nat. Genet. 18, 280–282 [DOI] [PubMed] [Google Scholar]
- 35. Pene-Dumitrescu T., Smithgall T. E. (2010) Expression of a Src family kinase in chronic myelogenous leukemia cells induces resistance to imatinib in a kinase-dependent manner. J. Biol. Chem. 285, 21446–21457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Meyn M. A., 3rd, Wilson M. B., Abdi F. A., Fahey N., Schiavone A. P., Wu J., Hochrein J. M., Engen J. R., Smithgall T. E. (2006) Src family kinases phosphorylate the Bcr-Abl SH3-SH2 region and modulate Bcr-Abl transforming activity. J. Biol. Chem. 281, 30907–30916 [DOI] [PubMed] [Google Scholar]
- 37. Plattner R., Kadlec L., DeMali K. A., Kazlauskas A., Pendergast A. M. (1999) c-Abl is activated by growth factors and Src family kinases and has a role in the cellular response to PDGF. Genes Dev. 13, 2400–2411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dai Z., Pendergast A. M. (1995) Abi-2, a novel SH3-containing protein, interacts with the c-Abl tyrosine kinase and modulates c-Abl transforming activity. Genes Dev. 9, 2569–2582 [DOI] [PubMed] [Google Scholar]
- 39. Shi Y., Alin K., Goff S. P. (1995) Abl-interactor-1, a novel SH3 protein binding to the carboxy-terminal portion of the Abl protein, suppresses v-abl transforming activity. Genes Dev. 9, 2583–2597 [DOI] [PubMed] [Google Scholar]
- 40. Xiong X., Cui P., Hossain S., Xu R., Warner B., Guo X., An X., Debnath A. K., Cowburn D., Kotula L. (2008) Allosteric inhibition of the nonmyristoylated c-Abl tyrosine kinase by phosphopeptides derived from Abi1/Hssh3bp1. Biochim. Biophys. Acta 1783, 737–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li Y., Clough N., Sun X., Yu W., Abbott B. L., Hogan C. J., Dai Z. (2007) Bcr-Abl induces abnormal cytoskeleton remodeling, β1 integrin clustering and increased cell adhesion to fibronectin through the Abl interactor 1 pathway. J. Cell Sci. 120, 1436–1446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sun X., Li Y., Yu W., Wang B., Tao Y., Dai Z. (2008) MT1-MMP as a downstream target of BCR-ABL/ABL interactor 1 signaling: polarized distribution and involvement in BCR-ABL-stimulated leukemic cell migration. Leukemia 22, 1053–1056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhuang C., Tang H., Dissanaike S., Cobos E., Tao Y., Dai Z. (2011) CDK1-mediated phosphorylation of Abi1 attenuates Bcr-Abl-induced F-actin assembly and tyrosine phosphorylation of WAVE complex during mitosis. J. Biol. Chem. 286, 38614–38626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cao X., Tanis K. Q., Koleske A. J., Colicelli J. (2008) Enhancement of ABL kinase catalytic efficiency by a direct binding regulator is independent of other regulatory mechanisms. J. Biol. Chem. 283, 31401–31407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wen S. T., Van Etten R. A. (1997) The PAG gene product, a stress-induced protein with antioxidant properties, is an Abl SH3-binding protein and a physiological inhibitor of c-Abl tyrosine kinase activity. Genes Dev. 11, 2456–2467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lin J., Sun T., Ji L., Deng W., Roth J., Minna J., Arlinghaus R. (2007) Oncogenic activation of c-Abl in non-small cell lung cancer cells lacking FUS1 expression: inhibition of c-Abl by the tumor suppressor gene product Fus1. Oncogene 26, 6989–6996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Glover R. T., Angiolieri M., Kelly S., Monaghan D. T., Wang J. Y., Smithgall T. E., Buller A. L. (2000) Interaction of the N-methyl-d-aspartic acid receptor NR2D subunit with the c-Abl tyrosine kinase. J. Biol. Chem. 275, 12725–12729 [DOI] [PubMed] [Google Scholar]
- 48. Engen J. R., Wales T. E., Chen S., Marzluff E. M., Hassell K. M., Weis D. D., Smithgall T. E. (2012) Partial cooperative unfolding in proteins as observed by hydrogen exchange mass spectrometry. Int. Rev. Phys. Chem. DOI 10.1080/0144235X.2012.751175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Marcsisin S. R., Engen J. R. (2010) Hydrogen exchange mass spectrometry: what is it and what can it tell us? Anal. Bioanal. Chem. 397, 967–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Engen J. R. (2009) Analysis of protein conformation and dynamics by hydrogen/deuterium exchange MS. Anal. Chem. 81, 7870–7875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Brier S., Engen J. R. (2008) in Mass Spectrometry Analysis for Protein-Protein Interactions and Dynamics (Chance M., ed) pp. 11–43, John Wiley & Sons, Inc., Hoboken, NJ [Google Scholar]
- 52. Wales T. E., Engen J. R. (2006) Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass Spectrom. Rev. 25, 158–170 [DOI] [PubMed] [Google Scholar]
- 53. Englander S. W., Kallenbach N. R. (1983) Hydrogen exchange and structural dynamics of proteins and nucleic acids. Q. Rev. Biophys. 16, 521–655 [DOI] [PubMed] [Google Scholar]
- 54. Zhang Z., Smith D. L. (1993) Determination of amide hydrogen exchange by mass spectrometry: a new tool for protein structure elucidation. Protein Sci. 2, 522–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hvidt A., Nielsen S. O. (1966) Hydrogen exchange in proteins. Adv. Protein Chem. 21, 287–386 [DOI] [PubMed] [Google Scholar]
- 56. Wales T. E., Engen J. R. (2006) Partial unfolding of diverse SH3 domains on a wide timescale. J. Mol. Biol. 357, 1592–1604 [DOI] [PubMed] [Google Scholar]
- 57. Chen S., Dumitrescu T. P., Smithgall T. E., Engen J. R. (2008) Abl N-terminal cap stabilization of SH3 domain dynamics. Biochemistry 47, 5795–5803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Engen J. R., Wales T. E., Hochrein J. M., Meyn M. A., 3rd, Banu Ozkan S., Bahar I., Smithgall T. E. (2008) Structure and dynamic regulation of Src family kinases. Cell. Mol. Life Sci. 65, 3058–3073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chen S., O'Reilly L. P., Smithgall T. E., Engen J. R. (2008) Tyrosine phosphorylation in the SH3 domain disrupts negative regulatory interactions within the c-Abl kinase core. J. Mol. Biol. 383, 414–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gorre M. E., Mohammed M., Ellwood K., Hsu N., Paquette R., Rao P. N., Sawyers C. L. (2001) Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science 293, 876–880 [DOI] [PubMed] [Google Scholar]
- 61. Shah N. P., Nicoll J. M., Nagar B., Gorre M. E., Paquette R. L., Kuriyan J., Sawyers C. L. (2002) Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell 2, 117–125 [DOI] [PubMed] [Google Scholar]
- 62. Azam M., Seeliger M. A., Gray N. S., Kuriyan J., Daley G. Q. (2008) Activation of tyrosine kinases by mutation of the gatekeeper threonine. Nat. Struct. Mol. Biol. 15, 1109–1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Iacob R. E., Pene-Dumitrescu T., Zhang J., Gray N. S., Smithgall T. E., Engen J. R. (2009) Conformational disturbance in Abl kinase upon mutation and deregulation. Proc. Natl. Acad. Sci. U.S.A. 106, 1386–1391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. McWhirter J. R., Galasso D. L., Wang J. Y. (1993) A coiled-coil oligomerization domain of Bcr is essential for the transforming function of Bcr-Abl oncoproteins. Mol. Cell. Biol. 13, 7587–7595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zhao X., Ghaffari S., Lodish H., Malashkevich V. N., Kim P. S. (2002) Structure of the Bcr-Abl oncoprotein oligomerization domain. Nat. Struct. Biol. 9, 117–120 [DOI] [PubMed] [Google Scholar]
- 66. Smith K. M., Van Etten R. A. (2001) Activation of c-Abl kinase activity and transformation by a chemical inducer of dimerization. J. Biol. Chem. 276, 24372–24379 [DOI] [PubMed] [Google Scholar]
- 67. Guo X. Y., Cuillerot J. M., Wang T., Wu Y., Arlinghaus R., Claxton D., Bachier C., Greenberger J., Colombowala I., Deisseroth A. B. (1998) Peptide containing the BCR oligomerization domain (AA 1–160) reverses the transformed phenotype of p210bcr-abl positive 32D myeloid leukemia cells. Oncogene 17, 825–833 [DOI] [PubMed] [Google Scholar]
- 68. Donato N. J., Wu J. Y., Stapley J., Gallick G., Lin H., Arlinghaus R., Talpaz M. (2003) BCR-ABL independence and LYN kinase overexpression in chronic myelogenous leukemia cells selected for resistance to STI571. Blood 101, 690–698 [DOI] [PubMed] [Google Scholar]
- 69. Panjarian S., Iacob R. E., Chen S., Wales T. E., Engen J. R., Smithgall T. E. (2013) Enhanced SH3/linker interaction overcomes Abl kinase activation by gatekeeper and myristic acid binding pocket mutations and increases sensitivity to small molecule inhibitors. J. Biol. Chem. 288, 6116–6129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Yang S., Blachowicz L., Makowski L., Roux B. (2010) Multidomain assembled states of Hck tyrosine kinase in solution. Proc. Natl. Acad. Sci. U.S.A. 107, 15757–15762 [DOI] [PMC free article] [PubMed] [Google Scholar]