

Background: Dystrophin deficiency leads to life-threatening cardiomyopathy, whereas cardiac p300 promotes cardiac hypertrophy/failure.
Results: The SIRT1 activator resveratrol inhibited p300 protein up-regulation and attenuated cardiomyopathy in dystrophin-deficient mice, and SIRT1-induced p300 down-regulation was mediated via its deacetylation and ubiquitination.
Conclusion: SIRT1 activation ameliorates dystrophic cardiomyopathy by targeting p300.
Significance: Negative regulation of p300 is a novel cardioprotective mechanism of SIRT1.
Keywords: Dystrophin, Heart, p300, Sirt1, Ubiquitination, Cardiomyopathy, Protein Deacetylation
Abstract
Cardiomyopathy is the main cause of death in Duchenne muscular dystrophy. Here, we show that oral administration of resveratrol, which leads to activation of an NAD+-dependent protein deacetylase SIRT1, suppresses cardiac hypertrophy and fibrosis and restores cardiac diastolic function in dystrophin-deficient mdx mice. The pro-hypertrophic co-activator p300 protein but not p300 mRNA was up-regulated in the mdx heart, and resveratrol administration down-regulated the p300 protein level. In cultured cardiomyocytes, cardiomyocyte hypertrophy induced by the α1-agonist phenylephrine was inhibited by the overexpression of SIRT1 as well as resveratrol, both of which down-regulated p300 protein levels but not p300 mRNA levels. In addition, activation of atrial natriuretic peptide promoter by p300 was inhibited by SIRT1. We found that SIRT1 induced p300 down-regulation via the ubiquitin-proteasome pathway by deacetylation of lysine residues for ubiquitination. These findings indicate the pathological significance of p300 up-regulation in the dystrophic heart and indicate that SIRT1 activation has therapeutic potential for dystrophic cardiomyopathy.
Introduction
Cardiomyopathy is frequently found in patients with muscular dystrophies as follows: disorders caused by loss-of-function mutations in genes encoding dystrophin, sarcoglycans, or other dystroglycan complex components. Duchenne muscular dystrophy (DMD),2 the most common and serious type of muscular dystrophy, is caused by a mutation in the dystrophin gene. Because mechanical ventilation prevents death from respiratory failure (1), heart failure is a major cause of mortality in DMD patients. Although treatment with angiotensin-converting enzyme inhibitors and β-blockers provide benefits in patients with cardiac dysfunction associated with DMD (2), it is still progressive under these medications. Therefore, it is important to analyze the mechanism underlying dystrophic cardiomyopathy and to develop new medical approaches.
Protein lysine acetylation/deacetylation is emerging as an important regulatory mechanism of cellular functions. The transcriptional co-activator p300 acetylates histones and transcription (co-)factors and controls physiological processes such as cell proliferation, development, and survival. Although p300 is essential for cardiac development (3), it also plays a key role in cardiac hypertrophy and heart failure by acetylating and activating myocyte enhancer factor-2 (4) and GATA4 (5) transcription factors. The dose of p300 seems to be critical for development of cardiac hypertrophy, because overexpression of p300 induces cardiomyocyte hypertrophy in vitro and in vivo, whereas down-regulating p300 ameliorated cardiomyopathy (4). In addition, cardiac fibrosis impairs myocardial diastolic function in hypertrophied and failing hearts, and p300 promotes tissue fibrosis by binding to and activating Smad3 (6). Therefore, p300 is a key contributor to cardiac hypertrophy and fibrosis, but the regulatory mechanism of p300 is not fully elucidated. It is also unclear whether p300 participates in the cardiac pathology of DMD.
SIRT1 is an NAD+-dependent class III protein/histone deacetylase, which regulates numerous cellular processes such as stress response, cell survival, cell cycle, development, and metabolism by deacetylating target proteins (7). Recently, SIRT1 was found to be an attractive therapeutic target for various diseases (7), and a polyphenol resveratrol was identified as a SIRT1 activator (8), although the mechanism of SIRT1 activation by resveratrol seems to be mediated by other molecules such as AMP-dependent protein kinase (7, 9–11). We have shown that SIRT1 is a nucleocytoplasmic shuttling protein (12, 13). Furthermore, we found that resveratrol increases superoxide dismutase 2, inhibits cardiomyocyte death, and prolongs the life span in TO-2 cardiomyopathic hamsters (14). We also recently reported that resveratrol reduces oxidative stress and ameliorates the skeletal muscle pathology in mdx mice (15). Therefore, SIRT1 activation by resveratrol may be a new potential treatment of cardiomyopathy related to dystrophin deficiency.
In this study, we report that p300 protein but not mRNA is up-regulated in the hearts of dystrophin-deficient mdx mice and that long term resveratrol administration to mdx mice suppresses cardiac p300 up-regulation and improves cardiomyopathy. We also show that the p300 protein level is reciprocally regulated by acetylation and deacetylation. Resveratrol down-regulates the p300 protein level via activation of SIRT1, which deacetylates p300 and promotes ubiquitin-dependent degradation. Our study reveals the significance of p300 regulation in dystrophic cardiomyopathy and also the therapeutic potential of SIRT1 activators in DMD.
EXPERIMENTAL PROCEDURES
mdx Mice
All in vivo experiments were conducted according to The Animal Guideline of Sapporo Medical University and approved by the Animal Use Committee of Sapporo Medical University. Male C57BL/10ScSn-Dmd mdx/J mice (mdx mice) and control C57BL/10 mice were purchased from the Oriental Yeast Co. Ltd. (Tokyo, Japan). Resveratrol (food grade, ChromaDex) was mixed with a powdered diet (4 g/kg meal) and orally administered ad libitum to mice for 32 weeks beginning at 9 weeks of age, after which the mice were sacrificed and the hearts examined.
Echocardiography
Echocardiography was performed under anesthesia with isoflurane, using Vivid-i ultrasound (GE Healthcare) with an 11.5-MHz probe. The left ventricle was assessed in the parasternal long axis view. The interventricular septal thickness, left ventricular posterior wall thickness, left ventricular dimension, and diastolic posterior wall velocity were measured from M-mode tracings of the left ventricles obtained at the mid-papillary muscle level with a sweep speed of 50 mm/s.
Tissue Analysis
Frozen heart tissue was prepared for the evaluation of cardiomyocyte cross-sectional area and immunostaining as described previously (15). To quantify the fluorescence-positive area, images were captured under the same conditions. To measure the myocyte minimal Feret's diameter as muscle fiber cross-sectional size (16), left ventricular sections were stained with FITC-conjugated wheat germ agglutinin (Sigma). Cardiomyocyte cell membrane images were captured digitally, and the minimal Feret's diameter was analyzed using ImageJ software (National Institutes of Health). For immunostaining, tissue sections were fixed with 4% paraformaldehyde, blocked, and incubated with antibodies against fibronectin and acetyl histone H3K9/K14. The percentage of the fibronectin-stained area was analyzed by using ImageJ software from eight independent images of sections of the left ventricle from 4 to 5 mice in each group. Immunoblot and quantitative RT-PCRs were performed as described in the supplemental material.
Constructs and Transfection
The expression constructs for wild-type SIRT1-EGFP and dominant-negative mutant (H355Y) SIRT1-EGFP were described previously (12). FLAG-p300 and p300-HA expression constructs were kindly provided by Dr. Richard Eckner (University of Zurich, Zurich, Switzerland) and Dr. Masaaki Ikeda (Tokyo Medical and Dental University, Tokyo, Japan). To generate the blank control vector of p300-HA (CMVβ(Δp300)), the p300-HA plasmid was digested with NotI and HindIII. The vector overhangs were then blunted by using T4 DNA polymerase (New England Biolabs), and the blunt ends were ligated with the DNA ligation kit Mighty Mix (Takara-Bio). The FLAG-PCAF (p300/CBP-associated factor) expression vector was kindly provided by Dr. Yoshihiro Nakatani (Dana Farber Cancer Institute, Boston). The GATA4-FLAG expression vector was kindly provided by Dr. Mona Nemer (University of Ottawa, Ottawa, Canada). The atrial natriuretic peptide (ANP)-luciferase reporter was constructed by subcloning PCR-amplified inserts corresponding to the promoter sequence of Nppa from mouse genomic DNA (−517 to +30) into the KpnI and XhoI sites of the pGL3 basic luciferase expression vector (Promega), using the following primers: 5′-GGTACCCGATGAATCAGGTGTGAAGCTA-3′ and 5′-CTCGAGCTCTCTCTCTCTCAGCTTTTGTCC-3′. Lipofectamine LTX with Plus Reagent (Invitrogen) was used to transfect plasmids into neonatal rat cardiomyocytes, COS7, and HEK293 cells. A QuikChange XL mutagenesis kit (Stratagene) was used for site-directed mutagenesis. The following primer sets were used for mutagenesis: p300-K1020R/K1024R_sense 5′-GAGAAGCACTGAGTTAAGAACTGAAATAAGAGAGGAGGAAGACC-3′ and K1020R/K1024R_antisense, 5′-GGTCTTCCTCCTCTCTAATTTCAGTTCTTAACTCAGTGCTTCTC-3′.
Luciferase Assay
Plasmid DNA (0.6 μg/well) was transfected with PRL-CMV (1 ng/well) into HEK293 cells on a 24-well dish using Lipofectamine LTX with Plus Reagent (Invitrogen). The amount of DNA transfected to a well was equalized by the addition of the control EGFP-N3 plasmid. Luciferase activity was assessed by normalization of transfection efficiency with Renilla luciferase with the Dual-Luciferase reporter assay system kit (Promega). Results were obtained from at least three independent experiments.
Analysis of p300 Acetylation and Ubiquitination
After FLAG-p300 transfection and experimental treatments, COS7 cells were harvested, and whole-cell extracts were prepared using lysis buffer (CelLytic M (Sigma) with protease inhibitor mixture (Nacalai Tesque)). The cell lysates were incubated with 2 μg of an anti-FLAG antibody or normal IgG at 4 °C overnight. To analyze p300 acetylation status in cardiac tissues, tissues were homogenized and lysed in lysis buffer (CelLytic MT (Sigma)) containing protease inhibitor mixture, trichostatin A (1 μm), and nicotinamide (10 mm). Heart lysates (1500 μg) were incubated with 4 μg of normal mouse IgG or mouse anti-p300 antibody at 4 °C overnight. The immunoprecipitates were bound to Protein A/G PLUS-agarose (Santa Cruz Biotechnology) and then analyzed by anti-acetylated lysine (Cell Signaling) or anti-ubiquitin antibody (Santa Cruz Biotechnology).
Statistics
Data are expressed as means ± S.E. Comparisons between multiple groups were made using one-way analysis of variance followed by a post hoc Student-Neuman-Keuls test. The difference was considered significant if the p was <0.05.
RESULTS
Resveratrol Prevents Cardiomyopathy and Restores Cardiac Function in Dystrophin-deficient mdx Mice
At the age of 41 weeks, both the heart weight and heart weight-to-body weight ratio were higher in the untreated mdx mice than in control C57BL/10 mice (Fig. 1A); these changes were completely suppressed in the resveratrol-treated mdx mice (Fig. 1A). The elevation of ANP mRNA, a marker of cardiac hypertrophy, in the mdx heart was also suppressed by resveratrol treatment (Fig. 1B). The minimal Feret's diameter of the cardiomyocyte in the left ventricle was significantly larger in the untreated mdx mice than in control mice, and resveratrol treatment significantly suppressed this increase (Fig. 1C). Echocardiography at 40 weeks old showed that the interventricular septal thickness at end diastole and left ventricular posterior wall thickness at end diastole in mdx mice was thicker than in control mice (Fig. 1D and supplemental Table S1). The interventricular septal thickness at end diastole in resveratrol-treated mdx mice was comparable with that in controls. Although the systolic function, measured by fractional shortening and ejection fraction, was comparable among the three groups (supplemental Table S1), the diastolic posterior wall velocity, an index of diastolic function, was significantly slower in the untreated mdx hearts than in control hearts (Fig. 1, D and E). The diastolic posterior wall velocity was restored in the resveratrol-treated mdx mice (Fig. 1, D and E).
FIGURE 1.

Long term resveratrol administration inhibits cardiac hypertrophy and fibrosis in mdx mice. A, heart weight (HW) and heart weight-to-body weight ratio (HW/BW) in control C57BL/10 (Control), untreated mdx mice (mdx), and resveratrol-treated mdx mice (RSV mdx). n = 5–6/groups. B, left ventricles were analyzed by quantitative RT-PCR for ANP. n = 5–6/groups. C, left panel, representative cross-sectional heart micrographs stained with FITC-conjugated wheat germ agglutinin. Scale bar, 10 μm. Right panel, average minimal Feret's diameter was determined from 4 to 5 hearts in each group by calculating 400–800 measurements from five sections per heart. D, representative echocardiograms (M-mode view) of mice. IVST, interventricular septal thickness. D and T indicate the distance and the time interval of the left ventricular posterior movement during diastole, respectively. E, average of the diastolic posterior wall velocities calculated by distance/time (mm/s). n = 5–6/group. F, left panel, representative immunofluorescence images of fibronectin (red) in heart sections. Scale bar, 10 μm. Right panel, percentage of fibronectin-stained area in each group was determined from eight images per heart. n = 4–5/groups. G, myocardial samples were subjected to quantitative RT-PCR analysis to quantify mRNA of Col1a1, Col1a2, Col3a1, and α-smooth muscle actin (αSMA). n = 5–6 in each group. H, representative immunofluorescence images of acetyl histone H3 at lysines 9 and 14 (AcH3K9/K14, red) and of Hoechst33342 staining (blue) in the heart. Scale bar, 10 μm. I, left panel, immunoblot analysis for p300 and SIRT1 in hearts. Right panel, quantification of myocardial p300 protein level. n = 4–5/group. J, myocardial levels of p300 mRNA determined by quantitative RT-PCR. n = 5–6/group. *, p < 0.05; NS, not significant.
Immunostaining showed the increased percentage of the fibronectin-stained area of the left ventricle, especially at the interstitial space, in the control mdx mice, and resveratrol treatment significantly suppressed this increase (Fig. 1F). The cardiac mRNA levels of the collagen genes Col1a1, Col1a2, and Col3a1, which mark fibrosis, were significantly up-regulated in untreated mdx mice compared with control mice, whereas cardiac collagen gene expression did not differ significantly between control C57BL/10 and resveratrol-treated mdx mice (Fig. 1G). In addition, the increase in mRNA of a myofibroblast marker α-smooth muscle actin in the mdx heart was significantly suppressed by resveratrol treatment (Fig. 1G).
Histone H3 acetylation at Lys-9 and Lys-14 (H3K9/K14) was elevated in the mdx hearts, suggesting that protein acetylation might be increased in mdx mice (Fig. 1H). Resveratrol administration decreased the acetyl-H3K9/K14 level in the mdx heart (Fig. 1H), indicating that resveratrol increased histone deacetylase activity and/or inhibited histone acetyltransferase activity. We next examined the histone acetyltransferase p300. Immunoblotting showed that the myocardial p300 protein level was up-regulated 2.8-fold in mdx mice compared with control mice, and this increase was completely suppressed by resveratrol treatment (Fig. 1I). In contrast, the myocardial p300 mRNA levels were comparable in the mdx and control mice (Fig. 1J). The SIRT1 protein and mRNA levels in the heart were unchanged in the mdx mice (Fig. 1I and supplemental Fig. S1A). Down-regulation of sarco(endo)plasmic reticulum calcium ATPase 2a (Serca2a) is a hallmark of failing hearts and contributes to abnormal cardiac function (17). We found that Serca2a mRNA level was significantly lower in the untreated mdx heart than the control heart, which was ameliorated by resveratrol administration (supplemental Fig. S1B). Resveratrol did not suppress the up-regulation of the NADPH oxidase subunits Nox2 and Nox4, mediators of cardiac hypertrophy and fibrosis (supplemental Fig. S1C). Phosphorylation of extracellular signal-regulated kinases (ERK) 1/2, other mediators of cardiac hypertrophy, was not suppressed by resveratrol in the mdx heart (supplemental Fig. S2, A and B). Although AMP-dependent protein kinase is reported to be a target of resveratrol (18), phosphorylation of AMP-dependent protein kinase α and its downstream target acetyl-CoA carboxylase was not modulated by resveratrol (supplemental Fig. S2, C and D).
Resveratrol Suppresses the Up-regulation of p300 Protein
To confirm the significance of p300 protein up-regulation in cardiac hypertrophy, we employed cultured neonatal rat cardiomyocytes. As reported previously (5), an α1-agonist phenylephrine significantly increased the p300 protein level (Fig. 2A) and cell surface area (Fig. 2B) in isolated cardiomyocytes. Phenylephrine also increased the level of α-tubulin, another marker of cardiac hypertrophy (Fig. 2, A and D). Knockdown of p300 by siRNA canceled the phenylephrine-induced increases in both α-tubulin level and cardiomyocyte surface area (Fig. 2, A and B). Furthermore, the ANP promoter activity was increased according to the p300 level, indicating that the p300 level regulates ANP expression (Fig. 2C). The vector control (CMVβ (Δp300)) did not affect ANP promoter activity (supplemental Fig. S3A). Similar to p300-siRNA, resveratrol significantly inhibited up-regulation of p300, α-tubulin, and ANP expression and also cellular hypertrophy induced by phenylephrine (Fig. 2, D, F, and G). Interestingly, phenylephrine did not change the p300 mRNA level (Fig. 2E). Chromatin immunoprecipitation experiments showed that phenylephrine increased p300 binding to the ANP promoter region at −197 to +14 (Fig. 2H), which contains the binding site of GATA4 (19), one of the partners of p300. Phenylephrine increased global acetyl-H3K9/K14 level (supplemental Fig. S3B) and also acetyl-H3K9 level at the ANP promoter (supplemental Fig. S3C). Resveratrol inhibited the increase of global acetyl-H3K9/K14 levels by phenylephrine (supplemental Fig. S3B). The levels of p300 and acetyl H3K9 at the ANP promoter region were also reduced by resveratrol treatment (Fig. 2H and supplemental Fig. S3C). These results indicate that phenylephrine up-regulated the p300 protein level, which induced the cardiomyocyte hypertrophic response, and suggest that resveratrol inhibited cardiomyocyte hypertrophy via inhibition of p300 protein up-regulation.
FIGURE 2.
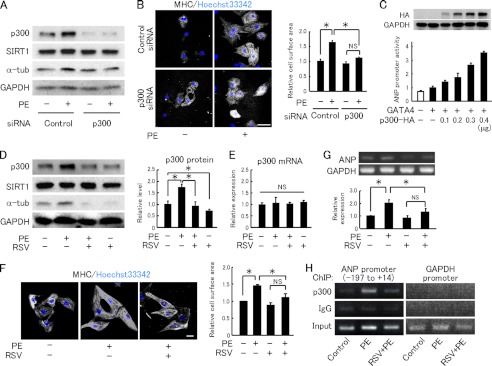
Resveratrol inhibits p300 protein up-regulation and hypertrophic responses in cardiomyocytes. A, immunoblot analysis for p300, SIRT1, and α-tubulin (α-tub) in neonatal rat cardiomyocytes transfected with control or p300-targeting siRNA followed by treatment with saline or 50 μm phenylephrine (PE). B, left panels, representative fields of neonatal rat cardiomyocytes stained with myosin heavy chain (MHC) and Hoechst 33342. Cardiomyocytes transfected with control or p300-targeting siRNA were treated with saline or PE for 48 h. Scale bar, 50 μm. Right panels, quantification of cell surface area. C, HEK293 cells were co-transfected with 0.2 μg of atrial natriuretic peptide (ANP) promoter-luc, 1 ng of CMV-pRL, and p300-HA at the indicated doses in the absence (white bar) or presence (black bars) of 0.5 ng of GATA4-FLAG. Expression of p300-HA and GAPDH is shown in upper panels. Relative promoter activities were calculated from the ratio of firefly to sea pansy luciferase activity. The data were averaged from three independent experiments. D, left panels, immunoblot analysis for p300, SIRT1, and α-tubulin (α-tub) in neonatal rat cardiomyocytes treated with vehicle or PE, with or without RSV (30 μm) pretreatment. Right panels, quantification of p300 protein levels normalized to GAPDH. E, quantification of p300 mRNA levels in neonatal rat cardiomyocytes treated with vehicle or PE, with or without RSV. n = 4/group. F, left panel, representative fields of neonatal rat cardiomyocytes stained with MHC and Hoechst 33342. Cardiomyocytes were stimulated with vehicle or PE in the absence or presence of RSV. Scale bar, 10 μm. Right panel, cell surface area quantification. G, ANP mRNA expression in neonatal rat cardiomyocytes. n = 3. H, chromatin immunoprecipitation (ChIP) assays were performed in neonatal rat cardiomyocytes treated with vehicle (Control), PE, or PE in the presence of resveratrol (RSV+PE) with antibodies against p300 and rabbit IgG. Input represents 1% of the total chromatin. ChIP samples were amplified using primers flanking the promoter region (−197 to +14) of the ANP gene and the GAPDH gene promoter region. The experiment was repeated three times and obtained similar results. *, p < 0.05; NS, not significant.
Resveratrol Down-regulates p300 through SIRT1
We next examined how resveratrol down-regulates the p300 protein level. Resveratrol decreased the p300 protein level in neonatal rat cardiomyocytes and C2C12 myoblast cells (Fig. 3, A and B); however, resveratrol did not reduce the p300 mRNA level (Fig. 3C). We next examined whether SIRT1 is involved in resveratrol's function. SIRT1 was knocked down by siRNA in C2C12 (Fig. 3D), and SIRT1-siRNA abolished resveratrol's down-regulation of p300 in C2C12 cells, showing that SIRT1 is involved in this process (Fig. 3D). SIRT1 knockdown (supplemental Fig. S4) also canceled the anti-hypertrophic effect of resveratrol in cardiomyocytes (Fig. 3E). However, in COS7 cells, inhibiting SIRT1 activity by nicotinamide increased the level of FLAG-tagged p300 protein (Fig. 3F). To examine whether SIRT1 suppresses the cardiac p300 level in vivo, we crossed SIRT1 conditional mutant mice (SIRT1flox/flox) with transgenic mice carrying a cre transgene with a mutated estrogen receptor ligand-binding domain under the α-myosin heavy chain promoter. Western blots of heart homogenates from cardiomyocyte-specific SIRT1 conditional knock-out mice (SIRT1HKO) showed a band of mutant SIRT1s with a lower molecular weight than that of wild-type SIRT1 (Fig. 3G). Cardiac p300 protein level was elevated in the SIRT1HKO compared with the SIRT1fl/fl (Fig. 3G). These data indicate that SIRT1 activation by resveratrol decreases p300 protein via its deacetylation activity.
FIGURE 3.
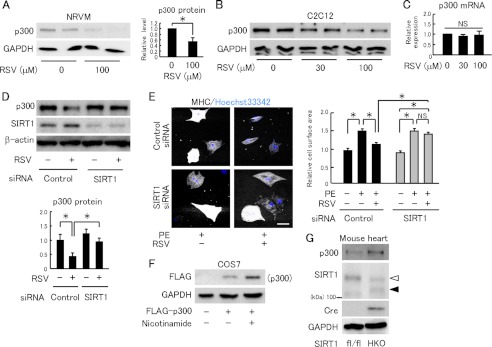
Resveratrol reduces p300 protein levels via SIRT1. A, left panel, immunoblot for p300 in neonatal rat ventricular myocytes treated with RSV for 12 h. Right panel, quantification from four independent experiments. B, immunoblot for p300 in C2C12 cells treated with RSV. C, quantification of p300 mRNA in C2C12 treated with RSV (n = 4). D, upper panel, representative immunoblots for p300 and SIRT1 in C2C12 transfected with control or SIRT1 siRNA, with or without 100 μm RSV treatment. Lower panel, quantification of p300 protein normalized by β-actin (n = 4). E, left panel, representative fields of neonatal rat cardiomyocytes stained with myosin heavy chain (MHC) and Hoechst 33342. Cardiomyocytes were transfected with control or SIRT1 siRNA and treated with PE (50 μm) for 48 h in the absence or presence of RSV (30 μm). Scale bar, 50 μm. Right panel, cell surface area quantification. F, immunoblot analysis in COS7 cells transfected with FLAG-p300 or a control vector, followed by treatment with vehicle or 10 mm nicotinamide for 12 h. G, representative immunoblots of heart lysates from control SIRT1fl/fl and SIRT1fl/fl mice with expressing tamoxifen-inducible cre recombinase administered tamoxifen (SIRT1HKO). Open and closed arrowheads denote wild type and a truncated mutant SIRT1, respectively. *, p < 0.05; NS, not significant.
We further examined whether SIRT1 down-regulates p300 and suppresses p300-induced ANP promoter activation. In COS7 cells, the level of p300-FLAG protein was reduced by co-expressing wild-type SIRT1 (SIRT1-WT) but not a deacetylase-inactive mutant SIRT1 (SIRT1-HY) (Fig. 4A). The increased ANP promoter activity by p300 overexpression was suppressed by co-expressing SIRT1-WT (Fig. 4B). SIRT1-HY failed to suppress ANP's transactivation by p300, indicating that SIRT1 inhibited the ANP promoter activity via its deacetylation activity (Fig. 4C). Alone, neither SIRT1-WT nor SIRT1-HY affected the ANP promoter activity (Fig. 4, B and C). Phenylephrine-induced cardiomyocyte hypertrophy was also completely inhibited by the overexpression of SIRT1-WT but not by SIRT1-HY (Fig. 4, D and E).
FIGURE 4.
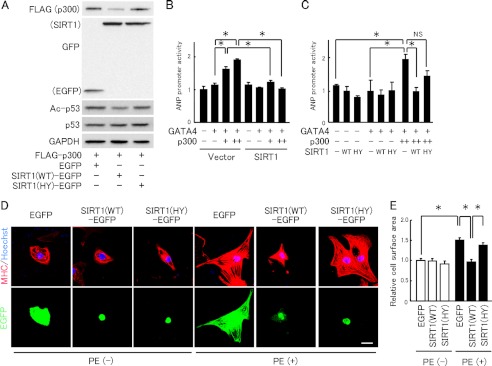
Overexpression of SIRT1 down-regulates p300 protein and inhibits cardiomyocyte hypertrophy and ANP transcription. A, immunoblot analysis in COS7 cells co-transfected with FLAG-p300 and wild-type SIRT1-EGFP (SIRT1(WT)-EGFP), the deacetylase-inactive H355Y mutant SIRT1-EGFP (SIRT1(HY)-EGFP), or a control vector (EGFP). B, HEK293 cells were co-transfected with ANP promoter-luc and CMV-pRL, in the absence (−) or presence (+) of GATA4-FLAG, 0.2 (+) or 0.4 μg (++) of p300-HA, and 0.1 μg of wild-type SIRT1. C, ANP promoter activity in HEK293 cells transfected as in B, except for wild type (WT) or H355Y SIRT1 (HY). D, representative fields of neonatal rat cardiomyocytes stained with MHC (red) and Hoechst 33342 (blue). Cardiomyocytes were transfected with EGFP-N3 (EGFP), SIRT1(WT)-EGFP, or SIRT1(HY)-EGFP, and then treated with vehicle or PE for 48 h. Scale bar, 20 μm. E, cell surface area quantification from experiments shown in the left panels, measured from 100 random EGFP-expressing cells in three independent experiments. *, p < 0.05; NS, not significant.
Down-regulation of p300 Is Associated with Its Deacetylation by SIRT1
Lys-1020 and Lys-1024 on p300 are reported to be targets for both sumoylation and deacetylation induced by SIRT1 activation (20). We therefore examined whether deacetylation of p300 by SIRT1 is associated with its down-regulation. Immunoprecipitation experiments showed that the acetylated p300 level was increased by nicotinamide (Fig. 5A) or the specific SIRT1 inhibitor EX527 (Fig. 5B). Trichostatin A, which inhibits class I and II histone deacetylases, also increased the acetylated p300 level but less potently than nicotinamide (Fig. 5A). SIRT1-WT but not SIRT1-HY greatly decreased the levels of acetylated p300 and total p300 (Fig. 5C). We also tested whether p300 acetylation might conversely up-regulate the p300 protein. Because p300 is acetylated by p300 itself (21), we co-expressed FLAG-tagged p300 and HA-tagged p300. HA-tagged p300 co-expression increased the expression level of FLAG-tagged p300 and vice versa (Fig. 5D). The p300 protein level was also markedly increased when co-expressed with PCAF that acetylates p300 and augments its activity (Fig. 5, E and F) (22). This increase was cancelled by co-expressing SIRT1 (Fig. 5F). However, anacardic acid, which inhibits protein acetylase activity of p300 and PCAF, reduced the p300 protein level in a dose-dependent manner (Fig. 5G). Anacardic acid also suppressed the up-regulation of p300 protein induced by phenylephrine in neonatal rat cardiomyocytes (Fig. 5G). To investigate whether p300 acetylation is associated with up-regulation of p300 protein in vivo, we analyzed hearts from cardiomyocyte-specific SIRT1 knock-out mice. Acetylated p300 was increased in the heart of SIRT1 knock-out mice, where p300 protein level was up-regulated (Fig. 5H). These data suggest that p300 acetylation status regulates the p300 protein level.
FIGURE 5.

Acetylation status of p300 regulates its protein level. A, analysis of p300 acetylation. COS7 cells were transfected with FLAG-p300, cultured for 36 h, and treated with vehicle, 10 mm nicotinamide, or 50 nm trichostatin A for 12 h. Cell lysates were immunoprecipitated with a mouse anti-FLAG antibody or mouse IgG. Acetylation of p300 was analyzed using an anti-acetyl-lysine (AcK) antibody. IP, immunoprecipitation. B, analysis of endogenous p300 acetylation in C2C12 cells. Lysates from cells treated with 3 μm Ex527 or vehicle for 12 h were immunoprecipitated with a rabbit anti-p300 or rabbit IgG. The acetylation status of p300 was analyzed using an anti-acetyl-lysine antibody. C, acetylation of p300 was analyzed in COS7 cells co-transfected with p300-FLAG and wild-type SIRT1-EGFP (WT), mutant H355Y SIRT1-EGFP (HY), or the control vector (EGFP). FLAG-p300 acetylation was analyzed as in A. Input samples were subjected to immunoblotting to confirm the SIRT1-EGFP expression. D, representative immunoblots to detect FLAG-tagged and HA-tagged p300 protein in COS7 cells transfected with either FLAG-p300 or p300-HA, or co-transfected with both constructs. E, immunoblots for HA and FLAG in COS7 cells transfected with p300-HA, FLAG-PCAF, or both. F, COS7 cells were transfected with p300-HA and/or FLAG-PCAF in the absence or presence of SIRT1-EGFP. The cell lysates were subjected to immunoblot analysis to determine the p300-HA level. To assess the p300 HAT activity, p53 acetylation was monitored. G, left panel, representative immunoblots for p300 and GAPDH in C2C12 cells treated with the indicated doses of anacardic acid (AnaA) for 12 h. Right panel, immunoblots for p300 and GAPDH in neonatal rat ventricular myocytes. Neonatal rat ventricular myocytes (NRVM) were treated with the indicated doses of anacardic acid for 1 h followed by incubation with 50 μm PE for 24 h. H, left panel, analysis of p300 acetylation in the heart from control SIRT1fl/fl and cardiomyocyte-specific SIRT1 knock-out SIRT1HKO mice. Right panel, immunoblots for SIRT1 and cre recombinase.
SIRT1 Down-regulates p300 through the Ubiquitin-Proteasome Pathway
Although p300 ubiquitination has been reported (23, 24), the relationship between p300 ubiquitination and acetylation has not been examined. We hypothesized that deacetylation of p300 by SIRT1 might induce degradation of p300 via the ubiquitin-proteasome pathway. A proteasome inhibitor MG132 blocked down-regulation of p300 by SIRT1 overexpression (Fig. 6A) and by resveratrol (Fig. 6B). MG132 did not affect deacetylation of p300 by SIRT1 (Fig. 6C). In the presence of MG132, SIRT1 overexpression promoted ubiquitination of p300 as well as p300 deacetylation (Fig. 6D). These findings suggest that SIRT1 deacetylates p300 and promotes ubiquitination and degradation of p300.
FIGURE 6.

Deacetylation of p300 by SIRT1 promotes the ubiquitin-proteasome-dependent p300 degradation. A, immunoblot analysis for FLAG-p300 in COS7 cells co-transfected with FLAG-p300 and a control vector or wild-type SIRT1 (SIRT1(WT)-EGFP) in the absence or presence of 10 μm MG132. B, immunoblot for p300 and SIRT1 in C2C12 cells treated with 10 μm MG132 or vehicle in the absence or presence of 100 μm resveratrol. C, acetylation of p300 in cells co-transfected with FLAG-p300 and SIRT1(WT)-EGFP, SIRT1(HY)-EGFP, or a control vector (EGFP). Cells were treated with MG132 for 12 h beginning 36 h after transfection. AcK, anti-acetyl-lysine; IP, immunoprecipitation. D, analysis of p300 ubiquitination (Ub). COS7 cells co-transfected with FLAG-p300 and wild-type SIRT1 (SIRT1-EGFP) or EGFP-N3 were cultured for 48 h. Cells were incubated with MG132 for the last 12 h of culture. Cell lysates were immunoprecipitated with the anti-FLAG antibody, and p300 ubiquitination and acetylation were analyzed using an anti-ubiquitin (Ub) and anti-acetyl-lysine (AcK) antibodies, respectively. E, diagram of the human p300 protein. The domain reported to contain potential lysine residues for ubiquitination contains amino acids 964–1194. Lysines (K) 1020 and 1024 of p300 are reported to be SIRT1 deacetylation targets. KIX, the CREB-interacting domain; Bromo, bromodomain; HAT, histone acetyltransferase. F, immunoblot analysis of FLAG-p300 in COS7 cells co-transfected with wild-type (WT), FLAG-p300, or K1020R/K1024R (KR) mutant FLAG-p300 and the control vector (EGFP-N3) or wild-type SIRT1-EGFP. G, analysis of mutant p300 ubiquitination. As in D, p300 ubiquitination was analyzed in cells co-transfected with SIRT1-EGFP, HA-ubiquitin, and wild-type or KR mutant FLAG-p300.
Deacetylation of p300 at Lys-1020 and Lys-1024 by SIRT1 facilitates p300 sumoylation in these lysine residues (20). These are in a potential ubiquitination region (amino acid residues 964–1194) (Fig. 6E) (23). Therefore, we tested whether p300 Lys-1020 and Lys-1024 are ubiquitination targets. Overexpressing SIRT1 did not reduce the level of a mutant FLAG-p300 in which the two lysine residues were replaced with arginine (p300-KR) (Fig. 6F). In the presence of MG132, the ubiquitination level was reduced in p300-KR compared with wild-type p300 (Fig. 6G). Immunoprecipitation showed that SIRT1 bound equally to the wild-type and mutant p300 (Fig. 6G), indicating that the lower ubiquitination level of the mutant p300 was not because of a lower affinity for SIRT1.
DISCUSSION
Our results provide the first evidence of the effectiveness of long term resveratrol treatment for cardiomyopathy in dystrophin-deficient mice. We found elevated p300 protein levels in mdx heart tissues. High p300 levels are found in the failing heart in humans and mice (4), and combination studies using cardiac p300 transgenic mice and p300 heterozygous knock-out mice showed that mortality and cardiac mass are directly regulated by myocardial p300 protein levels (4). Because a 1.5–3.7-fold overexpression of p300 causes prominent cardiac hypertrophy in p300-transgenic mice (4), the 2.8-fold increase in p300 protein level in the mdx heart is sufficient to induce cardiac hypertrophy. Resveratrol inhibited cardiac p300's up-regulation and hypertrophy in mdx mice (Fig. 1, A, C, and I), further indicating that down-regulation of p300 is responsible for the anti-hypertrophic function of resveratrol. This negative regulation of p300 is a novel mechanism for cardioprotection by resveratrol.
Resveratrol suppressed cardiac fibrosis (Fig. 1, F and G) in mdx mice. Previously, we showed that resveratrol represses skeletal muscle fibrosis in mdx mice (15) and cardiac fibrosis in cardiomyopathic TO2 hamsters (14). Down-regulation of α-smooth muscle actin expression in mdx hearts treated with resveratrol (Fig. 1G) suggests that inhibiting myofibroblast differentiation from fibroblasts contributes to the anti-fibrotic action of resveratrol. Smad transcription factors, key mediators of myofibroblast transformation and tissue fibrosis, form an active transcription complex with p300 (6). Thus, the findings suggest that p300 down-regulation by resveratrol also underlies inhibition of myofibroblast formation and fibrosis in the mdx heart. Resveratrol may act in fibroblasts in addition to cardiomyocytes.
Resveratrol preserved cardiac diastolic function in mdx mice (Fig. 1E). This seems to be attributed to inhibition of cardiac hypertrophy and fibrosis. In addition, to an extent, the maintenance of the cardiac Serca2a level (supplemental Fig. S1A) may also contribute to the improvement in cardiac function in resveratrol-treated mdx mice, because restoring cardiac Serca2a expression has a therapeutic impact in heart failure models (17), including the mdx heart (25). Overall, these findings indicate that resveratrol provides benefits that suppress or slow the development of heart failure in the dystrophin-deficient heart.
This is the first study demonstrating the relationship between p300 deacetylation and ubiquitination. In some proteins, acetylation of lysine residues is known to interfere with ubiquitin-dependent proteasomal degradation, whereas deacetylation of the protein by SIRT1 promotes its ubiquitination and degradation (26, 27). Our data strongly indicate that deacetylated lysine residues of p300 become the targets of ubiquitination, which leads to proteasomal degradation of p300. Furthermore, although SIRT1 and p300 reciprocally regulate cellular function by protein deacetylation and acetylation, respectively (26–28), this negative regulation of p300 by SIRT1 might enhance protein deacetylation induced by SIRT1.
It remains unclear how p300 protein up-regulates in the mdx heart. Our study strongly suggests that p300 acetylation is involved in the p300 up-regulation, although the p300 acetylation level in mdx hearts is unknown. Because cardiac SIRT1 expression level was not changed in mdx mice (Fig. 1I and supplemental Fig. S1A), the deacetylation rate of p300 by SIRT1 in mdx hearts might be comparable with that of control hearts. A previous study showed that pressure overload in the heart by transverse aortic constriction induced persistent p300 protein up-regulation in the heart, whereas the p300 mRNA was just transiently increased after transverse aortic constriction (4). Because autoacetylation activates p300 (21) and our data showed a mutual relationship between p300 acetylation and the p300 protein level, the transient increase in p300 activity or mRNA, induced by ERK1/2 (29) or other hypertrophic signals, may promote p300 autoacetylation, leading to p300 protein stabilization in the mdx hearts. Further study is necessary to investigate the mechanism of p300 up-regulation of mdx hearts.
These encouraging results show that resveratrol has the potential for translation into clinical situations. Evidence for the safety, pharmacokinetics, and clinical feasibility of resveratrol on obesity and metabolic disorders (30) is growing. Additionally, resveratrol has dual benefits in both cardiac and skeletal muscle (15) in mdx mice. At present, it is unknown whether resveratrol slows or reverses the progression of cardiac dysfunction administered even after the onset of cardiomyopathy in the dystrophin-deficient heart. Additional studies are needed to further establish the effectiveness of SIRT1 activation in dystrophinopathy.
This work was supported in part by Ministry of Education, Culture, Sports, Science, and Technology of Japan Grant-in-aid 22590245 (to Y. H.), the National Project of Knowledge Cluster Initiative of Second Stage, Sapporo Biocluster Bio-S (to Y. H.), the Regional R&D Proposal-based Program from the Northern Advancement Center for Science and Technology of Hokkaido (to Y. H.), the Tubasa Foundation (to Y. H.), the Hokkaido Heart Association grant for research (to A. K.), and research grants from the Fukuda Foundation for Medical Technology and the Akiyama Life Science Foundation (to A. K.).

This article contains supplemental Figs. S1–S4, Table S1, Materials and Methods, and additional references.
- DMD
- Duchenne muscular dystrophy
- PCAF
- p300/CBP-associated factor
- ANP
- atrial natriuretic peptide
- H3K9/K14
- histone H3 lysine 9 and lysine 14
- Serca2a
- sarco(endo)plasmic reticulum calcium ATPase 2a
- RSV
- resveratrol
- PE
- phenylephrine.
REFERENCES
- 1. Eagle M., Baudouin S. V., Chandler C., Giddings D. R., Bullock R., Bushby K. (2002) Survival in Duchenne muscular dystrophy. Improvements in life expectancy since 1967 and the impact of home nocturnal ventilation. Neuromuscul. Disord. 12, 926–929 [DOI] [PubMed] [Google Scholar]
- 2. McNally E. M. (2007) New approaches in the therapy of cardiomyopathy in muscular dystrophy. Annu. Rev. Med. 58, 75–88 [DOI] [PubMed] [Google Scholar]
- 3. Backs J., Olson E. N. (2006) Control of cardiac growth by histone acetylation/deacetylation. Circ. Res. 98, 15–24 [DOI] [PubMed] [Google Scholar]
- 4. Wei J. Q., Shehadeh L. A., Mitrani J. M., Pessanha M., Slepak T. I., Webster K. A., Bishopric N. H. (2008) Quantitative control of adaptive cardiac hypertrophy by acetyltransferase p300. Circulation 118, 934–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yanazume T., Hasegawa K., Morimoto T., Kawamura T., Wada H., Matsumori A., Kawase Y., Hirai M., Kita T. (2003) Cardiac p300 is involved in myocyte growth with decompensated heart failure. Mol. Cell. Biol. 23, 3593–3606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ghosh A. K., Varga J. (2007) The transcriptional coactivator and acetyltransferase p300 in fibroblast biology and fibrosis. J. Cell. Physiol. 213, 663–671 [DOI] [PubMed] [Google Scholar]
- 7. Horio Y., Hayashi T., Kuno A., Kunimoto R. (2011) Cellular and molecular effects of sirtuins in health and disease. Clin. Sci. 121, 191–203 [DOI] [PubMed] [Google Scholar]
- 8. Howitz K. T., Bitterman K. J., Cohen H. Y., Lamming D. W., Lavu S., Wood J. G., Zipkin R. E., Chung P., Kisielewski A., Zhang L. L., Scherer B., Sinclair D. A. (2003) Small molecule activators of sirtuins extend Saccharomyces cerevisiae life span. Nature 425, 191–196 [DOI] [PubMed] [Google Scholar]
- 9. Cantó C., Jiang L. Q., Deshmukh A. S., Mataki C., Coste A., Lagouge M., Zierath J. R., Auwerx J. (2010) Interdependence of AMPK and SIRT1 for metabolic adaptation to fasting and exercise in skeletal muscle. Cell Metab. 11, 213–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Park S. J., Ahmad F., Philp A., Baar K., Williams T., Luo H., Ke H., Rehmann H., Taussig R., Brown A. L., Kim M. K., Beaven M. A., Burgin A. B., Manganiello V., Chung J. H. (2012) Resveratrol ameliorates aging-related metabolic phenotypes by inhibiting cAMP phosphodiesterases. Cell 148, 421–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hosoda R., Kuno A., Hori Y. S., Ohtani K., Wakamiya N., Oohiro A., Hamada H., Horio Y. (2013) Differential cell-protective function of two resveratrol (trans-3,5,4′-trihydroxystilbene) glucosides against oxidative stress. J. Pharmacol. Exp. Ther. 344, 124–132 [DOI] [PubMed] [Google Scholar]
- 12. Tanno M., Sakamoto J., Miura T., Shimamoto K., Horio Y. (2007) Nucleocytoplasmic shuttling of the NAD+-dependent histone deacetylase SIRT1. J. Biol. Chem. 282, 6823–6832 [DOI] [PubMed] [Google Scholar]
- 13. Hisahara S., Chiba S., Matsumoto H., Tanno M., Yagi H., Shimohama S., Sato M., Horio Y. (2008) Histone deacetylase SIRT1 modulates neuronal differentiation by its nuclear translocation. Proc. Natl. Acad. Sci. U.S.A. 105, 15599–15604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tanno M., Kuno A., Yano T., Miura T., Hisahara S., Ishikawa S., Shimamoto K., Horio Y. (2010) Induction of manganese superoxide dismutase by nuclear translocation and activation of SIRT1 promotes cell survival in chronic heart failure. J. Biol. Chem. 285, 8375–8382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hori Y. S., Kuno A., Hosoda R., Tanno M., Miura T., Shimamoto K., Horio Y. (2011) Resveratrol ameliorates muscular pathology in the dystrophic mdx mouse, a model for Duchenne muscular dystrophy. J. Pharmacol. Exp. Ther. 338, 784–794 [DOI] [PubMed] [Google Scholar]
- 16. Bish L. T., Morine K. J., Sleeper M. M., Sweeney H. L. (2010) Myostatin is up-regulated following stress in an Erk-dependent manner and negatively regulates cardiomyocyte growth in culture and in a mouse model. PLoS ONE 5, e10230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shah A. M., Mann D. L. (2011) In search of new therapeutic targets and strategies for heart failure. Recent advances in basic science. Lancet 378, 704–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dasgupta B., Milbrandt J. (2007) Resveratrol stimulates AMP kinase activity in neurons. Proc. Natl. Acad. Sci. U.S.A. 104, 7217–7222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Morin S., Charron F., Robitaille L., Nemer M. (2000) GATA-dependent recruitment of MEF2 proteins to target promoters. EMBO J. 19, 2046–2055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bouras T., Fu M., Sauve A. A., Wang F., Quong A. A., Perkins N. D., Hay R. T., Gu W., Pestell R. G. (2005) SIRT1 deacetylation and repression of p300 involves lysine residues 1020/1024 within the cell cycle regulatory domain 1. J. Biol. Chem. 280, 10264–10276 [DOI] [PubMed] [Google Scholar]
- 21. Thompson P. R., Wang D., Wang L., Fulco M., Pediconi N., Zhang D., An W., Ge Q., Roeder R. G., Wong J., Levrero M., Sartorelli V., Cotter R. J., Cole P. A. (2004) Regulation of the p300 HAT domain via a novel activation loop. Nat. Struct. Mol. Biol. 11, 308–315 [DOI] [PubMed] [Google Scholar]
- 22. Karanam B., Jiang L., Wang L., Kelleher N. L., Cole P. A. (2006) Kinetic and mass spectrometric analysis of p300 histone acetyltransferase domain autoacetylation. J. Biol. Chem. 281, 40292–40301 [DOI] [PubMed] [Google Scholar]
- 23. Chen J., Halappanavar S., Th' ng J. P., Li Q. (2007) Ubiquitin-dependent distribution of the transcriptional coactivator p300 in cytoplasmic inclusion bodies. Epigenetics 2, 92–99 [DOI] [PubMed] [Google Scholar]
- 24. Poizat C., Sartorelli V., Chung G., Kloner R. A., Kedes L. (2000) Proteasome-mediated degradation of the coactivator p300 impairs cardiac transcription. Mol. Cell. Biol. 20, 8643–8654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shin J. H., Bostick B., Yue Y., Hajjar R., Duan D. (2011) SERCA2a gene transfer improves electrocardiographic performance in aged mdx mice. J. Transl. Med. 9, 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Min S. W., Cho S. H., Zhou Y., Schroeder S., Haroutunian V., Seeley W. W., Huang E. J., Shen Y., Masliah E., Mukherjee C., Meyers D., Cole P. A., Ott M., Gan L. (2010) Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron 67, 953–966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Guarani V., Deflorian G., Franco C. A., Krüger M., Phng L. K., Bentley K., Toussaint L., Dequiedt F., Mostoslavsky R., Schmidt M. H., Zimmermann B., Brandes R. P., Mione M., Westphal C. H., Braun T., Zeiher A. M., Gerhardt H., Dimmeler S., Potente M. (2011) Acetylation-dependent regulation of endothelial Notch signalling by the SIRT1 deacetylase. Nature 473, 234–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kemper J. K., Xiao Z., Ponugoti B., Miao J., Fang S., Kanamaluru D., Tsang S., Wu S. Y., Chiang C. M., Veenstra T. D. (2009) FXR acetylation is normally dynamically regulated by p300 and SIRT1 but constitutively elevated in metabolic disease states. Cell Metab. 10, 392–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gusterson R., Brar B., Faulkes D., Giordano A., Chrivia J., Latchman D. (2002) The transcriptional co-activators CBP and p300 are activated via phenylephrine through the p42/p44 MAPK cascade. J. Biol. Chem. 277, 2517–2524 [DOI] [PubMed] [Google Scholar]
- 30. Patel K. R., Scott E., Brown V. A., Gescher A. J., Steward W. P., Brown K. (2011) Clinical trials of resveratrol. Ann. N.Y. Acad. Sci. 1215, 161–169 [DOI] [PubMed] [Google Scholar]