

Background: Amino acid substitutions near the catalytic site of DNA polymerases can affect base discrimination.
Results: An A699Q substitution in the fingers domain of eukaryotic DNA polymerase δ (Pol δ) yields reduced fidelity via base misincorporation.
Conclusion: Intramolecular interactions involving the Pol δ fingers and N-terminal domains can affect base selectivity.
Significance: The structural determinants of fidelity intrinsic to replicative polymerases provide insight into general polymerase function.
Keywords: DNA Polymerase, DNA Synthesis, Enzyme Kinetics, Mutagenesis, Yeast Genetics
Abstract
DNA polymerase δ (Pol δ) is one of the major replicative DNA polymerases in eukaryotic cells, catalyzing lagging strand synthesis as well as playing a role in many DNA repair pathways. The catalytic site for polymerization consists of a palm domain and mobile fingers domain that opens and closes each catalytic cycle. We explored the effect of amino acid substitutions in a region of the highly conserved sequence motif B in the fingers domain on replication fidelity. A novel substitution, A699Q, results in a marked increase in mutation rate at the yeast CAN1 locus, and is synthetic lethal with both proofreading deficiency and mismatch repair deficiency. Modeling the A699Q mutation onto the crystal structure of Saccharomyces cerevisiae Pol δ template reveals four potential contacts for A699Q but not for A699. We substituted alanine for each of these residues and determined that an interaction with multiple residues of the N-terminal domain is responsible for the mutator phenotype. The corresponding mutation in purified human Pol δ results in a similar 30-fold increase in mutation frequency when copying gapped DNA templates. Sequence analysis indicates that the most characteristic mutation is a guanine-to-adenine (G to A) transition. The increase in deoxythymidine 5′-triphosphate-G mispairs was confirmed by performing steady state single nucleotide addition studies. Our combined data support a model in which the Ala-to-Gln substitution in the fingers domain of Pol δ results in an interaction with the N-terminal domain that affects the base selectivity of the enzyme.
Introduction
DNA in living cells is replicated with exceptionally high accuracy. The ensemble of eukaryotic DNA polymerases are classified typically as either translesion polymerases, which bypass sites of damage to prevent replication fork stalling and genomic instability, or the replicative polymerases, DNA polymerases δ, ϵ, and α (reviewed in Refs. 1–3). There is strong evidence that DNA polymerase δ (Pol δ)3 is responsible for lagging strand synthesis of the entire nuclear genome (4–6), and it has also been shown to function in recombination and repair. Thus, Pol δ is likely to be responsible for the majority of DNA synthesis in human cells.
The accuracy of synthesis by Pol δ is determined primarily by the configuration of the catalytic site, which creates a steric and electrostatic bias for the incorporation of correct nucleotides (reviewed in Ref. 7). Replicative fidelity is further enhanced by the intrinsic exonuclease activity (“proofreading”) of Pol δ, which catalyzes the preferential hydrolysis of non-complementary nucleotides at the 3′ terminus (4, 8). The active site of Pol δ, in the absence of exonucleolytic proofreading, catalyzes one base substitution error per 7,600 nucleotides polymerized in yeast (9), and one in 22,000 nucleotides in human (10). Proofreading activity decreases the error rate of the human enzyme by at least 10-fold, an average of one base substitution error per 220,000 nucleotides polymerized. The overall fidelity of cellular DNA replication can be further enhanced to a rate of 1.0 × 10−10 errors per base by extrinsic factors such as the mismatch repair system and by accessory proteins at the replication fork.
The structure of the Pol δ catalytic domain, similar with that of most DNA polymerases, resembles an open right hand, with the thumb domain angled across the palm, and one or more α-helical fingers projecting toward the polymerase active site (11, 12). The site of deoxynucleotide addition is in a central pocket, through which the template and elongating primer DNA strands are threaded. The addition of the nascent nucleotide is catalyzed by carbonyl groups on key amino acids of the palm domain, along with associated magnesium ions (13). However, the fingers domain is responsible for proper orientation of the incoming nucleotide (12, 14, 15). Residues in the fingers domain align the paired bases for hydrogen binding, promote base stacking interactions, and make the α-phosphate available for nucleophilic attack by the primer 3′-OH.
Alterations in the amino acid composition of the Pol δ catalytic subunit can affect the fidelity of DNA synthesis (for reviews, see Refs. 16 and 17). The regions that form the catalytic pocket are structurally and compositionally conserved across all known polymerase species, and very few naturally occurring catalytic site mutations in the human replicative polymerases have been reported. A number of variants have been generated in vitro to study Pol δ kinetics and base selectivity, many of them in critical positions that originally were identified in genetic screens from viral and bacterial studies (18, 19). These include the positions responsible for discrimination between the deoxyribo- and ribo forms of nucleotides, as well as variants of a conserved leucine in the palm domain that yield a spectrum of activities depending on the amino acid substituted at that position (20, 21).
Surprisingly, given the essential role of Pol δ, only one catalytic site mutation has been identified in a human disease: the R689W substitution, found in the DLD-2 colorectal cancer cell line (22). The R689W substitution confers a mutator phenotype and lethal error catastrophe when the analogous variant is expressed in yeast (23). Arg689 is located in motif B on the P-helix of the fingers domain, distal to the end that forms the active nucleotide binding site. Because this region of the fingers domain of Pol δ is not well explored, we searched for mutator mutants at the C-terminal end of motif B in Saccharomyces cerevisiae Pol δ by substituting each position with all possible amino acids. We identified a mutator mutation, A699Q, which appears to be dependent on a new interaction formed between the P-helix and the N-terminal domain. The cognate human mutation (hPol ΔA692Q) was introduced, and this enzyme was purified. We report that hPol δA692Q is a mutator polymerase that generates replicative errors, particularly base substitutions, and exhibits reduced proofreading capacity. These results highlight the critical role played by the fingers domain in maintaining high fidelity DNA synthesis.
EXPERIMENTAL PROCEDURES
Yeast Strains and Plasmids
The S. cerevisiae POL3 gene, which encodes the catalytic subunit of Pol δ, was cloned in the LEU2 containing vector YcpLac111. Variants were created at amino acid positions 696–699 on the S. cerevisiae DNA polymerase δ catalytic subunit (POL3) using the common reverse primer 5′-GGCAGCTTCGGTACCAAGATCCATAGCTTCCTT-3′, and the following randomized, site-specific forward primers: 5′-GATGAGAAGGATCCATTCAAAAGAGATGTTTTAAATGGTNNNCAATTGGCTTTGAAGATT-3′; 5′-GATGAGAAGGATCCATTCAAAAGAGATGTTTTAAATGGTAGANNNTTGGCTTTGAAGATTTCA-3′; 5′-GATGAGAAGGATCCATTCAAAAGAGATGTTTTAAATGGTAGACAANNNGCTTTGAAGATTTCAGCT-3′; and 5′-GATGAGAAGGATCCATTCAAAAGAGATGTTTTAAATGGTAGACAATTGNNNTTGAAGATTTCAGCTAAC-3′, where NNN indicates random nucleotides. Following PCR amplification, a library of plasmids harboring random substitutions at the designated positions on the LEU2 containing vector YcpLac111 was transformed into BY4741(pol3Δ0::kanMX) cells harboring YcpLac33-Pol3, a URA3 containing vector that provides wild type S. cerevisae Pol δ, and the cells were grown on Synthetic Dextrose medium (SD medium) lacking leucine and uracil. To remove the YcpLac33-Pol3 vector and thereby select for cells with functionally active polymerase variants, colonies were then transferred to plates lacking leucine, but containing 5-fluor-orotic acid (5-FOA). Surviving colonies from 5-FOA plates were plated onto both Synthetic Complete medium (SC medium) and SD medium lacking arginine and leucine, with canavanine sulfate (60 mg/liter) added for the CAN1 forward mutation assay.
CAN1 Forward Mutation Assay
Yeast colonies grown on 5-FOA-containing medium and expressing wild type or mutant polymerases were diluted in water. Most of each dilution was spread directly onto canavanine sulfate plates (SD-Leu-Arg+Can), and the remaining 5% was subjected to six 10-fold dilutions. The serial dilutions were plated in 10-μl drops onto SC medium. Eight colonies of each genotype were assayed. After 3 days of growth, the number of colony forming units on each SD-Leu-Arg+Can plate was estimated by assessing growth on SC medium. The mutation rate was determined through the MSS-Maximum Likelihood Estimator method using the web application FALCOR (24).
Purification of Human Pol δ
The four-subunit human DNA Pol δ was expressed and purified from bacteria as described (10, 25). Briefly, BL21-tRARE competent cells were co-transformed by electroporation with pET3-PolD1, harboring the Pol δ p125 catalytic subunit, and pCOLA-polD2,3,4, which harbors the accessory subunits of the holoenzyme. Expression was induced by overnight incubation of mid-log phase cells with isopropyl β-d-1 thiogalactopyranoside at 15 °C. Cells were pelleted and frozen, and the crude extract was obtained by 10 rounds of sonication in buffer containing 40 mm HEPES (pH 7.5), 200 mm NaCl, 30 mm imidazole, 10% (v/v) glycerol, 0.1% (v/v) Triton X-100, followed by centrifugation of the debris. The purification consisted of two stages: first, the extract was bound to a charged nickel column via a polyhistidine tag on the p12 subunit, and bound proteins were eluted in batch with increasing concentrations of imidazole (30–300 mm). Next, the 300 mm imidazole eluate was subjected to ion exchange purification on a HiTrap SP HP column (GE Healthcare) using a 0.2–1.0 m linear NaCl gradient. The peak fraction was identified by assaying each fraction for DNA polymerase activity by incorporation of [α-32P]ATP into activated calf thymus DNA. The presence of all four subunits in the active fraction was confirmed by SDS-PAGE and Flamingo fluorescent protein staining (Bio-Rad) (data not shown).
M13mp2 Forward Mutagenesis Assay
M13mp2 gapped DNA substrate was extended by Pol δ variants at 37 °C for 1 h in a 25-μl reaction containing 86 nmol of enzyme, 1.5 nmol of gapped plasmid, 0.2 mg/ml BSA, 50 mm KCl, 40 mm Tris-HCl, pH 7.5, 2.5 mm MgCl2, 0.1% (v/v) Triton X-100, and 10% (v/v) glycerol. To separate the enzyme and DNA, 0.5 μl of 20 mg/ml proteinase K was added to each reaction, followed by incubation for an additional 15 min at 37 °C. Most of each reaction was run on a 0.8% TAE agarose gel (Invitrogen Ultra Pure) overnight at 80 V to verify gap filling. MC1061 cells were transformed by electroporation with a 1:10 dilution of the filled DNA substrate, and the transformed cells were combined in soft agar with mid-log CSH50 α-complementation cells, 0.75 mm isopropyl β-d-1 thiogalactopyranoside, 4.4 μg/ml 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside and subsequently plated onto agar plates containing VB salts (0.2 mg/ml MgSO4-7H20, 2.0 mg/ml citric acid, 10 mg/ml K2HPO4, 3.5 mg/ml Na(NH4)HPO4-4H2O), 0.4% glucose (w/v), and 0.5 μg of thiamine-HCl.
Plaques were visually evaluated for degree of blue pigment, and any clear or light blue plaques were marked. Mutation frequency was determined by dividing the number of mutant plaques by the total number of plaques. Mutant plaques were picked into 0.9% NaCl (w/v) and were used to infect fresh liquid cultures of CSH50 cells to obtain sufficient DNA for sequencing. Mutation rates for base substitutions and ±1 insertions and deletions were calculated using the formula ((N1/N) × MF)/(D × 0.6), where N1 is the number of mutations in that category, N is the total number of mutations, MF is the mutation frequency, and D is the number of detectable sites in that category (26).
Exonuclease Activity Assay
The substrate poly(dA)-oligo(dT) (12–18) was labeled with [α-32P]dTTP in a 10-μl reaction containing 5 units of exonuclease-deficient Klenow fragment for 15 min at 37 °C. Unincorporated [α-32P]dTTP was removed by Sephadex G-50 filtration, followed by ethanol precipitation. The exonuclease assays contained 0.036–0.2 pmol of polymerase in 40 mm Tris-HCl (pH 7.5), 50 mm KCl, 2.5 mm MgCl2, 0.1% (v/v) Triton X-100, 10% (v/v) glycerol, 2 μg of BSA, 1 mm dithiothreitol. Reactions were performed at 37 °C for 20 min. The reaction was stopped by the addition of a buffer containing 1 mm EDTA. The product was ethanol precipitated, and the supernatant was assayed for released radioactivity by scintillation counting.
Single Nucleotide Kinetics, Mismatch Extension, Exonuclease Assay
Steady state nucleotide addition experiments were performed using a 46-bp template, 5′-GCGCGGAAGCTTGGCTGCAGAAGATTGCTAGCGGGAATTCGGCGCG-3′ and a 23-bp primer, 5′-CGCGCCGAATTCCCGCTAGCAAT-3′, or a 24-bp primer, 5′-CGCGCCGAATTCCCGCTAGCAATT-3′ for mismatch extension and degradation studies. Enzyme concentration and time of incubation at 37 °C was established to be linear for each enzyme. The indicated concentrations of enzymes were incubated in a 10-μl reaction with 40 mm Tris-HCl (pH 7.4), 4 mm MgCl2, 0.1 mg/ml bovine serum albumin, 10 nm primer template, and 5 mm dithiothreitol. Reactions were initiated by the addition of nucleotides, run at 37 °C, and then placed on ice and stopped by the addition of an equal volume of 95% (v/v) formamide/20 mm EDTA. After resolution on 14% polyacrylamide gel, the bands were developed on a PhosphorImager device and quantified. Km and Vmax were obtained by curve-fitting the density data to the Michaelis-Menten equation using Kaleidagraph (Synergy software). Frequency of misincorporation (finc) was calculated by ((kcat/Km)incorrect/(kcat/Km)correct) (27).
RESULTS
Mutagenesis of Yeast Pol δ Residues Arg696-Ala699
Motif B of Pol δ is highly conserved at the amino acid level from humans to yeast. To determine the ability of residues Arg696-Ala699 in motif B of S. cerevisiae Pol δ to tolerate substitutions, we undertook a PCR mutagenesis strategy wherein a set of forward PCR primers with randomly substituted nucleotides at the three positions of each codon was used to modify the region, codon by codon. The mixture of primers in a single reaction should have representation of all 64 possible trinucleotides within the codon. Because many codons are degenerate, there was an overrepresentation of more commonly coded amino acids, such as arginine (six possible codons) over more rarely coded amino acids, such as methionine (one codon). The products of the PCR mutagenesis were subcloned into YcpLac111-POL3E (LEU2) and transformed into the yeast BY4741, whose chromosomal copy of POL3, encoding the catalytic subunit of Pol δ, was deleted. As Pol δ is an essential protein, these cells harbor an extrachromosomal copy of POL3 (YcpLac33-POL3 (ura3)). This library of mutant polymerases underwent purifying selection for activity by plasmid shuffling between media lacking leucine and uracil but containing 5-FOA. Colonies that grow on 5-FOA are under the direct control of the product of the PCR mutagenesis reactions, as 5-FOA is toxic to cells that harbor the URA3 cassette. We picked colonies and sequenced motif B to determine the spectrum of surviving substitutions at the first four positions of motif B.
Mutagenesis of the 696–699 region of S. cerevisiae Pol δ revealed a large tolerance for single amino acid substitutions (Table 1). At the Arg696 site alone, we identified 13 amino acids representing polar, nonpolar, charged, and hydrophobic residues that could substitute for alanine. This is in contrast to previous work on Escherichia coli Pol I and TaqDNA polymerase using random mutagenesis of the entire polymerase molecule which suggested that this position is very highly conserved (28, 29). The previously described mutation, R689W, which was identified in the human colorectal cancer cell line, DLD-1, has been demonstrated to be a strong mutator that is non-viable in haploid yeast (23). Consistent with this work, the analogous R696W substitution did not appear in our screen. Among the other positions, Gln697 permitted 10 amino acid substitutions; Leu698 was highly permissive, with 16 possible substitutions; and Ala699 allowed 10 substitutions.
TABLE 1.
Allowable substitutions at amino acids 696–699 of S. cerevisiae POL3
The wild type amino acid was also present in all cases at >50% of plasmids sequenced.
| Arg696 | Gln | Asn | Glu | Asp | Val | Ile | Leu | Ala | Tyr | Thr | Ser | Gly | Cys | ||
| Gln697 | Asn | Trp | Phe | Cal | Ile | Leu | Ala | Tyr | Thr | ||||||
| Leu698 | Gln | Asn | Glu | Asp | Val | Ile | Ala | Tyr | Thr | Ser | Gly | Cys | His | Arg | Lys |
| Ala699 | Gln | Met | His | Cys | Val | Gly | Leu | Ser | Tyr | Thr |
The large number of active substituted proteins served as a pool from which to screen for alterations in DNA polymerase function. We screened the variant strains for increased mutagenesis in a forward mutation assay that is based upon inactivation of the CAN1 gene. A few of the variants demonstrated an increase in mutation frequency, and their mutation rates were subsequently established by fluctuation analysis (Fig. 1A). We identified the A699Q substitution as our strongest mutator, with a mutation rate ∼20-fold higher than wild type Pol δ. By comparison, the exonuclease-deficient Pol δ variant, pol3-01, which lacks the ability to undergo proofreading, has only a 7-fold higher rate than wild type Pol δ in the CAN1 assay.
FIGURE 1.
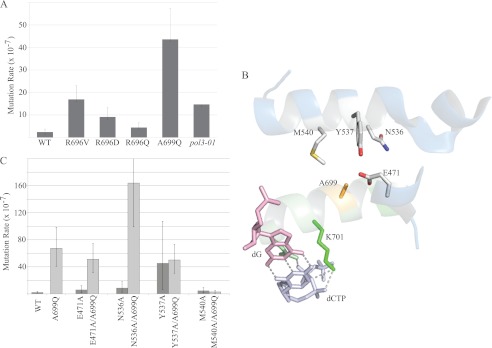
Mutation rates of wild type and mutant DNA Pol δ yeast. A, haploid yeast expressing mutant Pol δ were subjected to fluctuation analysis in the canavanine sulfate (CAN1) forward mutagenesis assay, and mutation rates were calculated using MSS maximum likelihood. The error bars indicate the 95% confidence interval. B, schematic representation of the region proximal to Ala699 (yellow) showing the P-helix (green) harboring motif B, and N-terminal domain helices (blue). The four putative interacting residues Met540, Tyr537, Asn536, and Glu471 are represented as gray sticks. The incoming dCTP (silver) is shown interacting with Lys701 and Asn705 of motif B and the template G (pink) and is provided for context. The image was produced in PyMOL using Protein Data Bank code 3IAY (The PyMOL Molecular Graphics System, version 1.2r3pre, Schrödinger, LLC.). C, mutation rates of single and double mutants at the CAN1 locus. The M540A substitution suppresses the mutator effect of A699Q. Tyr536 and Asn537 show evidence of interaction with A699Q (see “Results” and “Discussion”).
Rescue of the Mutator Phenotype of Pol δA699Q
Modeling the A699Q mutation onto the crystal structure of S. cerevisiae Pol δ, consisting of a closed ternary complex containing the catalytic subunit of Pol δ, the template primer, and the incoming complementary nucleotide, reveals four potential contacts that A699Q could make, but that the wild type Ala699 cannot (Fig. 1B) (30). The four positions are located on the N-terminal domain, separated from the P-helix of the fingers domain by an aqueous channel. To explore whether a new intramolecular contact was being made by the A699Q mutation, we constructed Pol δ mutants in which alanine was substituted for these four potential positions of contact: Glu471, Asn536, Tyr537, and Met540. These were made as individual substitutions, as well as in combination with the A699Q mutation. If interaction at any of these target sites substantially contributes to the mutator phenotype of Pol δA699Q, then substituting that target for an alanine residue could suppress the mutator phenotype of A699Q.
There was no reduced growth phenotype with E471A, N536A, or M540A, nor was there any abnormal growth phenotype with these mutations in the A699Q background. However, the Y537A mutant grew poorly, producing only microcolonies after several days. As shown in Fig. 1C, fluctuation analysis of the singly and doubly mutated Pol δ species revealed that the single mutants had a base-line rate of mutation similar to wild type. A possible exception is the Y537A variant, which was difficult to assess via canavanine resistance due to its severe growth phenotype.
Analysis of the double mutants presents a complicated picture. Three of the double mutants (E471A/A699Q, N536A/A699Q, and Y537A/A699Q) exhibited elevated rates of mutation that were similar, within experimental error, to A699Q alone. The M540A/A699Q variant, however, exhibited a wild type mutation rate, strongly suggesting that an interaction between A699Q and Met540 is responsible for the mutator phenotype of A699Q. The combination of A699Q and N536A produced an apparent increase in mutation rate over A699Q alone, but with high variance. This high level of noise within three independent replicates may reflect a transient interaction with Asn536. Furthermore, the rescue of the Y537A growth phenotype by A699Q suggests a potential interaction between these sites. Alternatively, it may represent the ability of A699Q to compensate for the Y537A substitution via some other mechanism than direct contact because the presence or absence of Tyr537 does not seem to affect the mutation rate of Pol δA699Q. Further analysis of these apparent interactions and how they might affect fidelity is presented in the “Discussion.”
Synthetic Lethality of Proofreading and Mismatch Repair Deficiency with Pol δA699Q
Given the dynamic nature of the fingers domain, the effect of A699Q on mutagenesis could be due to a change in the kinetics of nucleotide addition or in the efficiency of proofreading. If proofreading was directly affected by the mutation in question, then one would expect only an additive effect (or less) in mutation rate if Pol δ exonuclease activity were also abolished. We abolished exonuclease activity of the A699Q variant by mutating two conserved residues that have been determined to be essential for exonuclease activity (D421A and E423A). The A699Q exonuclease-deficient mutant was non-viable, demonstrating a synthetically lethal relationship between the substitution in the fingers domain and the absence of exonuclease activity (Fig. 2). The profound effect of eliminating exonuclease activity implies that the mutations introduced by Pol δA699Q are normally corrected by intrinsic exonuclease activity. Similarly, there was synthetic lethality when the Pol δA699Q variant was introduced into haploid cells that were deficient for mismatch repair, owing to the absence of the MSH2 gene (Fig. 2).
FIGURE 2.

Synthetic lethality of Pol δA699Q with exonuclease and mismatch repair deficiency. Haploid yeast cells that are positive or negative for the mismatch repair protein Msh2 are plated on 5-fluororotic acid plates to select for an expression plasmid containing either wild type Pol δ, Pol δA699Q, or exonuclease-deficient Pol δA699Q. Wild type Pol δ permits growth in both Msh2-containing and Msh2-deficient cells. Pol δA699Q, however, exhibits growth in Msh2-containing cells but not in the context of mismatch repair deficiency and also requires exonuclease activity for survival.
Purification of Human Pol δA692Q
To more directly assess the role of the A699Q mutation in polymerase fidelity, the cognate mutation, A692Q, was introduced into human Pol δ. An exonuclease-deficient version of the mutant protein (A692Q/D400A) also was made. There is nearly 90% amino acid identity in the three catalytic motifs between human and budding yeast Pol δ and 100% identity within motif B. Thus, we considered the human polymerase to be a reasonable model for investigating the biochemical basis of the in vivo mutation rates of the S. cerevisiae Pol δ mutants. We expressed the four-subunit holoenzyme in E. coli and purified the complex via sequential affinity purification and ion exchange chromatography as described previously (10, 25). The major protein fraction correlated with the peak of DNA polymerase activity, and all four subunits were shown to be present via Flamingo fluorescent staining on an SDS-PAGE gel (data not shown). A scintillation-based assay for release of radioac tivity on 3′-labeled poly(dA)-oligo(dT) substrate demonstrated that exonuclease-deficient Pol δA692Q (referred to as hPol δDQ) lacks the ability to degrade the substrate (data not shown).
Mutation Rate of hPolδA692Q
We used the M13mp2-lacZ gap-filling assay to assess the mutation rate of hPol δA692Q. In this assay, a double-stranded M13 construct is prepared, which includes a single stranded portion within the coding sequence of the LacZ α-fragment. The single-stranded portion of the substrate is copied by the DNA polymerase in vitro, and the products are introduced into E. coli, followed by plating on medium containing X-Gal and isopropyl β-d-1 thiogalactopyranoside. Accurate replication by the polymerase results in dark blue plaques, whereas polymerase errors that inactivate LacZ-α result in light blue or colorless plaques. Transformation of the substrate alone yielded a background frequency of 1 × 10−3, consistent with prior reports (10). The A692Q variant yielded a mutant plaque frequency of 3 × 10−2, a 30-fold increase, which indicates that hPolδA692Q is highly error prone. Fig. 3B summarizes the rates of mutation for single base substitutions and ±1 frame shifts. No complex or large deletions were observed, although there were four independent dinucleotide deletions, all at different sites in the lacZ gene (Fig. 3A).
FIGURE 3.

Mutation rates and spectrum of hPol δA692Q at the lacZ locus on M13mp2 gapped plasmid. A, mutations detected in the gapped plasmid filling assay are mapped on the scorable region of the gene. Base substitutions appear above the line, whereas −1 deletions (Δ) or +1 insertions (+N) are found below the line. B, mutation rates from 188 mutants sequenced, including individual base substitutions, ±1 frameshifts and overall base substitution rate, calculated as described under “Experimental Procedures.”
The spectrum of base substitutions differs markedly from those generated by wild type and exonuclease-deficient human Pol δ (Fig. 4). Specifically, we observed a large increase in the incorporation of dTTP opposite both template C and template G (Fig. 4). In particular, the G to A transitions (G:dT) occurred at 12/22 sites in the gapped sequence, with only one hot spot, and no clear preference for a sequence context. Overall, nearly 60% of the mutant plasmids analyzed harbored single base substitutions, compared with the 47–48% reported for wild type and exonuclease-deficient hPol δ (10), indicating that the A692Q substitution predisposes Pol δ to errors in base selection.
FIGURE 4.

Comparison of mutation rates between Pol δA692Q and Pol δD400A. Mutation rates for individual base substitutions from the gapped filling assay using purified Pol δA692A (black bars, values on left axis) are compared with published values (10) for Pol δD400A (gray bars, values on right axis). Pol δA692Q displays an elevated base substitution rate overall, relative to Pol δD400A, with the largest increase in mutagenesis by the A692Q variant occurring in the setting of G→A and C→A substitutions.
Single Nucleotide Incorporation Kinetics
Because hPol δA692Q appears to exhibit a base selection phenotype in the gap-filling assay, we examined the kinetics of correct and incorrect incorporation of nucleotides using the purified polymerase. hPol δDQ was used to assess the base-selection properties of the enzyme to avoid the confounding influence of proofreading on estimating catalytic efficiency. hPol δD400A, which is wild type Pol δ with an inactivating mutation in the exonuclease domain, was used as a control. The experimental conditions were independently determined for each enzyme by determining the kinetics of incorporation of the correct nucleotide on a 23/46-bp primer/template duplex. In experiments examining incorporation of dCTP across from template G, the hPol δDQ variant had a 26-fold lower kcat/Km value, suggesting that the Ala692 mutation results in decreased catalytic activity (Table 2).
TABLE 2.
hPol δ catalysis of nucleotide addition during correct and incorrect incorporation
Primer extension experiments measured incorporation of dCTP or dTTP across from template G. S.D. reflect three replicate experiments.
| Pol δD400A | Pol δDQ | Fold (DQ:D400A) | |
|---|---|---|---|
| Correct (dCTP-G)a | |||
| Vmax (fmol/min) | 18 ± 0.13 | 26 ± 1.2 | |
| Km (μm) | 0.25 ± 0.01 | 2.5 ± 0.4 | |
| kcat (min−1) | 0.51 ± 0.004 | 0.19 ± 0.009 | |
| kcat/Km | 2 | 0.076 | −26 |
| Incorrect (dTTP-G)b | |||
| Vmax (fmol/min) | 38 ± 11 | 9.1 ± 0.98 | |
| Km (μm) | 340 ± 180 | 180 ± 43 | |
| kcat (min−1) | 0.39 ± 0.11 | 0.027 ± 0.002 | |
| kcat/Km | 0.001 | 0.0001 | −10 |
| finc*c | 0.0005 | 0.0013 | 2.6 |
a Correct incorporation experiments used 0.14 pmol hPol δDQ and 0.036 pmol hPol δD400A.
b Incorrect incorporation experiments used 0.336 pmol hPol δDQ and 0.099 pmol hPol δD400A.
c finc given by (kcat/Km)incorrect/(kcat/Km)correct.
Incorporation of an incorrect nucleotide was performed using the same primer template duplex but with dTTP as the substrate. The choice of a dTTP·G mispair was based on the observation that hPol δA692Q exhibited a proportionally high level of G→A base substitutions in the gap filling assay, whereas the same substitution was not strongly represented in previously reported spectra from hPol δ. We report a 2.6-fold increase in the misincorporation efficiency (finc) for Pol δDQ over that of Pol δD400A, which partially confirms the increase in mutagenesis seen in the gap-filling assay (Table 2).
Mismatch Extension and Exonuclease Assays
An increased base substitution rate could be due to reduced base selectivity, reduced proofreading, and/or a greater capacity for extending mispaired substrate. We assayed hPol δDQ and hPol δD400A for the ability to extend a 24/46-bp primer:template duplex with a 3′-terminal T:G mismatch. hPol δDQ extended this mismatch with a slightly lower efficiency than hPol δD400A, so it is unlikely that this mechanism contributes to the increased base substitution rate of hPol δA692Q (data not shown).
Next, we compared hPol δA692Q and wild type hPol δ for their ability to recognize and degrade a terminal G:C base pair or a terminal G:T mismatch on duplex DNA. Using concentrations of protein that were normalized for equivalent polymerase activity, hPol δA692Q catalyzed degradation of both matched and mismatched substrate at 79% of the rate of wild type enzyme (data not shown). These results suggest that a general deficiency in proofreading may also contribute to the increased base substitution rate of hPol δA692Q.
DISCUSSION
The Pol δ catalytic domain undergoes rapid, large-scale conformational changes as it engages in DNA polymerization, translocation along the template, and transfer of the primer end for exonucleolytic proofreading. In particular, the fingers domain is a highly mobile region whose opening and closing precedes each nucleotide addition step. A change in the mechanics or kinetics of fingers domain movement could affect the rate of nucleotide addition or the efficiency of active site switching. Additionally, the polymerase active site is a highly conserved structure that is highly sensitive to perturbations induced by changes in the composition of surrounding residues.
The amino acid positions that directly bind and position the incoming nucleotide are located along the helix of motif B, as determined by site-directed mutagenesis of these positions in the B-family polymerase Pol α (14). The previously reported, cancer cell-associated R689W mutation is located four turns of an α-helix away from the active site residues Tyr708 and Gly709 and is not one of the residues thought to bind the incoming nucleotide. When this mutation was introduced into yeast Pol δ, it was found to be lethal in haploid cells, most likely due to an accumulation of point mutations. The position of R689W (R696W in yeast) in the catalytic subunit was suggested to interfere with exonuclease partitioning, due to the disruption of a putative interaction between the fingers and exonuclease domains. This suggests that the other C-terminal sites in motif B may have similar effects that could elucidate the role of the fingers domain in DNA polymerization and proofreading.
We examined the role of the C-terminal residues in motif B in yeast Pol δ by determining the spectrum of allowable substitutions in amino acids 696–699. We found evidence for a high tolerance of substitution at these positions. Further characterization of variants with increased mutagenic capacity identified A699Q as a highly mutagenic substitution that requires proofreading and mismatch repair for survival in haploid yeast. We interrogated potential interacting amino acid positions by substituting each one for alanine and discovered a complex interaction between the N-terminal domain and the fingers domain that affects replication fidelity. For further characterization, we introduced this substitution into human Pol δ at Ala692. In the M13-LacZ forward mutation assay, hPol δA692Q generated an increase in base substitution rates and overall mutation frequency when compared with exonuclease-deficient hPol δ. Steady state kinetics results confirm an increase in the efficiency of nucleotide misincorporation by hPol δDQ and indicate that the A692Q substitution may lower the overall rate of exonuclease activity.
We used the crystal structure of yeast Pol δ in a ternary complex with duplex DNA and an incoming nucleotide (12) to determine positions that could interact with the A699Q substitution in the finger-closed conformation. Despite our identification of a critical interaction with Met540, we lack information on how this substitution could affect the structure of the region in the open state or the transition between open to closed states. However, the complete suppression of the A699Q-induced increase in mutation rate by M540A demonstrates the importance of interactions between the fingers domain and the N-terminal domain.
How could such an interaction affect polymerase fidelity? Modeling A699Q onto the yeast Pol δ structure suggests a possible mechanism. The residues that are close enough to A699Q to potentially interact are located on a loop (Glu471) or on the J helix in the N-terminal domain (Asn536, Tyr537, Met540). The single substitution, Y537A, leads to a severe growth defect, resulting in microcolonies that take many days to form. The double mutant, Pol δY537A/A699Q, grows normally but has a mutation rate similar to Pol δA699Q. One possible explanation is that the polar Tyr537 side chain participates in regional stability and that A699Q but not Ala699 can restore that stability. We introduced all nine identified substitutions at position 699 on the available structure of Pol δ and found that some degree of steric clash always occurs between the β-carbon hydrogens at position 699, when present, and Met540. The clash can be relieved by displacement of Met540, as suggested by models, leaving the side chains of amino acids at 699 pointing toward Tyr537 and Glu471. Although most of the Ala699-substituted amino acids found in our screen would clash with Met540, A699Q is the only one with a polar group within range to interact with the hydrogen bonding network between Tyr537 and Glu471. Replacement of Met540 with alanine relieves the steric clash, and A699Q could be expected to adopt an orientation toward M540A. Therefore, the major contributor to the mutagenicity of Pol δA699Q may be the interaction with Tyr537-Glu471 hydrogen bonding network, directly or through water molecules. Alternatively, the steric clash with Met540 could displace the highly mobile P-helix, which could change the orientations of critical residues in the catalytic pocket.
Gap-filling experiments on a LacZ reporter plasmid using hPol δA692Q confirmed its higher mutation frequency over wild type hPol δ (30-fold) and exonuclease-deficient hPol δ (3-fold). A comparison of mutation rates and spectra with Pol δD400A identifies base substitutions as the overwhelming source of this 3-fold increase. dTTP misincorporations accounted for nearly 40% of all substitutions, despite representing only 20% of possible scorable outcomes in the assay. Single nucleotide incorporation experiments confirmed that hPolδDQ incorporates incorrect nucleotides at a greater frequency than hPol δD400A, and exonuclease assays reveal a reduced rate of proofreading on both matched and mismatched primer-template termini. Altogether, the kinetics data suggest that multiple aspects of Pol δ activity are compromised by A692Q.
The A692Q substitution and other local mutations could serve as important tools to ascertain the role of the fingers domain in eukaryotic B family polymerases. Future work aimed at defining additional intramolecular interactions at the mobile end of the fingers domain would be a step toward better understanding how the Pol δ active site discriminates between a correct and incorrect incorporation, how the fingers domain contributes to proofreading, and how Pol δ commits to the nucleotide addition step.
Acknowledgments
We thank Ranga Venkatesan for critical reagents, ideas, and advice and Ashwini Kamath Loeb for critical reading of the manuscript.
Addendum
Since this article was submitted, a mutation in human Pol δ affecting the proofreading domain has been reported for the first time in human cancer (31), underscoring the importance of replication fidelity in the prevention of human disease.
This work was supported, in whole or in part, by National Institutes of Health Grants CA15802, CA102029, and POLCA77852.
- Pol δ
- DNA polymerase δ
- 5-FOA
- 5-fluor-orotic acid
- dTTP
- deoxythymidine 5′-triphosphate.
REFERENCES
- 1. Hubscher U., Maga G., Spadari S. (2002) Eukaryotic DNA polymerases. Annu. Rev. Biochem. 71, 133–163 [DOI] [PubMed] [Google Scholar]
- 2. Lange S. S., Takata K., Wood R. D. (2011) DNA polymerases and cancer. Nat. Rev. Cancer 11, 96–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sale J. E., Lehmann A. R., Woodgate R. (2012) Y-family DNA polymerases and their role in tolerance of cellular DNA damage. Nat. Rev. Mol. Cell Biol. 13, 141–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Morrison A., Sugino A. (1994) The 3′–>5′ exonucleases of both DNA polymerases δ and ϵ participate in correcting errors of DNA replication in Saccharomyces cerevisiae. Mol. Gen. Genet. 242, 289–296 [DOI] [PubMed] [Google Scholar]
- 5. Nick McElhinny S. A., Gordenin D. A., Stith C. M., Burgers P. M., Kunkel T. A. (2008) Division of labor at the eukaryotic replication fork. Mol. Cell 30, 137–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Larrea A. A., Lujan S. A., Nick McElhinny S. A., Mieczkowski P. A., Resnick M. A., Gordenin D. A., Kunkel T. A. (2010) Genome-wide model for the normal eukaryotic DNA replication fork. Proc. Natl. Acad. Sci. U.S.A. 107, 17674–17679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kunkel T. A., Bebenek K. (2000) DNA replication fidelity. Annu. Rev. Biochem. 69, 497–529 [DOI] [PubMed] [Google Scholar]
- 8. Morrison A., Bell J. B., Kunkel T. A., Sugino A. (1991) Eukaryotic DNA polymerase amino acid sequence required for 3′—-5′ exonuclease activity. Proc. Natl. Acad. Sci. U.S.A. 88, 9473–9477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fortune J. M., Pavlov Y. I., Welch C. M., Johansson E., Burgers P. M., Kunkel T. A. (2005) Saccharomyces cerevisiae DNA polymerase δ: high fidelity for base substitutions but lower fidelity for single- and multi-base deletions. J. Biol. Chem. 280, 29980–29987 [DOI] [PubMed] [Google Scholar]
- 10. Schmitt M. W., Matsumoto Y., Loeb L. A. (2009) High fidelity and lesion bypass capability of human DNA polymerase δ. Biochimie 91, 1163–1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Joyce C. M., Steitz T. A. (1995) Polymerase structures and function: variations on a theme? J. Bacteriol. 177, 6321–6329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Swan M. K., Johnson R. E., Prakash L., Prakash S., Aggarwal A. K. (2009) Structural basis of high-fidelity DNA synthesis by yeast DNA polymerase δ. Nat. Struct. Mol. Biol. 16, 979–986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Steitz T. A. (1998) A mechanism for all polymerases. Nature 391, 231–232 [DOI] [PubMed] [Google Scholar]
- 14. Ogawa M., Limsirichaikul S., Niimi A., Iwai S., Yoshida S., Suzuki M. (2003) Distinct function of conserved amino acids in the fingers of Saccharomyces cerevisiae DNA polymerase α. J. Biol. Chem. 278, 19071–19078 [DOI] [PubMed] [Google Scholar]
- 15. Yang G., Franklin M., Li J., Lin T. C., Konigsberg W. (2002) Correlation of the kinetics of finger domain mutants in RB69 DNA polymerase with its structure. Biochemistry 41, 2526–2534 [DOI] [PubMed] [Google Scholar]
- 16. Herr A. J., Williams L. N., Preston B. D. (2011) Antimutator variants of DNA polymerases. Crit. Rev. Biochem. Mol. Biol. 46, 548–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Prindle M. J., Loeb L. A. (2012) DNA polymerase δ in DNA replication and genome maintenance. Environ. Mol. Mutagen. 53, 666–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Reha-Krantz L. J., Nonay R. L. (1994) Motif A of bacteriophage T4 DNA polymerase: role in primer extension and DNA replication fidelity. Isolation of new antimutator and mutator DNA polymerases. J. Biol. Chem. 269, 5635–5643 [PubMed] [Google Scholar]
- 19. Schaaper R. M. (1998) Antimutator mutants in bacteriophage T4 and Escherichia coli. Genetics 148, 1579–1585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pavlov Y. I., Shcherbakova P. V., Kunkel T. A. (2001) In vivo consequences of putative active site mutations in yeast DNA polymerases α, ϵ, δ, and ζ. Genetics 159, 47–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Venkatesan R. N., Hsu J. J., Lawrence N. A., Preston B. D., Loeb L. A. (2006) Mutator phenotypes caused by substitution at a conserved motif A residue in eukaryotic DNA polymerase δ. J. Biol. Chem. 281, 4486–4494 [DOI] [PubMed] [Google Scholar]
- 22. Flohr T., Dai J. C., Büttner J., Popanda O., Hagmüller E., Thielmann H. W. (1999) Detection of mutations in the DNA polymerase δ gene of human sporadic colorectal cancers and colon cancer cell lines. Int. J. Cancer 80, 919–929 [DOI] [PubMed] [Google Scholar]
- 23. Daee D. L., Mertz T. M., Shcherbakova P. V. (2010) A cancer-associated DNA polymerase δ variant modeled in yeast causes a catastrophic increase in genomic instability. Proc. Natl. Acad. Sci. U.S.A. 107, 157–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hall B. M., Ma C. X., Liang P., Singh K. K. (2009) Fluctuation analysis CalculatOR: a web tool for the determination of mutation rate using Luria-Delbruck fluctuation analysis. Bioinformatics 25, 1564–1565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fazlieva R., Spittle C. S., Morrissey D., Hayashi H., Yan H., Matsumoto Y. (2009) Proofreading exonuclease activity of human DNA polymerase δ and its effects on lesion-bypass DNA synthesis. Nucleic Acids Res. 37, 2854–2866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bebenek K., Kunkel T. A. (1995) Analyzing fidelity of DNA polymerases. Methods Enzymol. 262, 217–232 [DOI] [PubMed] [Google Scholar]
- 27. Bloom L. B., Chen X., Fygenson D. K., Turner J., O'Donnell M., Goodman M. F. (1997) Fidelity of Escherichia coli DNA polymerase III holoenzyme. The effects of β, γ complex processivity proteins and epsilon proofreading exonuclease on nucleotide misincorporation efficiencies. J. Biol. Chem. 272, 27919–27930 [DOI] [PubMed] [Google Scholar]
- 28. Suzuki M., Avicola A. K., Hood L., Loeb L. A. (1997) Low fidelity mutants in the O-helix of Thermus aquaticus DNA polymerase I. J. Biol. Chem. 272, 11228–11235 [DOI] [PubMed] [Google Scholar]
- 29. Loh E., Choe J., Loeb L. A. (2007) Highly tolerated amino acid substitutions increase the fidelity of Escherichia coli DNA polymerase I. J. Biol. Chem. 282, 12201–12209 [DOI] [PubMed] [Google Scholar]
- 30. Leaver-Fay A., Tyka M., Lewis S. M., Lange O. F., Thompson J., Jacak R., Kaufman K., Renfrew P. D., Smith C. A., Sheffler W., Davis I. W., Cooper S., Treuille A., Mandell D. J., Richter F., Ban Y. E., Fleishman S. J., Corn J. E., Kim D. E., Lyskov S., Berrondo M., Mentzer S., Popović Z., Havranek J. J., Karanicolas J., Das R., Meiler J., Kortemme T., Gray J. J., Kuhlman B., Baker D., Bradley P. (2011) ROSETTA3: an object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol. 487, 545–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Palles C., Cazier J. B., Howarth K. M., Domingo E., Jones A. M., Broderick P., Kemp Z., Spain S. L., Almeida E. G., Salguero I., Sherborne A., Chubb D., Carvajal-Carmona L. G., Ma Y., Kaur K., Dobbins S., Barclay E., Gorman M., Martin L., Kovac M. B., Humphray S., Thomas H. J., Maher E., Evans G., Lucassen A., Cummings C., Stevens M., Walker L., Halliday D., Armstrong R., Paterson J., Hodgson S., Homfray T., Side L., Izatt L., Donaldson A., Tomkins S., Morrison P., Goodman S., Brewer C., Henderson A., Davidson R., Murday V., Cook J., Haites N., Bishop T., Sheridan E., Green A., Marks C., Carpenter S., Broughton M., Greenhalge L., Suri M., Donnelly P. C., Bell J., Bentley D., McVean G., Ratcliffe P., Taylor J., Wilkie A., Broxholme J., Buck D., Cornall R., Gregory L., Knight J., Lunter G., Tomlinson I., Buck D. L., Kingsbury Z., McVean G. L., Donnelly P., Grocock R., Hatton E., Holmes C. C., Hughes L., Humburg P., Kanapin A., Murray L., Rimmer A., Petridis C., Roylance R., Sawyer E. J., Kerr D. J., Clark S., Grimes J., Kearsey S. E., Houlston R. S., Tomlinson I. (2012) Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat. Genet. 10.1038/ng.2503 [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]