

Background: The 5-hydroxytryptamine receptor (5-HT3R) is a prototypical pentameric ligand-gated ion channel.
Results: The receptor's thermal stability was investigated in native plasma membranes, in detergent solution, and in reconstituted lipid bilayers.
Conclusion: Unfolding of the 5-HT3R occurs via hierarchical, consecutive structural transitions, which can be distinguished experimentally.
Significance: Our findings serve as an important base to create receptors of increased stability for structural/functional studies.
Keywords: Circular Dichroism (CD), Fluorescence, Membrane Proteins, Membrane Reconstitution, Plasma Membrane, Membrane Protein Expression, Detergents, Ligand Binding, Protein Aggregation, Thermal Stability
Abstract
Pentameric ligand-gated ion channels (LGICs) play an important role in fast synaptic signal transduction. Binding of agonists to the β-sheet-structured extracellular domain opens an ion channel in the transmembrane α-helical region of the LGIC. How the structurally distinct and distant domains are functionally coupled for such central transmembrane signaling processes remains an open question. To obtain detailed information about the stability of and the coupling between these different functional domains, we analyzed the thermal unfolding of a homopentameric LGIC, the 5-hydroxytryptamine receptor (ligand binding, secondary structure, accessibility of Trp and Cys residues, and aggregation), in plasma membranes as well as during detergent extraction, purification, and reconstitution into artificial lipid bilayers. We found a large loss in thermostability correlating with the loss of the lipid bilayer during membrane solubilization and purification. Thermal unfolding of the 5-hydroxytryptamine receptor occurred in consecutive steps at distinct protein locations. A loss of ligand binding was detected first, followed by formation of different transient low oligomeric states of receptor pentamers, followed by partial unfolding of helical parts of the protein, which finally lead to the formation receptor aggregates. Structural destabilization of the receptor in detergents could be partially reversed by reconstituting the receptor into lipid bilayers. Our results are important because they quantify the stability of LGICs during detergent extraction and purification and can be used to create stabilized receptor proteins for structural and functional studies.
Introduction
The 5-HT3R2 is a homopentameric ligand-gated ion channel (LGIC) responsible for rapid transmission of serotonin signals between synapses of nerve cells (1–4). It belongs to the large family of Cys loop LGICs that has considerable therapeutic interest because of its important role in signal transduction (3, 5–7). Present structural models of Cys loop receptors rely on high resolution structures of nonvertebrate homologs (8–11). According to these models, a typical Cys loop LGIC is composed of three distinct structural and functional entities as follows: a large β-structured extracellular domain comprising the ligand-binding site(s); a helical transmembrane channel-forming part, and an intracellular domain that probably interacts with the cellular cytoskeleton. How binding of an agonist on the extracellular side gates the intramembranous channel and finally transmits an electrochemical signal across the plasma membrane remains an important open question. Between ligand binding and channel opening, the receptor probes a large number of sequential conformational transitions (12, 13). However, directly determined high resolution structures of vertebrate Cys loop receptors are still missing due to difficulties in production, extraction, and crystallization of these proteins. Investigating the thermodynamic stability of these membrane proteins will contribute to overcoming some of these hurdles.
Thermodynamics of α-helical membrane protein folding and unfolding have been reviewed elsewhere (14–20). Classical work concentrated on model membrane proteins like bacteriorhodopsin to obtain Gibbs free energies for their unfolding/refolding. This has resulted in the proposal of a two-stage mechanism (21): during membrane insertion the α-helices form first and subsequently assemble into compact functional transmembrane protein structures. Since then, this model has been further refined and extended by taking into consideration the intermediate insertion steps such as helical protein parts at the membrane interface eventually switching to a transmembrane structure, as well as membrane protein oligomerization within the lipid bilayer (17, 18, 22).
In vivo, most α-helical membrane proteins are assumed to fold and insert into the membrane via a similar mechanism, but enzymatically regulated and catalyzed by the translocon machinery (23, 24), which is very difficult to replicate in vitro. Therefore, in the absence of such sophisticated machinery, membrane proteins are extracted and purified so that biochemical, biophysical, and structural studies (including x-ray crystallography) can be performed on proteins in detergent solution (25). However, the particular composition of the lipid bilayer plays an important role in the activity of LGICs (26); removal of the lipid bilayer by detergent solubilization usually results in a decreased thermodynamic stability and sometimes in loss of functional activity (27–29).
The thermostability of a protein is typically characterized by its melting temperature (Tm), the temperature at which 50% of thermal unfolding occurred. It has been found that proteins denaturing at higher temperatures generally have a greater free energy of maximal stability (30). It is widely assumed that increasing protein thermostability improves the quality of protein crystals, which is why candidates for crystallization are often selected according to their thermostability (31). For example, homologous proteins coming from thermophilic bacteria have often yielded crystals of better quality and in turn higher resolution than their less stable counterparts from mesophiles (32). Screening for stabilizing mutations has become standard for water-soluble proteins in x-ray crystallography and is now slowly introduced for membrane proteins (29, 33–37). Recently, engineered mutants of G protein-coupled receptors (GPCR) that have been screened for thermostability resulted in several high resolution three-dimensional structures (38, 39). Despite the demonstrated importance of thermostability for protein crystallization, little is known about its mechanism in case of membrane proteins (40).
In this study, we analyze the thermostability of the 5-HT3R in the native plasma membrane, during the process of detergent extraction from the plasma membrane of its host cell, in its detergent-solubilized purified state, and after lipid bilayer reconstitution. We use different methods (ligand binding, secondary structure, accessibility of Trp and Cys residues, and aggregation) to determine membrane protein thermostability to probe and distinguish different functions and in turn different structural domains of the receptor. Up until now, most of the unfolding/refolding studies have been performed on much simpler membrane proteins than the presently studied multimeric, multifunctional, and multidomain 5-HT3R.
EXPERIMENTAL PROCEDURES
Materials
Materials for cell medium like DMEM/F-12 with GlutaMAX, fetal bovine serum (FBS), trypsin/EDTA, Dulbecco's PBS, and blasticidin were purchased from Invitrogen. Hygromycin and complete protease inhibitors were from Roche Applied Science. Tetracycline, sodium butyrate, d-desthiobiotin, polyethyleneimine, asolectin, Coomassie Brilliant Blue G-250, acrylamide, 6-aminohexanoic acid, and N-acetyl-l-tryptophanamide (NATA) were from Sigma. Nonaoxyethylene n-dodecyl ether (C12E9) was from Affymetrix. Quipazine and granisetron were from Tocris Bioscience. [3H]GR-65630 (3-(5-methyl-1H-imidazol-4-yl)-1-(1-[3H]methyl-1H-indol-3-yl)propanone) had a specific activity of 79 Ci/mmol and was from PerkinElmer Life Sciences. N-[4-(7-Diethylamino-4-methyl-3-coumarinyl)-phenyl]-maleimide (CPM) was from Invitrogen. 1,2,3,9-Tetrahydro-3-[(5-methyl-1H-imidazol-4-yl)-methyl]-9-(3-amino(N-fluorescein-thiocarbamoyl)-propyl)-4H-carbazol-4-one) was obtained from Ruud Hovius; synthesis has been described elsewhere (41).
Cell Lines and Cell Maintenance
Mouse 5-HT3R cDNA with an N-terminal Strep tag was inserted into a tetracycline-inducible CHO cell line. Cells were maintained at 37 °C under a humidified 5% CO2 atmosphere in tissue culture-treated flasks (TPP, Switzerland) containing DMEM/F-12 medium with GlutaMAX and 10% FBS. Cells were passaged by trypsin or PBS/EDTA treatment (final EDTA concentration 1 mm) when they reached 80% confluence and reseeded by a 20–50-fold dilution into fresh medium. To keep selection pressure on stable cell lines, 5-HT3R-expressing cells were cultivated in the presence of 200 μg/ml hygromycin and 10 μg/ml blasticidin.
Production of Purified 5-HT3R
5-HT3R-expressing cells were grown in roller bottles containing DMEM/F-12 medium with GlutaMAX and 10% FBS. After 8 days 5-HT3R expression was induced with 2 μg/ml tetracycline and 4 mm sodium butyrate, and 40 h later cells were harvested with Dulbecco's PBS/EDTA, pelleted, frozen in liquid nitrogen, and stored at −80 °C.
Membrane preparation and solubilization of 5-HT3R were performed as described before (42). All steps were performed at 4 °C. Membranes were prepared by thawing cells and resuspending them in 10 mm HEPES, pH 7.4, containing 1 mm EDTA and protease inhibitors. The suspension was kept on ice and homogenized with an Ultra-Turrax T25 (IKA, Germany) for 2 min. The homogenate was centrifuged at 110,000 × g for 1 h and resuspended in 50 mm Tris/HCl, 500 mm NaCl, pH 7.4. Membranes were frozen in liquid nitrogen and stored at −80 °C until further use.
C12E9 was added to 1% (w/v), and membranes were solubilized by gentle agitation for at least 1 h. The solution was cleared by centrifugation (110,000 × g for 30 min) and filtration (0.8 μm) and then applied to a StrepTactin high capacity affinity column (IBA, Germany). 5-HT3R was allowed to attach to the column overnight by continuous flow, then washed with 2 column volumes of 50 mm Tris/HCl, 125 mm NaCl, 0.010% (w/v) C12E9, pH 7.4, and eluted in 3 column volumes of the same buffer supplemented with 5 mm d-desthiobiotin. For most applications, purified 5-HT3R was concentrated to 2 mg/ml using a 100,000 molecular weight cutoff ultrafiltration unit (Millipore). Protein concentration was determined by A280 absorbance (Nanodrop 2000C, Thermo Scientific) assuming an A280 of 1 = 1 mg/ml protein and/or with a BCA protein assay (Thermo Scientific) containing 0.1% Triton X-100, using BSA as a standard. Phospholipid concentration was determined with the phospholipid C assay kit (Wako Diagnostics) using choline as a standard (assuming 0.54 mg/ml choline = 3 mg/ml phospholipids).
Reconstitution of 5-HT3R into Proteoliposomes
5-HT3R was reconstituted from a solution of C12E9 into asolectin proteoliposomes using Bio-Beads to remove detergent (43). All steps were performed at 4 °C or on ice. A ternary mixture of 5-HT3R, detergent, and lipids was created by mixing C12E9, asolectin, and purified 5-HT3R using a detergent concentration of 1% (w/v), a detergent-to-lipid ratio of 27 (w/w), and a lipid-to-protein ratio of 8 (w/w). Bio-Beads SM-2 were added to the ternary mixture with a bead-to-detergent ratio of 10 in three steps as follows: (i) 2 volumes of Bio-Beads were added followed by incubation of 30 min at room temperature; (ii) 4 volumes of Bio-Beads were added followed by incubation overnight at 4 °C; (iii) 1 volume of Bio-Beads was added followed by incubation for 1 h at 4 °C. Proteoliposomes were collected by filtration over a disposable column, centrifuged at 110,000 × g, and resuspended at 8 mg/ml in 50 mm Tris/HCl, 125 mm NaCl, pH 7.4.
To test which components (type of lipids, the bilayer itself, or cytosolic proteins like chaperones) contribute to 5-HT3R thermostability, we also recreated conditions of extraction and purification using cytosolic extract and lipids from native membranes. A cell homogenate from nonexpressing cells was made as described above (production of purified 5-HT3R), using 15 ml of buffer/g of cells. After centrifugation, the membrane fraction (containing native lipids) and supernatant (named cytosolic extract) were collected. The cytosolic extract was filtered through a 0.8-μm diameter membrane filter before further use. Using the same ratios as described above, a ternary mixture and proteoliposomes were reconstituted with native membranes and lipids. A reconstituted cell homogenate was created by mixing 1 volume of proteoliposomes with 3 volumes of CHO cell extract.
Thermostability Probed by Radioligand Binding
A radioligand binding-based thermal stability assay was adapted according to Serrano-Vega et al. (38). Samples were kept on ice, unless stated otherwise. Aliquots of 100 μl were incubated for 30 min at temperatures between 25 and 80 °C in increments of 5 °C. A control sample was kept on ice all the time. The remaining activity was determined by radioligand binding at 4 °C (described below). Radioligand binding data were normalized by comparing with the untreated sample. Data analysis was done by plotting the fraction of normalized ligand binding versus the incubation temperature using the Igor Pro software package (Wavemetrics) and fitting the data with a sigmoid curve, assuming the apparent Tm equals the inflection point of the curve as shown in Equation 1,
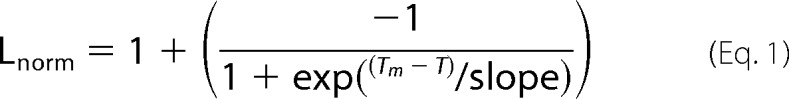 |
In which Lnorm is normalized (radio-) ligand binding; T is the incubation temperature, and Tm is the apparent melting temperature.
Radioligand Binding
Radioligand binding was performed as described elsewhere (42), with minor changes. Samples were diluted in 1 ml of 10 mm HEPES, pH 7.4, in the presence of 0.4 nm [3H]GR65630 and equilibrated for 1 h at 4 °C. For solubilized samples, 0.015% (w/v) C12E9 was included. Free radioligand was removed from the samples by rapid filtration under vacuum through GF/B glass fiber filters (Whatman), pretreated for 15 min in 0.5% (w/v) polyethyleneimine, followed by 3-fold washing with 3 ml of ice-cold 10 mm HEPES, pH 7.4. Filters were transferred to scintillation vials, filled with 4 ml of Ultima Gold (PerkinElmer Life Sciences), and radioactivity was counted with a Tri-Carb 3100 TR liquid scintillation analyzer (Packard Instrument Co.). Nonspecific binding was determined in the presence of 1 μm quipazine. Radioligand binding assays were performed in duplicate or triplicate.
Fluorescence Spectroscopy
Fluorescence spectra were recorded on a Cary Eclipse spectrophotometer (Varian) using 2 × 10-mm2 quartz cuvettes. Sample temperature was controlled with a Peltier heating element. For tryptophan fluorescence, purified 5-HT3R was diluted to 150 μg/ml in KPi-buffer (10 mm potassium phosphate, pH 7.4, with 0.025% C12E9), and in a closed cuvette the remaining air was replaced by purging with argon. Samples were excited at 290 nm, and emission spectra were taken from 300 to 500 nm with a 5-nm slit width. Spectra were taken from 5 to 80 °C in 5 °C increments. The temperature was set manually, and samples were allowed to equilibrate for 5 min before measuring a new spectrum. All spectra were corrected for background fluorescence and Raman scattering by subtracting a buffer-only spectrum. A control sample containing NATA in an equimolar amount to 5-HT3R tryptophan residues was recorded at the same time. Tryptophan quenching experiments were performed in the presence of 100 mm acrylamide. Data analysis was done using Igor Pro. Temperature-dependent changes of fluorescence intensity at the maximum emission wavelength of 5-HT3R and NATA were determined by taking a ratio (R) of the intensities at λmax of 5 and 80 °C, R = Iλ max/Iλ max +5 nm (I340/I345 nm for 5-HT3R and I356/I361 nm for NATA). When indicated, the area under the curve was determined using the areaXY function of Igor Pro.
CPM fluorescence was recorded as described elsewhere (33). Samples containing 5-HT3R at 150 μg/ml in Tris buffer were excited at 387 nm, and emission was recorded at 463 nm on a Cary Eclipse spectrophotometer (Varian). The sample was heated using a Peltier element at 2 °C/min from 4 to 80 °C in 500-μl quartz cuvettes. The inflection point of a sigmoid curve was not a good approximation for the melting temperature midpoint. Therefore, the apparent Tm was determined by taking the temperature corresponding to the half-maximum intensity.
Far-UV Circular Dichroism
Purified 5-HT3R was diluted in KPi-buffer (10 mm potassium phosphate, pH 7.4, with 0.025% (w/v) C12E9) to a concentration of 150 μg/ml. When the stock concentration of the protein was smaller than 2 mg/ml, the sample was dialyzed for several hours against the same buffer to prevent high UV absorption of contaminating buffer components. Circular dichroism was measured on a 62DS CD spectrometer (Aviv Biomedical) in 1-mm pathlength quartz cuvettes. Spectra were recorded from 260 to 190 nm at a sampling interval of 1 nm, a bandwidth of 1.5 nm, and an averaging time of 5 s. Sample temperature was controlled with a Peltier heating element, and temperature dependence was measured from 5 to 80 °C in 5 °C increments. A 5-min equilibration time was sufficient after experimental observation that the signal no longer changed thereafter. All spectra were recorded in triplicate and were background-corrected with buffer. Spectra were smoothed by Savitzky-Golay filtering with 7 points and a second order polynomial using Igor Pro and converted to molar ellipticity ϴ (44).
Near-UV Circular Dichroism
Purified 5-HT3R was diluted in KPi-buffer to a concentration of 325 μg/ml, and near-UV CD spectra were recorded on a Peltier-controlled 62DS CD spectrometer in 1-cm pathlength quartz cuvettes at temperatures ranging from 10 to 80 °C in 10 °C increments. Twenty spectra were summed and averaged, each recorded from 320 to 250 nm with a sampling interval of 2 nm and an averaging time of 5 s. Samples were equilibrated for at least 5 min each time a new temperature set point was reached. All spectra were corrected for background and were smoothed by Savitzky-Golay filtering with 7 points and a second order polynomial in Igor Pro software.
Blue Native-PAGE
Blue native-PAGE was performed as described elsewhere (45–47). Pre-casted 5–20% gradient gels (Bio-Rad) were loaded with 10 μg of 5-HT3R in blue native sample buffer (final concentrations 10% glycerol, 0.2% Coomassie Brilliant Blue G-250, and 20 mm 6-aminohexanoic acid). Electrophoresis cathode buffer consisted of 50 mm Tricine, 15 mm BisTris/HCl, 0.002% Coomassie, pH 7, and anode buffer of 50 mm BisTris/HCl, pH 7. Electrophoresis was done at 4 °C, starting at 25 V for 20 min, until all samples entered into the gel, and continued at 125 V for at least 2 h, until the bands were practically immobilized by the gel matrix. Excess Coomassie dye from detergent micelles without protein was removed by destaining, and protein bands were visualized again by normal staining. Oligomers of BSA were used as a molecular weight marker. Densitometric analysis of BN-PAGE bands was performed using ImageJ and fitted using Igor Pro.
Transmission Electron Microscopy
For EM, samples of untreated and heat-treated 5-HT3R were diluted to 20 μg/ml. 5 μl was adsorbed for 60 s to glow-discharged Parlodion carbon-coated copper grids. The grids were then blotted, washed on five drops of double distilled water, and negatively stained on two droplets of 2% uranyl acetate, pH 4.3, solution. Samples were imaged at a nominal magnification of ×64,000 and ×92,000 using a Philips CM10 electron microscope (Philips, Eindhoven, the Netherlands) operating at 80 kV. Electron micrographs were recorded on a 2000 × 2000 pixel charge-coupled device camera (Veleta, Olympus soft imaging solutions GmbH, Münster, Germany) mounted in the 35-mm port of the electron microscope, resulting in a final pixel size of 0.74 and 0.52 nm on the specimen level.
RESULTS
Thermostability at Different Stages of 5-HT3R Purification
Thermostability of 5-HT3R was measured at different stages during solubilization, purification, and reconstitution in lipid bilayers. The function of the receptor was probed by a thermostability assay based on radioligand binding (38), which allows heat perturbation of small samples with a different composition of membranes, lipids, and detergents. The binding activity of 5-HT3R for a particular antagonist at different incubation temperatures could be fitted with a sigmoid curve describing the thermotropic transition of the receptor by a two-state model from a folded to an unfolded state (Fig. 1). From the inflection of the sigmoid curve, an apparent Tm can be determined, signifying the temperature at which 50% of ligand binding is lost.
FIGURE 1.

Thermostability of 5-HT3R at different stages of purification measured by radioligand binding. Samples containing 5-HT3R were collected after extraction with C12E9, purification, and reconstitution into lipid bilayers, and after a 30-min thermal challenge, the remaining activity was determined by radioligand binding at 4 °C. A, thermostability during purification from cell homogenate, membrane fraction, and solubilized membranes to purified 5-HT3R. B, thermostability during reconstitution (Rec.) from purified 5-HT3R, ternary mixture, and proteoliposomes to cell homogenate. C, apparent Tm of indicated samples.
The Tm of 5-HT3R in plasma membranes is 70 °C and drops by 17 °C when the receptor is detergent-solubilized and affinity-purified (Fig. 1A). Addition of detergent alone leads to a drop of 11 °C; washing away lipids and other membrane components leads to a further drop in Tm by 6 °C. This process is only partly reversible; recreating the membrane environment by first adding asolectin lipids (ΔTm = 3 °C) and then removing detergent (ΔTm = 8 °C) resulted in 5-HT3R proteoliposomes with a final Tm of 63 °C (Fig. 1C).
To rule out any influence of cytosolic chaperones on protein stability, the thermostability of 5-HT3R in the membrane and proteoliposome fractions was also determined in the presence of a cytosolic cell extract. We found that addition of a cellular cytosolic fraction had no influence on the Tm of 5-HT3R in proteoliposome or membrane fractions. The effect of the lipid composition on 5-HT3R thermostability was tested by replacing asolectin lipids for native lipids from a CHO membrane fraction (not containing 5-HT3R). However, this detergent-solubilized extract of native lipids did not change the melting temperature either of the ternary mixture (5-HT3R, lipids, and detergent), or of the proteoliposomes, or of the reconstituted homogenate fractions (results not shown).
Thermostability of Purified 5-HT3R Investigated by Spectroscopic Methods
Detergents are typically used to keep membrane proteins in solution. C12E9 was used here because it was reported to be the optimal detergent for 5-HT3R solubilization, while retaining ligand binding (42). Most detergents have a cloud point (the temperature at which detergent separates into a detergent-rich and a detergent-poor phase, giving the solution a turbid, cloud-like appearance) that could interfere with thermal unfolding. The documented cloud point of C12E9 is 88 °C (48, 49), which is lowered by a few degrees in the presence of physiological concentrations of sodium chloride (49). We routinely found the cloud point between 80 and 85 °C, after visually confirming a sudden increase of sample turbidity, effectively setting the upper limit of heating detergent-containing solutions to 80 °C. Additionally, the critical micelle concentration and the aggregation number of detergents per micelle are dependent on temperature. We determined that the critical micelle concentration of C12E9 increased from ≈10 to ≈20 μm during heating from room temperature to 65 °C (results not shown). In this study, the concentration of C12E9 was kept always ≥250 μm, well above the critical micelle concentration threshold.
A single 5-HT3R subunit contains 13 Trp residues of which 9 are located in the extracellular domain, 2 in the transmembrane domain, and 2 in the intracellular domain. During thermal unfolding of the 5-HT3R, aromatic tryptophan residues buried inside the protein are expected to become water-accessible, leading to a shift of the protein's intrinsic fluorescence spectrum toward long wavelengths. This is best visualized by the intensity-normalized spectra and their difference with respect to the 5 °C spectrum, taking the fluorescence spectra of N-acetyltryptophanamide (NATA) as reference (Fig. 2, A and B). Because we were interested in a red shift of the wavelength at maximum intensity, λmax, we have plotted the temperature dependence of the fluorescence intensity ratio Rrec = I340/I345 of the receptor in Fig. 2C, which corresponds to the λmax at 5 and 80 °C (i.e. 340 and 345 nm). Rrec started to decrease around 50 °C until it reached a plateau at around 70 °C; fitting these changes by a two-state unfolding model resulted in an apparent Tm of 63.7 ± 2.8 °C. Contrary to the temperature-induced red shift of the fluorescence spectra of 5-HT3R, the shape of the intensity-normalized fluorescence spectra of the water-soluble small NATA molecule remained almost constant with a maximum at 360 nm and only tiny intensity changes below 350 nm. If we take an analogy to the case of 5-HT3R, an intensity ratio RNATA = I356/I361 surrounding λmax and plot it as a function of temperature, the NATA ratio remains constant over the entire temperature range (Fig. 2C).
FIGURE 2.

Thermostability of 5-HT3R and NATA in C12E9 observed by intrinsic fluorescence. A and B, spectra (290 nm excitation) were measured after a 5-min incubation at increasing temperatures (insets). Intensity (maximum amplitude)-normalized spectra of 5-HT3R and NATA, respectively, are shown in different colors for 5–80 °C in 5 °C increments. The difference spectra were obtained by subtracting from an intensity-normalized spectrum of a certain temperature the spectrum of 5 °C. C, temperature dependence of the wavelength shift of λmax monitored by the intensity ratio Rrec = I340/I345 for the 5-HT3R and RNATA = I356/I361 for NATA. The sigmoidal two-state fit yields an apparent Tm = 63.7 ± 2.8 °C for 5-HT3R unfolding. D, left axis, relative development of fluorescence intensity (spectral area 300–500 nm) of 5-HT3R and NATA with temperature from the original data shown in the inset of A and B normalized to 1 at 5 °C; right axis, ratio of normalized intensities (5-HT3R/NATA, data from left axis).
Next, we investigated the temperature dependence of the intrinsic fluorescence of 5-HT3R and NATA. In both cases, the fluorescence intensity decreases with increasing temperature (Fig. 2, A and B). To extract the influence of the receptor's structure and structural changes on the fluorescence intensity temperature profile, we first plotted the fluorescence intensities (total area under the peak) of 5-HT3R and NATA as a function of temperature, normalized to identical intensities at 5 °C (Fig. 2D). Although the intensity of NATA decreases hyperbolically with increasing temperature, the intensity profile of 5-HT3R shows a clear break between 50 and 60 °C. This becomes most obvious when plotting the temperature dependence of the ratio of intensities of the receptor relative to NATA, Ri = Irec/INATA. First, this ratio shows that the Trp residues (and to a minor extent the Tyr residues) giving rise to the receptor's intrinsic fluorescence are always more shielded than the small fluorophore NATA dissolved in water. The temperature profile of Ri shows two distinct regimes as follows: one linear increase with temperature between 5 and 50 °C, followed by a clear break, and then further a steeper, continuous, and slightly hyperbolic increase with temperature. The break correlates with the onset of protein unfolding detected in Fig. 2C. Apparently, the receptor's Trp and Tyr residues are better shielded to temperature-dependent quenching effects in the unfolded than in the folded state.
This correlates with the result we have obtained from an additional experiment where the intrinsic fluorescence of the 5-HT3R was quenched with acrylamide (Fig. 3, A and B). Acrylamide is often used to probe the accessibility of aromatic residues within proteins by collisional quenching (50). For 5-HT3R, the efficiency of fluorescence quenching at a particular acrylamide concentration continuously increases with elevating temperatures between 10 and 40 °C. At higher temperatures, where the protein starts to unfold, the quenching efficiency is distinctly decreased, until this transition is completed at around 70 °C, at which point the quenching efficiency increases as between 10 and 40 °C (Fig. 3B).
FIGURE 3.

Quenching of the intrinsic fluorescence of 5-HT3R. A, spectra (290 nm excitation) were measured after a 5-min incubation at increasing temperatures in the presence of 100 mm actrylamide (inset). Intensity-normalized spectra are indicated with different colors ranging from 5 to 80 °C in 5 °C increments. B, temperature dependence of the intrinsic fluorescence intensity of the receptor in the absence (I0, continuous line) and presence (Iq, dashed line) of acrylamide (left axis, data from A). Temperature dependence of the Stern-Vollmer fluorescence intensity ratio I0/Iq (right axis) is shown (points).
As is the case for tryptophan residues, cysteine residues buried in the interior of a protein (two in the ECD, two in the TMD, and three in the ICD) become more accessible upon thermal unfolding. The fluorescence intensity of the thiol-specific probe CPM increases substantially upon reacting with cysteine residues and is therefore suited for following thermal unfolding of proteins (33). 5-HT3R, which contains five free cysteine residues in the membrane and intracellular regions per receptor subunit, was heated while CPM fluorescence was monitored (Fig. 4). From the thermal profile, we determined an apparent Tm of 61 °C by taking the temperature at half-maximum fluorescence intensity.
FIGURE 4.

Thermostability of 5-HT3R in C12E9 measured by fluorescence of the cysteine-reactive CPM dye. Taking the temperature at half-maximum intensity a Tm of 61 °C was found.
Information about changes in the secondary structure of proteins during thermal unfolding can be obtained by far-UV CD spectroscopy. α-Helices, β-sheets, turns, and unordered residues contribute in different proportions to the molar ellipticities ϴ of the CD spectrum between 190 and 240 nm. The temperature dependence of the 5-HT3R CD spectrum between 190 and 240 nm was recorded in 5 °C increments (Fig. 5A). The α-helical structure, observed preferentially at 222 nm (51), was unaffected by temperature until about 40 °C, when a slow transition started, which reached a plateau around 80 °C (Fig. 5B). The presence of an isosbestic point at 201 nm indicates a two-state unfolding process. Fitting the transition with a two-state sigmoidal model results in a Tm = 60.4 ± 1.4 °C for ϴ at 222 nm in Fig. 5B and a Tm of 61.5 ± 1.9 °C for the change of ϴ at 210 nm in Fig. 5C (α-helix and β-sheet content contribute in roughly equal amounts to ϴ210). The temperature-induced changes of ϴ at 195 nm (Fig. 5D) also show a transition between 50 and 70 °C (a decrease of ϴ195 indicates a loss of both α-helix and β-sheet content and a concomitant increase of unordered conformations), which we did not fit to a two-state model due to increased scattering of data values at wavelengths lower than 200 nm.
FIGURE 5.

Thermostability of 5-HT3R in C12E9 measured by far-UV circular dichroism. A, after a 10-min equilibration, CD spectra were recorded between 5 and 80 °C in 5 °C increments. B–D, temperature-induced change of the molar ellipticity ϴ at 222, 210, and 195 nm, respectively. Transition temperatures, Tm = 60.4 ± 1.4 °C (ϴ222) and 61.5 ± 1.9 °C (ϴ210), were obtained by fitting the corresponding thermograms with a two-state unfolding model.
To probe the tertiary structure of 5-HT3R, we measured near-UV CD spectra of the protein between 250 and 320 nm (Fig. 6) originating from the protein's aromatic amino acid side chains (Trp, Tyr, and Phe). At temperatures between 10 and 50 °C, several distinct peaks were observed (around 264, 280–282, and 296 nm; Fig. 6A), which suggest a defined tertiary structure of our receptor protein (52, 53). At temperatures T ≥ 60 °C, the spectra have lost these peaks, indicating a loss of defined tertiary structure (Fig. 6A). The major optical changes occur at temperatures between 50 and 70 °C (Fig. 6B), coincident with the changes observed before in the far-UV CD spectra (Fig. 5).
FIGURE 6.

Temperature dependence of 5-HT3R in C12E9 measured by near-UV circular dichroism. A, after a 5-min equilibration, CD spectra were recorded between 10 and 80 °C in 10 °C increments. B, development of CD signal with increasing temperature at three different wavelengths (264, 280, and 296 nm), corresponding roughly to the signal of phenylalanine, tyrosine, and tryptophan, respectively.
It has been shown that ligand binding can improve the thermostability of certain receptor proteins (33). Here, we used CD to investigate the influence of the antagonist granisetron (54) on the thermostability of 5-HT3R. At 5 °C, binding of granisetron to 5-HT3R did not change the receptor's CD spectrum (Fig. 7A). But when melting of 5-HT3R was observed at ϴ222 and fitted with a sigmoid curve, the resulting Tm indicated a stabilization of 5-HT3R by granisetron of about 2 °C (Fig. 7B).
FIGURE 7.

Thermostability of 5-HT3R in C12E9 in the absence or presence of granisetron measured by circular dichroism. A, CD spectra of nonliganded and granisetron-liganded 5-HT3R taken at 5 °C are identical. B, change in ϴ222 upon heating of 5-HT3R yields a Tm = 60.7 ± 1.6 °C and 62.4 ± 1.5 °C for nonligand and liganded 5-HT3R, respectively.
Aggregation of 5-HT3R after Thermal Perturbation
Melting of the 5-HT3R results in the irreversible loss of function when measured by ligand binding, and clear changes in its structure can be deduced from spectroscopic measurements. Of considerable interest for membrane protein thermodynamics is the reversibility of the unfolding process and, related to that, the aggregation of unfolded proteins. Large aggregates of unfolded protein can usually be detected by an increase of sample turbidity or can be collected after centrifugation. But when 5-HT3R was melted, the turbidity of the samples did not change visually (until the cloud point of C12E9 was reached), and no pellet was detected after centrifugation.
In this context, we analyzed the state of aggregation of the 5-HT3R by EM and BN-PAGE. Samples of negatively stained 5-HT3R were compared with transmission electron microscopy before and after a 30-min incubation at 80 °C. Before heating, the sample appeared homogeneous in EM images, with a clearly detectable pentameric 5-HT3R top view, and it contained only a tiny amount of aggregates (Fig. 8A). After heating, the pentameric top views of 5-HT3R were lost, and 20–50-nm-sized protein aggregates were formed, containing several 5-HT3R pentamers (Fig. 8B, inset).
FIGURE 8.

Temperature-induced aggregation of 5-HT3R observed by transmission electron microscopy of negative-stained samples. Preparations containing 20 μg/ml 5-HT3R in C12E9 were imaged before (A) and after (B) incubation for 30 min at 80 °C. The insets show enlarged versions of images (A and B). White arrows in the inset of A indicate top views of 5-HT3R, and arrows in the inset of B show aggregated 5-HT3R. Scale bar represents 200 nm (50 nm for insets).
Aggregation of 5-HT3R was investigated with BN-PAGE, which has the potential to detect up to megadalton-sized objects. Samples containing 10 μg of 5-HT3R were incubated at increasing temperatures (similar as Fig. 1) and separated by BN-PAGE (Fig. 9A). When the incubation temperature increased above 50 °C, first a transient increase of dimers was observed, followed by trimers and tetramers of 5-HT3R. At more elevated temperatures, the fraction of mono- and di-pentameric species of 5-HT3R was further reduced, and larger oligomers were formed that reached a size that effectively excluded them from entering the gel matrix. By using densitometric analysis of BN-PAGE bands, a transition temperature for aggregation could be calculated at 54.3 °C for the loss of mono-pentameric 5-HT3R and at 58.5 °C for the formation of large aggregates (Fig. 9B).
FIGURE 9.

Temperature-induced aggregation of 5-HT3R visualized by BN-PAGE. A, 40-μl samples containing 10 μg of 5-HT3R in C12E9 were incubated for 30 min at temperatures ranging from 25 to 80 °C and subsequently applied to a 5–20% gradient gel. At T ≥50 °C, oligomers of 5-HT3R pentamers are formed. Oligomers of BSA were used as size markers (left axis); the calculated molecular mass of the 5-HT3R pentamer is 263 kDa. B, densitometric analysis of the mono-pentameric and higher aggregate fractions of 5-HT3R pentamers of the BN-PAGE in A. The fractions of monomers (1-mer) or higher aggregates (2-, 3-, 4-, and >4-mers) of the total amount of 5-HT3R pentamers were plotted against the temperature. Fits to a two-state model yield a Tm = 54.3 and 58.5 °C for loss of monomers and formation of aggregates, respectively.
DISCUSSION
In this study, several methods were combined to measure the stability of a pentameric ligand-gated ion channel, the 5-HT3 receptor. The results presented here show a unique picture of how stability is dependent on external membrane-related factors as well as internal structure (and ultimately function)-related factors.
Ligand Binding Activity of the 5-HT3R Is Modulated by the State and Composition of the Lipid Bilayer Membrane
Although the ligand binding site(s) of the 5-HT3R is (are) predicted to be located at the interfaces between the β-sheet-structured extracellular regions of the subunits (3), it is striking that the thermostability of the receptor's ligand binding activity very sensitively depends on the particular environment of its helical transmembrane regions. In the cellular plasma membrane, ligand binding of the receptor remains active up to 70 °C (Fig. 1 and Table 1). Already the solubilization of the plasma membrane by adding detergent shifts the ligand binding thermostability by 10 °C to lower temperatures. Purification of the receptor in a detergent-solubilized form (i.e. removing all native lipids and other membrane components) finally shifts the ligand binding thermostability to 53 °C, in total down by 17 °C from its state in native plasma membranes. The thermostability can be partly restored to 63 °C by reconstituting the receptor into membranes, either from the purified form into artificial lipid bilayers or from the detergent-solubilized plasma membrane mixture. This shows that the lipid bilayer contributes by about 10 °C to the thermostability of both down- and upward shifts. The fact that thermostability was not fully reconstituted, even from a plasma membrane detergent solution comprising all original membrane lipids, indicates that further contributions to thermostability originate from interactions of the 5-HT3R with other membrane components in the highly crowded cellular environment, which is impossible to reconstitute under the conditions used (maximum 11% 5-HT3R by weight).
TABLE 1.
Melting temperatures (Tm) of 5-HT3R unfolding under different conditions
Tm values (except for CPM fluorescence) indicating a 50% unfolding are obtained by fitting corresponding thermograms with a two-state model. Standard deviations are indicated when relevant.
| Method of analysis | 5-HT3R preparation | Tm |
|---|---|---|
| °C | ||
| Radioligand bindinga | Cell homogenate | 69.5 ± 0.8 |
| Radioligand bindinga | Plasma membranes | 69.4 ± 0.5 |
| Radioligand bindinga | Solubilized plasma membranes | 59.2 ± 0.6 |
| Radioligand bindinga | Purifiedb | 53.2 ± 0.3 |
| Radioligand bindinga | Ternary mixturec | 55.7 ± 0.6 |
| Radioligand bindinga | Proteoliposomesd | 62.9 ± 1.0 |
| Radioligand bindinga | Reconstituted homogenatee | 62.8 ± 0.5 |
| Intrinsic protein fluorescencef | Purifiedb | 63.7 |
| CPM fluorescenceg | Purifiedb | 61 |
| Circular dichroism, ϴ208 | Purifiedb | 59.5 ± 1.3 |
| Circular dichroism, ϴ210h | Purifiedb | 61.5 ± 1.9 |
| Circular dichroism, ϴ215 | Purifiedb | 61.4 ± 2.2 |
| Circular dichroism, ϴ222h | Purifiedb | 60.4 ± 1.4 |
| BN-PAGE, detecting monomersi | Purifiedb | 54.3 |
| BN-PAGE, detecting aggregatesi | Purifiedb | 58.5 |
a See Fig. 1C.
b Purified receptor was solubilized in C12E9.
c Purified receptor in mixed micelles composed of C12E9 and asolectin.
d Purified receptor was reconstituted in asolectin lipid vesicles.
e Data shown are after removing C12E9 from the solubilized plasma membranes.
f See Fig. 2.
g See Fig. 4.
h See Fig. 5.
i See Fig. 9.
From the study of the thermostability of the 5-HT3R, the following important lessons can be learned. (i) The thermal stability of the receptor's extracellular ligand binding region is coupled via the receptor's transmembrane helical region to the lipid bilayer membrane. Apparently, the transmembrane part serves as a structural template for the extracellular portion of the LGIC. (ii) The contribution of the lipid membrane can be distinguished from other influences such as receptor-protein interactions.
5-HT3R Shows a Higher Thermal Stability than Most Other Membrane Proteins Investigated
As the lipid bilayer and the detergent micelle serve like a solvent for the transmembrane part, the situation of the LGIC's function can be compared with the situation of water-soluble proteins, for which structural fluctuations important for folding and function follow solvent fluctuations (55). This view is further supported by our investigations of the thermal unfolding of the 5-HT3R probed by the structure-sensitive techniques discussed below.
The role of distinct lipids for the allosteric activation of LGIC is well documented for several forms of the homologous nAChR (26). Although it is generally known that detergents form a poor substitute of the lipid bilayer in terms of retaining the protein's stability (27), the experimental proofs thereof are poorly documented. For instance, from differential scanning calorimetry thermograms, a stabilization of 20 °C was found for bacteriorhodopsin in its trimeric state in native purple membranes (Tm = 100 °C) compared with its detergent-solubilized monomeric state (14). In another case, the structural stability of the β-barrel bacterial outer membrane protein OmpA was coupled to the properties of a lipid bilayer (56). The thermal stability of the ligand binding activity of the 5-HT3R is quite high compared with that of other membrane protein classes such as G protein-coupled receptors that typically are observed in a temperature range of 23–40 °C in detergent-solubilized form (34, 36).
As a consequence of the destabilizing effects of detergents on membrane proteins, the first and most important step in extraction and purification of membrane proteins is the choice of detergent. Hence, considerable attention has been given elsewhere for finding detergents that keep membrane proteins stable (33, 57–59). Here, we have used C12E9 because it was found before to be the most suitable detergent for 5-HT3R extraction while retaining the receptor's ligand binding activity (42). For the homologous nAChR, C12E9 was also found to be among the most lipid-retaining detergents (57). Here, we found that lipids stabilize 5-HT3R directly (Fig. 1 and Table 1), suggesting that lipid retention is an important factor for stabilizing membrane proteins in detergent micelles. In fact, a specific composition of lipids was found to stabilize the nAChR (60), which is routinely purified in the presence of small amounts of lipids.
Apparent melting temperatures of Tm = 60–62 °C have been reported for nAChR from calorimetric thermograms and bungarotoxin binding (61, 62). Using Fourier transform infrared spectroscopy, a melting temperature of 56 °C was found for nAChR in phosphatidylcholine/phosphatidic acid/cholesterol lipids that was sensitive to the lipid composition and the presence of agonist carbamylcholine (60). Circular dichroism studies found melting transitions of acetylcholine-binding proteins (AChBP), at Tm = 65 °C for Lymnaea stagnalis AChBP and at Tm = 50–55 °C for Bulinus truncatus AChBP (63). For purified 5-HT3R in C12E9, we determined a Tm of 61 °C using circular dichroism and 64 °C from intrinsic protein fluorescence (Table 1). For the ligand binding activity of detergent-solubilized 5-HT3R, we found a Tm of 59 °C, which dropped to 53 °C after plasma membrane lipids were removed by affinity purification of the receptor. It is interesting to compare our Tm values for the 5-HT3R with those reported elsewhere for dodecyl maltoside-solubilized, nonpurified GPCRs as follows: Tm = 23 °C for human adenosine A2A receptor, 32 °C for turkey β1-adrenergic receptor, 40 °C for the neurotensin receptor (29 °C for apo-neurotensin), and 55 °C for rhodopsin (34, 36).
5-HT3R Increases Conformational Flexibility with Temperature before Unfolding
For thermostability determined by CPM fluorescence, only few examples have been reported, with Tm values of 42 °C for the human adenosine A2A receptor (64) and 45 °C for the human apelin receptor (33). Under comparable circumstances, our CPM fluorescence melting curve for the 5-HT3R delivers a Tm of 61 °C (Fig. 4). However, the continuous melting curve does not resemble a sigmoid-like transition, as is common in other experiments reported here and elsewhere. Instead, next to a sigmoid-like transition centered around 60 °C, a second component can be observed that increases continuously with temperature until about 55 °C. This continuous component resembles the broad thermal conformational changes below the Tm observed by CD and intrinsic protein fluorescence. The increase of cysteine accessibility and the decrease of helical content must be correlated with increased structural fluctuations that parallel the continuous thermal volume expansion of proteins (65). A correlation between internal conformational fluctuations and CD helix content was shown for short transmembrane polypeptides elsewhere (66). A further increase of temperature above 55 °C induces a discontinuous volume change correlated with structural unfolding of 5-HT3R, which resembles a sigmoid transition scaling with the loss of ligand binding (Fig. 1A) and the formation of higher oligomers (Fig. 9). Obviously, with increasing temperatures, the receptor explores an enlarging conformational landscape, which eventually induces the protein to unfold.
During Unfolding the 5-HT3R Adopts Consecutively Distinct Structural States
Heating of 5-HT3R ultimately leads to the formation of irreversible aggregates (Figs. 8 and 9). With increasing temperature, aggregates, in the form of higher oligomers, can be distinguished both visually (Fig. 8B) and quantitatively from the densitometric analysis (Fig. 9B). The loss of monomeric 5-HT3R pentamers is accompanied by a transient increase of di-pentameric 5-HT3R, reaching a maximal concentration around 50 °C. At incubation temperatures higher than 50 °C, there is a further loss of the monomeric form of 5-HT3R pentamers as well as a loss of the dimeric form of 5-HT3R pentamers, but now trimeric and tetrameric forms of 5-HT3R pentamers appear transiently with maximal concentration around 55 °C. At even higher temperatures, high molecular mass aggregates appear at the cost of disappearing monomeric and lower oligomeric forms of the 5-HT3R pentamers. Still, as shown by electron microscopy, a large fraction of aggregates remain at a small size (inset, Fig. 8B), probably due to the presence of detergent. These small aggregates (around 20 nm) do not influence spectroscopic measurements in the form of light-scattering artifacts.
For the 5-HT3R in detergent solution, intrinsic protein fluorescence, CPM fluorescence, and circular dichroism, which all report on changes of the protein structure, show a melting temperature around 61 °C, with changes occurring over a temperature range of more than 20 °C. Contrastingly, the radioligand binding thermostability assays reflect an irreversible loss of binding function within a small temperature range of about 5 °C with a Tm of 53 °C for 5-HT3R in C12E9. Apparently, subtle structural changes are sufficient to lead to the receptor's loss of ligand binding activity at a relatively low temperature in detergent solution, and the conformation-sensitive probes sample a mean overall structural change composed of successive different structural transitions. As the ligand-binding site is proposed to be located at the interface between protein subunits in the extracellular region, the thermal stability of ligand binding intrinsically reflects interactions between subunits of the 5-HT3R.
The loss of ligand binding during thermal unfolding of 5-HT3R (Fig. 1 and Table 1) is highly correlated with a sharp loss of monomeric 5-HT3R pentamers (Fig. 9B), whereas the appearance of large aggregates correlates with Tm values detected by structure-sensitive optical techniques (CD, intrinsic protein fluorescence, and CPM reactivity for cysteine residues). Radioligand binding assays and BN-PAGE clearly detect distinct and sharp consecutive structural transitions of 5-HT3R during thermal unfolding, whereas the structure-sensitive optical techniques sampled over these transitions reveal the following: (i) the overall average conformational changes are spread over a broad temperature range, and (ii) the major conformational change of the α-helical transmembrane region occurs at a higher temperature than the loss of ligand binding.
Of interest in this context is a recent study suggesting a loose packing of the extracellular ligand binding domains of pentameric. LGIC; this structural evolution is supposed to lead to increased flexibility of the extracellular domains, which should facilitate agonist-induced channel activation (63, 67). Another study shows the important role of the C-terminal helix (TM4) in the global stability of pentameric LGIC's: glycine and 5-HT3 receptors lacking the TM4 form oligomers of random stoichiometry and can only be rescued by the presence of a TM4 peptide (68). The membrane-spanning helical bundles might act as a template to keep the loosely packed extracellular domain together (Fig. 10), as indicated in our case by the increased thermostability of the ligand binding activity of the 5-HT3R in native and reconstituted membranes.
FIGURE 10.

Topological model of the 5-HT3AR subunit folding. Highlighted are antiparallel β-sheets (blue arrows), α-helices (red boxes) including membrane-spanning helices (M1 to M4), N-glycosylation sites (brown) (70), pore-lining residues (red) (3), cysteine residues, including the predicted Cys loop disulfide bridge (orange), the tryptophan residues (magenta), and the location of the lipid bilayer (gray). Secondary structure is assigned based on prediction algorithm PROMALS3D (71), including helix X. Helix MA is predicted to extend transmembrane helix M4 (with analogy to the electron microscopy-based structural model of the nAChR) (11).
Taken together, our results show an uncoupling between unfolding of the extracellular domain and the membrane-spanning domain of the 5-HT3R in detergents. Unfolding would start at the β-sheet-rich extracellular domain where the ligand-binding site is located (3, 69), with a subsequent onset of aggregation when the extent of unfolding proceeds toward the α-helical membrane domain. It should be noted that even at 80 °C protein unfolding is not complete, as 80% of the α-helices are still intact; a similar trend is often observed during thermal unfolding of helical membrane proteins (14, 20). A recent study showed this exceptional thermostability by using a helical polyleucine peptide inserted into synthetic lipid vesicles. No loss of helical structure was observed by CD over a temperature range between 45 and 85 °C (22). Our CD data of the 5-HT3R are consistent with a folding model of the protein subunit in a lipid membrane depicted in Fig. 10. It is reasonable to speculate that a major part of the 20% decrease of the helical content seen in the change of the CD signal at ϴ222 during thermal unfolding concerns the predicted extra-membranous helical portions of the receptor.
Conclusion
In the last decade, protein thermostability is increasingly appreciated for its role in structure determination by crystallization (27, 30). Especially for proteins that have proven difficult to crystallize, like membrane proteins, the use of thermostable proteins could improve the chance of successful crystallization (31, 34, 38). Our study concerns the effect of detergent solubilization and purification on the thermostability of the 5-HT3R. We show that a lower thermostability can be attributed in a major part to loss of the lipid bilayer and additionally to the loss of individual constituents of the lipid bilayer, like certain lipids and other membrane components. These results restate the importance of detergent screening for membrane proteins or, vice versa, the thermostabilization of membrane proteins for detergents.
It is also shown that a majority of the methods used for the analysis of 5-HT3R thermostability in detergent solution result in similar melting temperatures. In comparison with other membrane proteins in their native state, for example GPCRs, the 5-HT3R (and by extrapolation other pentameric LGICs) shows increased thermostability. Because the ligand-binding domain of 5-HT3R proceeds to unfold before the helical membrane domain (this study), and because extracellular domains of pentameric LGICs are predicted to have a loose hydrophobic packing (67), we propose that the origin of the increased stability of LGICs over GPCRs must be searched in the superior packing of the transmembrane helices of LGICs compared with GPCRs. Another conclusion that can be drawn is that in the presence of detergent 5-HT3R is more likely to aggregate irreversibly at higher temperatures.
This study shows a correlation between the presence of detergents, the irreversible formation of aggregates, and protein thermostability; this points to the importance of screening for enhanced thermostability for the crystallizability of membrane proteins (29, 33–37). Here, we reveal for the first time that thermal unfolding of a multisubunit, multidomain, and multifunctional membrane protein such as the 5-HT3R occurs via hierarchical consecutive structural transitions, which can be distinguished experimentally using different approaches to probe different properties of the receptor.
This work was supported by the European Community's Seventh Framework Program FP7/2007-2013 under Grant 211800, the Swiss National Science Foundation (the SystemsX program and SNF Grant 31003A-118148), and internal funds of the Ecole Polytechnique Fédérale de Lausanne.
- 5-HT3R
- 5-hydroxytryptamine (serotonin) receptor type 3A
- nAChR
- nicotinic acetylcholine receptor
- LGIC
- ligand-gated ion channel
- BN-PAGE
- blue-native-PAGE
- NATA
- N-acetyl-l-tryptophanamide
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- CPM
- N-[4-(7-diethylamino-4-methyl-3-coumarinyl)-phenyl]-maleimide
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine
- GPCR
- G protein-coupled receptor
- AChBP
- acetylcholine-binding protein.
REFERENCES
- 1. Barnes N. M., Hales T. G., Lummis S. C., Peters J. A. (2009) The 5-HT3 receptor–the relationship between structure and function. Neuropharmacology 56, 273–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Walstab J., Rappold G., Niesler B. (2010) 5-HT(3) receptors: role in disease and target of drugs. Pharmacol. Ther. 128, 146–169 [DOI] [PubMed] [Google Scholar]
- 3. Thompson A. J., Lester H. A., Lummis S. C. (2010) The structural basis of function in Cys loop receptors. Q. Rev. Biophys. 43, 449–499 [DOI] [PubMed] [Google Scholar]
- 4. Lummis S. C. (2012) 5-HT3 receptors. J. Biol. Chem. 287, 40239–40245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Miller P. S., Smart T. G. (2010) Binding, activation, and modulation of Cys loop receptors. Trends Pharmacol. Sci. 31, 161–174 [DOI] [PubMed] [Google Scholar]
- 6. Tsetlin V., Kuzmin D., Kasheverov I. (2011) Assembly of nicotinic and other Cys loop receptors. J. Neurochem. 116, 734–741 [DOI] [PubMed] [Google Scholar]
- 7. Machu T. K. (2011) Therapeutics of 5-HT3 receptor antagonists. Current uses and future directions. Pharmacol. Ther. 130, 338–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bocquet N., Nury H., Baaden M., Le Poupon C., Changeux J.-P., Delarue M., Corringer P.-J. (2009) X-ray structure of a pentameric ligand-gated ion channel in an apparently open conformation. Nature 457, 111–114 [DOI] [PubMed] [Google Scholar]
- 9. Hibbs R. E., Gouaux E. (2011) Principles of activation and permeation in an anion-selective Cys loop receptor. Nature 474, 54–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hilf R. J., Dutzler R. (2008) X-ray structure of a prokaryotic pentameric ligand-gated ion channel. Nature 452, 375–379 [DOI] [PubMed] [Google Scholar]
- 11. Unwin N. (2005) Refined structure of the nicotinic acetylcholine receptor at 4 Å resolution. J. Mol. Biol. 346, 967–989 [DOI] [PubMed] [Google Scholar]
- 12. Purohit P., Auerbach A. (2010) Energetics of gating at the apo-acetylcholine receptor transmitter binding site. J. Gen. Physiol. 135, 321–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schmauder R., Kosanic D., Hovius R., Vogel H. (2011) Correlated optical and electrical single-molecule measurements reveal conformational diffusion from ligand binding to channel gating in the nicotinic acetylcholine receptor. ChemBioChem 12, 2431–2434 [DOI] [PubMed] [Google Scholar]
- 14. Haltia T., Freire E. (1995) Forces and factors that contribute to the structural stability of membrane proteins. Biochim. Biophys. Acta 1228, 1–27 [DOI] [PubMed] [Google Scholar]
- 15. Mackenzie K. R. (2006) Folding and stability of α-helical integral membrane proteins. Chem. Rev. 106, 1931–1977 [DOI] [PubMed] [Google Scholar]
- 16. Minetti C. A., Remeta D. P. (2006) Energetics of membrane protein folding and stability. Arch. Biochem. Biophys. 453, 32–53 [DOI] [PubMed] [Google Scholar]
- 17. Popot J.-L., Engelman D. M. (2000) Helical membrane protein folding, stability, and evolution. Annu. Rev. Biochem. 69, 881–922 [DOI] [PubMed] [Google Scholar]
- 18. White S. H., Wimley W. C. (1999) Membrane protein folding and stability. Physical principles. Annu. Rev. Biophys. Biomol. Struct. 28, 319–365 [DOI] [PubMed] [Google Scholar]
- 19. Heyden M., Freites J. A., Ulmschneider M. B., White S. H., Tobias D. J. (2012) Assembly and stability of α-helical membrane proteins. Soft Matter 8, 7742–7752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Harris N. J., Booth P. J. (2012) Folding and stability of membrane transport proteins in vitro. Biochim. Biophys. Acta 1818, 1055–1066 [DOI] [PubMed] [Google Scholar]
- 21. Popot J.-L., Engelman D. M. (1990) Membrane protein folding and oligomerization. The two-stage model. Biochemistry 29, 4031–4037 [DOI] [PubMed] [Google Scholar]
- 22. Ulmschneider J. P., Smith J. C., White S. H., Ulmschneider M. B. (2011) In silico partitioning and transmembrane insertion of hydrophobic peptides under equilibrium conditions. J. Am. Chem. Soc. 133, 15487–15495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rapoport T. A. (2007) Protein translocation across the eukaryotic endoplasmic reticulum and bacterial plasma membranes. Nature 450, 663–669 [DOI] [PubMed] [Google Scholar]
- 24. White S. H., von Heijne G. (2008) How translocons select transmembrane helices. Annu. Rev. Biophys. 37, 23–42 [DOI] [PubMed] [Google Scholar]
- 25. Hong H., Joh N. H., Bowie J. U., Tamm L. K. (2009) Methods for measuring the thermodynamic stability of membrane proteins. Methods Enzymol. 455, 213–236 [DOI] [PubMed] [Google Scholar]
- 26. Baenziger J. E., Corringer P.-J. (2011) 3D structure and allosteric modulation of the transmembrane domain of pentameric ligand-gated ion channels. Neuropharmacology 60, 116–125 [DOI] [PubMed] [Google Scholar]
- 27. Bowie J. U. (2001) Stabilizing membrane proteins. Curr. Opin. Struct. Biol. 11, 397–402 [DOI] [PubMed] [Google Scholar]
- 28. Tate C. G. (2010) Practical considerations of membrane protein instability during purification and crystallisation. Methods Mol. Biol. 601, 187–203 [DOI] [PubMed] [Google Scholar]
- 29. Bill R. M., Henderson P. J., Iwata S., Kunji E. R., Michel H., Neutze R., Newstead S., Poolman B., Tate C. G., Vogel H. (2011) Overcoming barriers to membrane protein structure determination. Nat. Biotechnol. 29, 335–340 [DOI] [PubMed] [Google Scholar]
- 30. Rees D. C., Robertson A. D. (2001) Some thermodynamic implications for the thermostability of proteins. Protein Sci. 10, 1187–1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mancusso R., Karpowich N. K., Czyzewski B. K., Wang D.-N. (2011) Simple screening method for improving membrane protein thermostability. Methods 55, 324–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jenney F. E., Jr., Adams M. W. (2008) The impact of extremophiles on structural genomics (and vice versa). Extremophiles 12, 39–50 [DOI] [PubMed] [Google Scholar]
- 33. Alexandrov A. I., Mileni M., Chien E. Y., Hanson M. A., Stevens R. C. (2008) Microscale fluorescent thermal stability assay for membrane proteins. Structure 16, 351–359 [DOI] [PubMed] [Google Scholar]
- 34. Magnani F., Shibata Y., Serrano-Vega M. J., Tate C. G. (2008) Co-evolving stability and conformational homogeneity of the human adenosine A2a receptor. Proc. Natl. Acad. Sci. U.S.A. 105, 10744–10749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tate C. G., Schertler G. F. (2009) Engineering G protein-coupled receptors to facilitate their structure determination. Curr. Opin. Struct. Biol. 19, 386–395 [DOI] [PubMed] [Google Scholar]
- 36. Schlinkmann K. M., Hillenbrand M., Rittner A., Künz M., Strohner R., Plückthun A. (2012) Maximizing detergent stability and functional expression of a GPCR by exhaustive recombination and evolution. J. Mol. Biol. 442, 414–428 [DOI] [PubMed] [Google Scholar]
- 37. Hattori M., Hibbs R. E., Gouaux E. (2012) A fluorescence-detection size-exclusion chromatography-based thermostability assay for membrane protein precrystallization screening. Structure 20, 1293–1299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Serrano-Vega M. J., Magnani F., Shibata Y., Tate C. G. (2008) Conformational thermostabilization of the β1-adrenergic receptor in a detergent-resistant form. Proc. Natl. Acad. Sci. U.S.A. 105, 877–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Warne T., Serrano-Vega M. J., Baker J. G., Moukhametzianov R., Edwards P. C., Henderson R., Leslie A. G., Tate C. G., Schertler G. F. (2008) Structure of a β1-adrenergic G-protein-coupled receptor. Nature 454, 486–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Derewenda Z. S. (2010) Application of protein engineering to enhance crystallizability and improve crystal properties. Acta Crystallogr. D Biol. Crystallogr. 66, 604–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tairi A. P., Hovius R., Pick H., Blasey H., Bernard A., Surprenant A., Lundström K., Vogel H. (1998) Ligand binding to the serotonin 5HT3 receptor studied with a novel fluorescent ligand. Biochemistry 37, 15850–15864 [DOI] [PubMed] [Google Scholar]
- 42. Hovius R., Tairi A.-P., Blasey H., Bernard A., Lundström K., Vogel H. (1998) Characterization of a mouse serotonin 5-HT3 receptor purified from mammalian cells. J. Neurochem. 70, 824–834 [DOI] [PubMed] [Google Scholar]
- 43. Geertsma E. R., Nik Mahmood N. A., Schuurman-Wolters G. K., Poolman B. (2008) Membrane reconstitution of ABC transporters and assays of translocator function. Nat. Protoc. 3, 256–266 [DOI] [PubMed] [Google Scholar]
- 44. Greenfield N. J. (2006) Using circular dichroism spectra to estimate protein secondary structure. Nat. Protoc. 1, 2876–2890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Heuberger E. H., Veenhoff L. M., Duurkens R. H., Friesen R. H., Poolman B. (2002) Oligomeric state of membrane transport proteins analyzed with blue native electrophoresis and analytical ultracentrifugation. J. Mol. Biol. 317, 591–600 [DOI] [PubMed] [Google Scholar]
- 46. Nicke A., Bäumert H. G., Rettinger J., Eichele A., Lambrecht G., Mutschler E., Schmalzing G. (1998) P2X1 and P2X3 receptors form stable trimers. A novel structural motif of ligand-gated ion channels. EMBO J. 17, 3016–3028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Schägger H., von Jagow G. (1991) Blue native electrophoresis for isolation of membrane protein complexes in enzymatically active form. Anal. Biochem. 199, 223–231 [DOI] [PubMed] [Google Scholar]
- 48. Chen M. L., Wang Z. W., Zhang G. X., Gu J., Cun Z., Tao F. (2007) Studies on the cloud points of nonionic surfactants with QSPR. Chem. Res. Chin. Univ. 23, 715–719 [Google Scholar]
- 49. Sharma K. S., Patil S. R., Rakshit A. K. (2003) Study of the cloud point of C12En nonionic surfactants. Effect of additives. Colloids and Surfaces A 219, 67–74 [Google Scholar]
- 50. Zelent B., Kuśba J., Gryczynski I., Johnson M. L., Lakowicz J. R. (1998) Time-resolved and steady-state fluorescence quenching of N-acetyl-l-tryptophanamide by acrylamide and iodide. Biophys. Chem. 73, 53–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Vogel H. (1987) Comparison of the conformation and orientation of alamethicin and melittin in lipid membranes. Biochemistry 26, 4562–4572 [DOI] [PubMed] [Google Scholar]
- 52. Garriga P., Liu X., Khorana H. G. (1996) Structure and function in rhodopsin. Correct folding and misfolding in point mutants at and in proximity to the site of the retinitis pigmentosa mutation Leu-125 → Arg in the transmembrane helix C. Proc. Natl. Acad. Sci. U.S.A. 93, 4560–4564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kelly S. M., Price N. C. (2000) The use of circular dichroism in the investigation of protein structure and function. Curr. Protein Pept. Sci. 1, 349–384 [DOI] [PubMed] [Google Scholar]
- 54. Maksay G. (1996) Distinct thermodynamic parameters of serotonin 5-HT3 agonists and antagonists to displace [3H]granisetron binding. J. Neurochem. 67, 407–412 [DOI] [PubMed] [Google Scholar]
- 55. Frauenfelder H., Fenimore P. W., Chen G., McMahon B. H. (2006) Protein folding is slaved to solvent motions. Proc. Natl. Acad. Sci. U.S.A. 103, 15469–15472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hong H., Tamm L. K. (2004) Elastic coupling of integral membrane protein stability to lipid bilayer forces. Proc. Natl. Acad. Sci. U.S.A. 101, 4065–4070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Asmar-Rovira G. A., Asseo-García A. M., Quesada O., Hanson M. A., Cheng A., Nogueras C., Lasalde-Dominicci J. A., Stevens R. C. (2008) Biophysical and ion channel functional characterization of the Torpedo californica nicotinic acetylcholine receptor in varying detergent-lipid environments. J. Membr. Biol. 223, 13–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sonoda Y., Newstead S., Hu N.-J., Alguel Y., Nji E., Beis K., Yashiro S., Lee C., Leung J., Cameron A. D., Byrne B., Iwata S., Drew D. (2011) Benchmarking membrane protein detergent stability for improving throughput of high resolution x-ray structures. Structure 19, 17–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Yeh A. P., McMillan A., Stowell M. H. (2006) Rapid and simple protein-stability screens. Application to membrane proteins. Acta Crystallogr. D Biol. Crystallogr. 62, 451–457 [DOI] [PubMed] [Google Scholar]
- 60. daCosta C. J., Baenziger J. E. (2009) A lipid-dependent uncoupled conformation of the acetylcholine receptor. J. Biol. Chem. 284, 17819–17825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Artigues A., Villar M. T., Ferragut J. A., Gonzalez-Ros J. M. (1987) Thermal perturbation studies of membrane-bound acetylcholine receptor from Torpedo. Effects of cholinergic ligands and membrane perturbants. Arch. Biochem. Biophys. 258, 33–41 [DOI] [PubMed] [Google Scholar]
- 62. Perez-Ramirez B. (1994) Thermal stability of Torpedo californica acetylcholine receptor in a cholesterol lipid environment. Mol. Cell. Biochem. 132, 91–99 [DOI] [PubMed] [Google Scholar]
- 63. Celie P. H., Klaassen R. V., van Rossum-Fikkert S. E., van Elk R., van Nierop P., Smit A. B., Sixma T. K. (2005) Crystal structure of acetylcholine-binding protein from Bulinus truncatus reveals the conserved structural scaffold and sites of variation in nicotinic acetylcholine receptors. J. Biol. Chem. 280, 26457–26466 [DOI] [PubMed] [Google Scholar]
- 64. O'Malley M. A., Naranjo A. N., Lazarova T., Robinson A. S. (2010) Analysis of adenosine A2a receptor stability: effects of ligands and disulfide bonds. Biochemistry 49, 9181–9189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Frauenfelder H., Hartmann H., Karplus M., Kuntz I. D., Jr., Kuriyan J., Parak F., Petsko G. A., Ringe D., Tilton R. F., Jr., Connolly M. L. (1987) Thermal expansion of a protein. Biochemistry 26, 254–261 [DOI] [PubMed] [Google Scholar]
- 66. Vogel H., Nilsson L., Rigler R., Voges K. P., Jung G. (1988) Structural fluctuations of a helical polypeptide traversing a lipid bilayer. Proc. Natl. Acad. Sci. U.S.A. 85, 5067–5071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Dellisanti C. D., Hanson S. M., Chen L., Czajkowski C. (2011) Packing of the extracellular domain hydrophobic core has evolved to facilitate pentameric ligand-gated ion channel function. J. Biol. Chem. 286, 3658–3670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Haeger S., Kuzmin D., Detro-Dassen S., Lang N., Kilb M., Tsetlin V., Betz H., Laube B., Schmalzing G. (2010) An intramembrane aromatic network determines pentameric assembly of Cys loop receptors. Nat. Struct. Mol. Biol. 17, 90–98 [DOI] [PubMed] [Google Scholar]
- 69. Schreiter C., Hovius R., Costioli M., Pick H., Kellenberger S., Schild L., Vogel H. (2003) Characterization of the ligand-binding site of the serotonin 5-HT3 receptor: the role of glutamate residues 97, 224, and 235. J. Biol. Chem. 278, 22709–22716 [DOI] [PubMed] [Google Scholar]
- 70. Quirk P. L., Rao S., Roth B. L., Siegel R. E. (2004) Three putative N-glycosylation sites within the murine 5-HT3A receptor sequence affect plasma membrane targeting, ligand binding, and calcium influx in heterologous mammalian cells. J. Neurosci. Res. 77, 498–506 [DOI] [PubMed] [Google Scholar]
- 71. Pei J., Kim B.-H., Grishin N. V. (2008) PROMALS3D. A tool for multiple protein sequence and structure alignments. Nucleic Acids Res. 36, 2295–2300 [DOI] [PMC free article] [PubMed] [Google Scholar]