

Background: Family 7 cellulases exhibit significant hydrolytic potential in cellulose degradation.
Results: We report the Heterobasidion irregulare GH7 structure and compare it with other GH7 cellobiohydrolases with simulation.
Conclusion: H. irregulare Cel7A exhibits intermediate dynamical and structural properties between Phanerochaete chrysosporium Cel7D and Hypocrea jecorina Cel7A.
Significance: These results highlight regions of family 7 cellobiohydrolases important for carbohydrate processivity and association-dissociation rates on cellulose.
Keywords: Biofuel, Cellulase, Crystallography, Glycoside Hydrolases, Molecular Dynamics, Heterobasidion annosum, Hypocrea jecorina, Phanerochaete chrysosporium, Trichoderma reesei, Cellobiohydrolase
Abstract
Root rot fungi of the Heterobasidion annosum complex are the most damaging pathogens in temperate forests, and the recently sequenced Heterobasidion irregulare genome revealed over 280 carbohydrate-active enzymes. Here, H. irregulare was grown on biomass, and the most abundant protein in the culture filtrate was identified as the only family 7 glycoside hydrolase in the genome, which consists of a single catalytic domain, lacking a linker and carbohydrate-binding module. The enzyme, HirCel7A, was characterized biochemically to determine the optimal conditions for activity. HirCel7A was crystallized and the structure, refined at 1.7 Å resolution, confirms that HirCel7A is a cellobiohydrolase rather than an endoglucanase, with a cellulose-binding tunnel that is more closed than Phanerochaete chrysosporium Cel7D and more open than Hypocrea jecorina Cel7A, suggesting intermediate enzyme properties. Molecular simulations were conducted to ascertain differences in enzyme-ligand interactions, ligand solvation, and loop flexibility between the family 7 glycoside hydrolase cellobiohydrolases from H. irregulare, H. jecorina, and P. chrysosporium. The structural comparisons and simulations suggest significant differences in enzyme-ligand interactions at the tunnel entrance in the −7 to −4 binding sites and suggest that a tyrosine residue at the tunnel entrance of HirCel7A may serve as an additional ligand-binding site. Additionally, the loops over the active site in H. jecorina Cel7A are more closed than loops in the other two enzymes, which has implications for the degree of processivity, endo-initiation, and substrate dissociation. Overall, this study highlights molecular level features important to understanding this biologically and industrially important family of glycoside hydrolases.
Introduction
Basidiomycete fungi of the Heterobasidion annosum sensu lato complex are among the most serious pathogens in conifer forests throughout the world. The annual economic losses (∼1995) caused by tree mortality and wood decay as a result of this pathogen have been estimated at €790 million within the European Union alone (1). Heterobasidion fungi infect trees through wounds and newly cut stumps and grow within the trunk and roots of live and dead trees causing white rot decay. Most importantly, the fungus is capable of infecting new trees via root to root contact, which is a major factor for its high pathogenicity. The complete genome of Heterobasidion irregulare, formerly H. annosum North American P-type, which mainly infects pine, was recently sequenced (2). More than 280 carbohydrate-active enzyme genes are predicted in the genome, nearly two-thirds of which are glycoside hydrolases (GH)4 (3).
Generally, plant- and wood-degrading microorganisms require multiple enzymatic activities for degradation of the various polysaccharides that comprise plant cell walls, including GHs and lytic polysaccharide monooxygenases (4–7). Because cellulose is the main structural component of terrestrial plants, cellulase enzymes are often found as both the largest number of GH genes and the majority of proteins by mass in the secretomes of biomass degrading organisms. In several cellulolytic fungi, such as the soft rot ascomycete Hypocrea jecorina (8) and the white rot basidiomycete Phanerochaete chrysosporium (9), the major proteins produced under cellulase-inducing conditions are glycoside hydrolase family 7 (GH7) cellobiohydrolases (CBHs) (3). In addition to fungi, GH7 genes have also been found in crustaceans (e.g., Daphnia magna and Limnoria quadripunctata), stramenopiles (e.g., Phytophthora infestans), slime molds (e.g., Dictyostelium discoideum), and parabasilian protists (e.g., Pseudotrichonympha grassii) but not in bacteria and archaea (10). In H. jecorina, the enzyme Cel7A (HjeCel7A) constitutes nearly half of total secreted protein, and gene knock-out studies have shown that it is a rate-limiting factor in cellulose degradation (8), pointing at a key role for GH7 enzymes in biomass degrading fungi. The HjeCel7A enzyme is also a key component in industrial conversion of biomass to soluble sugars for production of biofuels, because H. jecorina is the predominant commercial source of enzymes for such applications (11).
Several GH7 enzymes, including HjeCel7A and P. chrysosporium Cel7D (PchCel7D), are bimodular proteins with a family 1 carbohydrate-binding module (CBM) connected to the GH7 catalytic domain by a glycosylated linker peptide (12, 13), typically ∼30 amino acids long (14), that is extended and flexible (15). Two distinct groups can be recognized within GH family 7: endoglucanases (EG) and CBHs. Structures of catalytic domains are known from both groups, five CBHs: H. jecorina Cel7A (HjeCel7A) (16, 17), P. chrysosporium Cel7D (also called CBH58; PchCel7D) (18), Melanocarpus albomyces Cel7B (19), Talaromyces emersonii Cel7A (20), and Trichoderma harzianum Cel7A (21), and three EGs: H. jecorina Cel7B (22), Humicola insolens Cel7B (23), and Fusarium oxysporum Cel7B (24). These structures share a common β-jelly roll fold with two largely antiparallel β-sheets packing face to face to form a curved β-sandwich. Long loops extend the edges of the β-sandwich and form a 40–50-Å-long substrate-binding groove along the entire GH7 module. In CBHs, several loops are further elongated and effectively enclose the cellulose chain in a tunnel, whereas EG enzymes exhibit a more open cleft.
The enclosed tunnel in CBHs likely imparts higher ligand binding free energy than EGs (25, 26), which enables the enzyme to bind to a cellulose chain and processively cleave off many cellobiose residues from the end of a chain before dissociation (27–29). This processive action is preferentially from the reducing end toward the nonreducing end in GH7 CBHs (30). Moreover, GH7s cleave β-1,4-glycosidic bonds via a retaining mechanism (16, 24). Both the catalytic nucleophile and the catalytic acid/base are glutamic acid residues and are positioned close to each other on the same β-strand in the conserved EXDXXE sequence motif. The conserved aspartic acid residue between the two glutamates forms a hydrogen bond with the catalytic nucleophile that is important for catalysis (31).
Structural studies of cellodextrin binding to HjeCel7A revealed that the active site can bind up to 11 glucose residues of a cellulose chain, with seven subsites, numbered −1 to −7, from the catalytic center toward the nonreducing end of the chain, and four subsites, +1 to +4, toward the reducing end (17, 32). Four tryptophan residues form glucosyl-binding platforms at subsites −7, −4, −2, and +1 (Trp-40, Trp-38, Trp-367, and Trp-376, respectively). These tryptophans, as well as most protein-sugar interactions, are highly conserved among the known GH7 CBHs. However, differences in length and sequence of loops along the cellulose-binding path vary the accessibility of the active site and will affect the dynamics of loop movements, which will influence processivity, product inhibition, probability of endo-initiation, and release of nonproductively bound enzyme.
Additionally, of the known GH7 CBH structures, HjeCel7A exhibits the most closed tunnel, whereas PchCel7D displays the most open active site because of several deletions and reductions in size of residues at the tip of tunnel enclosing loops. As a result, PchCel7D shows weaker cellobiose inhibition and faster degradation of microcrystalline cellulose (but only a slight increase in kcat on soluble substrates) (32). A recent study comparing the processive action on cellulose between these two enzymes showed a higher probability of endo-initiation and 3.6-fold higher rate of enzyme release for PchCel7D on bacterial cellulose, whereas the apparent processivity was lower with an average of ∼50 hydrolytic events per chain initiation for PchCel7D compared with ∼60 for HjeCel7A (28). The structural basis for these types of observations can now be examined for these two enzymes, but there is a clear need to develop a larger set of structure-function relationships for GH7 CBHs, such that the molecular level basis for processivity and activity can be more clearly established and such that GH7s can be more readily engineered for biofuels processes.
Lastly, we note that x-ray crystallography studies provide limited information about the flexibility of proteins in solution. Computational approaches offer complementary insights into the protein dynamics, enzyme-ligand interactions, and interactions with solvent and substrate (26, 33). Recent computational studies on HjeCel7A cast new light on the role of the CBM (34–37) and the linker (14, 15). Additionally, computational work on the energetics of cellobiose binding to the product sites has suggested that processive and nonprocessive GH7 enzymes from H. jecorina vary significantly, which has implications for product inhibition (38, 39).
In this study, we report the identification, biochemical characterization, crystallization, and three-dimensional structure of HirCel7A, which is the most abundant protein in the culture supernatant when H. irregulare was grown on spruce powder. Using this new structure, we conduct molecular dynamics (MD) simulations to compare the flexibility of tunnel-enclosing loops with HjeCel7A, which has a more closed tunnel, and PchCel7D, where the tunnel is more open. These results highlight several key differences in multiple loops around the active sites of GH7s that potentially give rise to differences in dissociation from the substrate and the likelihood of endo-initiation by these key biomass-degrading enzymes.
EXPERIMENTAL PROCEDURES
Protein Preparation and Identification
The recently sequenced H. irregulare strain TC-32-1 (2) was maintained on Hagem agar (40) and was cultivated at ambient temperature (20–25 °C) using the minimal medium (pH 5.0) of Kremer and Wood (41), with microcrystalline cellulose, milled spruce heart wood, or Aspen sawdust as carbon sources. Methods for cultivation, protein identification, protein purification, and crystallization are described in further detail in the supplemental text.
Protein identification was done with broth from a static culture on spruce wood powder. The fungus was grown for 3 weeks on the surface of 15 g of wood powder wetted with 150 ml of medium. An additional 400 ml of medium was added, and the culture was shaken for 2 days before centrifugation and sterile filtration. The culture filtrate was concentrated and passed through a cation exchange column at pH 5.0, and the nonadsorbed proteins were fractionated by anion exchange chromatography at pH 6.5. Selected fractions were analyzed by SDS-PAGE, and the major protein band was subjected to trypsin digestion (42). Tryptic peptides were then analyzed by MALDI-TOF mass spectrometry and peptide mass fingerprint search (MASCOT) against predicted proteins in the H. irregulare genome database (43). From the gene model of the best hit, protein ID 38802, the predicted sequence of the secreted protein (440 amino acids) was used to calculate the peptide molecular mass (46,978.5 Da), theoretical isoelectric point (pI = 4.51), and molar extinction coefficient (ϵ = 60,485 m−1 cm−1), using the ProtParam tool at the ExPASy Proteomics Server.
The HirCel7A enzyme was purified from two static cultures with Aspen sawdust as carbon source, as above, but with 15 g of sawdust, 90 ml of medium, 4 weeks of cultivation, and the addition of 500 ml of 10 mm sodium acetate, pH 5.0 (instead of medium), prior to the 2 days of shaking before harvest. The purification was done by anion exchange (two steps at pH 6.5) and size exclusion chromatography. The purified protein in 10 mm sodium acetate, pH 5.0, was concentrated to 9.0 mg/ml and sterile-filtered (0.2 μm). The protein concentration was determined by UV absorbance at 280 nm.
Crystallization and X-ray Data Collection
The initial search for crystallization conditions was done with the JCSG+ suite sparse matrix screen (Qiagen) by sitting drop vapor diffusion using an Oryx-8 crystallization robot (Douglas Instruments). The crystals used for data collection were grown in hanging drop vapor diffusion experiments (44) at 20 °C, for the apo structure with 8 mg/ml HirCel7A enzyme mixed 1:1 with precipitant (20 mm MgCl2, 0.1 m HEPES, pH 7.5, 22% polyacrylic acid 5100 sodium salt). Crystals appeared within 24 h without seeding and could be flash frozen in liquid nitrogen without prior cryo-protectant soaking. For the thio-β-d-xylopentaose (SX5) structure (described below), 4 mg/ml HirCel7A was used with precipitant (20 mm MgCl2, 0.1 m HEPES, pH 7.7, 15–22% polyethylene glycol 3350) and microseeding after 24 h of equilibration. SX5 (∼0.8 mm) was added to drops with grown crystals. After 48 h of soaking, single crystals were picked up with ready-made cryo loops and dipped for a few seconds in cryoprotectant solution (mother liquor with 22.5% PEG 3350 + 10% glycerol) prior to flash freezing.
Synchrotron radiation diffraction data sets were collected from single crystals at 100 K using Beamlines I911-2 and I911-5 at MAX-Lab (Lund, Sweden). Indexing and spot integration of the diffraction data were done with XDS, and then reflections were scaled and merged with programs of the CCP4 package (45). 5% of the reflections were set aside for Rfree factor calculation during structure refinement.
Structure Solution, Refinement, and Analysis
The HirCel7A structure was solved by molecular replacement with Phaser (46) using the apo data set and the structure of PchCel7D as a search model (Protein Data Bank entry code 1GPI (18)). Initial phases were then improved by rigid body refinement in REFMAC5 (47). Before further refinement, a homology model was made by threading the HirCel7A sequence onto the PchCel7D structure solution using Swiss PDBViewer (48). Further model building and refinement, including the addition of ligand and water molecules, was done by alternating cycles of restrained refinement with REFMAC5 (47), and manual inspection and structure adjustments in COOT (49) against 2Fo − Fc and Fo − Fc electron density maps. A starting model for the SX5 structure (thio-β-d-xylopentaose soak) was obtained by fitting the apo structure to the new space group by molecular replacement.
Swiss PDBViewer (48) was used for superimposing the structure models. Interpretation and comparison of structures and preparation of figures were done with COOT or PyMOL (DeLano W.L, 2002). Atomic coordinates and structure factor files have been deposited at the Protein Data Bank with accession codes 2YG1 for apo and 2XSP for SX5.
Activity Measurements
The pH dependence was monitored by mixing 1 mm substrate (p-nitrophenyl-β-d-lactopyranoside; Sigma) and 1 μm HirCel7A enzyme in 160 μl of citrate phosphate-NaCl buffers with constant ionic strength (I = 20 mm) at pH 2.7, 3.05, 3.35. 3.6, 4.0, 4.55, 5.0, 6.0, and 7.0. After incubation at 30 °C for 50 min, the reaction was stopped by the addition of 150 μl of 0.5 m sodium carbonate, and released p-nitrophenol was measured spectrophotometrically at 405 nm. The temperature dependence was monitored at pH 4.0 using the same assay at 30, 40, 45, 50, and 60 °C.
Computational Methods
MD simulations were conducted for three GH7 enzymes: HirCel7A, HjeCel7A, and PchCel7D, each with a cellononaose ligand. Each system was constructed to model the catalytically active complex, and the full cellononaose ligand from the H. jecorina 8CEL structure (17) was docked into HirCel7A and PchCel7D via a structural alignment of the proteins from the −7 to +2 subsites. The PchCel7D structure was taken from 1GPI (18), except for the rotamer of Arg-240, which was taken from 1Z3W (50), because it is in the correct position to hydrogen bond to the cellodextrin ligand. In all cases, crystallographic water molecules that did not overlap with the docked ligand were retained. The protonation states of the catalytic triad for each enzyme were taken from the proposed retaining mechanism of GH7 enzymes (16). Each enzyme was solvated in a box of explicit water molecules ∼84 × 84 × 84 Å, and ions were added (Na+) to ensure that the systems were charge neutral. The total system size for each case was ∼60,000 atoms. The details of the MD simulations and simulation analyses are described in detail in the supplemental text.
RESULTS
Identification and Preparation of HirCel7A
The recently sequenced strain TC-32-1 of H. irregulare (2) was cultivated with Avicel, spruce heart wood powder, or Aspen sawdust as a carbon source. The fungus grew better in static cultures, which provided the highest protein yields. The protein concentrations in the culture filtrates after 3–4 weeks of growth at room temperature were on average 60 mg/liter with Avicel, 90 mg/liter with Spruce heart wood powder, and 130 mg/liter with Aspen sawdust. However, all three substrates were only partially consumed, and thus some enzymes likely remained bound to the residual substrate.
SDS-PAGE analysis of fractions from ion exchange chromatography of filtrate from a culture on spruce wood powder revealed that a protein at ∼50 kDa was by far the most abundant in the culture filtrate (Fig. 1). Trypsin digestion and peptide mapping by MALDI-TOF identified the protein as a product of the only gene in the H. irregulare genome coding for a GH7 enzyme (protein ID 38802) that was annotated as a “candidate [reducing end-acting] cellobiohydrolase” (43). The predicted protein consists of an 18-amino acid signal peptide and a single GH7 catalytic module of 440 residues that lacks a linker and CBM. The enzyme was designated HirCel7A. Approximately 10 mg of purified and concentrated HirCel7A was obtained from a culture with Aspen sawdust as a carbon source.
FIGURE 1.
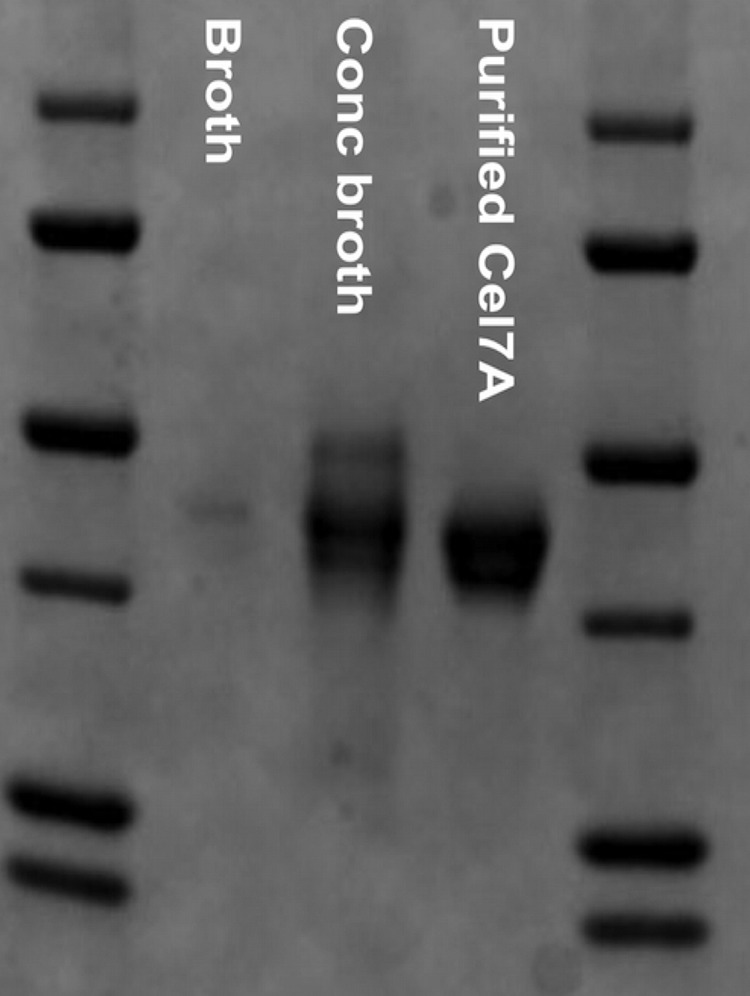
SDS-PAGE gel showing that HirCel7A is the major protein in the H. irregulare culture filtrate.
Temperature and pH profiles of activity on the chromogenic substrate p-nitrophenyl-β-d-lactopyranoside are shown in Fig. 2. The pH optimum is between pH 3.6 and 4.5, with a gradual decrease toward neutral pH (71% at pH 5; 23% at pH 6) and a sharp drop in the acidic direction (1% at pH 2.7). At pH 4.0, the activity was highest at 40–45 °C and dropped at 50 °C to 19% of maximum.
FIGURE 2.

Top panel, temperature profile of HirCel7A activity on p-nitrophenyl-β-d-lactopyranoside from 30 to 60 °C at pH 4.0. Bottom panel, pH profile of HirCel7A activity on p-nitrophenyl-β-d-lactopyranoside at 30 °C.
Crystallization, Structure Solution, and Quality of the Structure Models
Two crystallization data sets were collected: without addition of ligand (apo) and after soaking crystals with 0.8 mm SX5. The details and statistics of data collection and structure refinement are summarized in Table 1.
TABLE 1.
Crystallization data and refinement statistics for the H. irregulare Cel7A structures
| Apo | SX5 soak | |
|---|---|---|
| Protein Data Bank code | 2YG1 | 2XSP |
| Beamline | I911-2 | I911-5 |
| Space group | P21 | C2 |
| Cell dimensions (Å) | 60.5, 84.1, 75.9 | 134.8, 49.5, 73.5 |
| Molecules/asymmetric unit | 2 | 1 |
| Wavelength (Å) | 1.04 | 0.91 |
| Resolution (Å) | 29.4 (1.9) | 29.5 (1.7) |
| Unique reflections | 54,342 | 45,525 |
| Multiplicity | 4.1 | 6.5 |
| Completeness (%) | 98.6 (96.5) | 98.2 (96.1) |
| I/σI | 30.1 (5.5) | 33.1 (5.7) |
| Rmerge | 0.026 (0.232) | 0.043 (0.331) |
| Rwork | 20.0 | 15.0 |
| Rfree | 25.3 | 17.7 |
| Bond angle root mean square deviation (°) | 1.0995 | 1.418 |
| Bond length root mean square deviation (Å) | 0.0074 | 0.0124 |
| Ramachandran outliers | 1.7% | 0.5% |
The HirCel7A structure was solved by molecular replacement using the apo data set and the structure of P. chrysosporium Cel7D as the search model (PchCel7D; Protein Data Bank entry code 1GPI (18)). The apo and SX5 structure models were refined at 1.9 and 1.7 Å resolution and Rfree of 25.3 and 17.7%, respectively (Table 1). In the single protein chain present in the asymmetric unit of the SX5 crystal, all 440 amino acid residues could be fit to electron density. The apo structure contained two molecules in the asymmetric unit, which both displayed regions with somewhat poor electron density and elevated temperature factors (that correlate with higher mobility in the MD simulations). In chain A of the apo structure, all residues could be fit to density, but in chain B, there were three loop regions with insufficient density, and the following amino acid residues have been omitted in the deposited model: 45–47 (3 amino acids), 52–55 (4 amino acids), and 197–203 (7 amino acids).
In all the structures, the N-terminal glutamine residue is cyclized to pyroglutamate (PCA1), Pro-390 is cis-proline, all 18 cysteines form disulfide bonds, and N-glycosylation is evident at Asn-270 with density for two N-acetylglucosamine residues. There is also one hexa-coordinated metal ion-binding site and an additional metal ion in the apo molecule B not present in the other protein molecule. The metal ions were assumed to be magnesium, which was present in the precipitant solution. Furthermore, the SX5 structure model contains one molecule of HEPES at subsite −2 and one xylose residue at subsite +1 in the active site. Both molecules show relatively weak electron density and elevated temperature factors, suggesting only partial occupancy and/or some flexibility of binding. The xylose residue binds in the same position and orientation as the +1 glucosyl unit in the structures of HjeCel7A E212Q mutant in complex with cellobiose and cellotetraose (respectively 3CEL (31) and 5CEL (17)) but is in the form of the α-anomer and is of course lacking the 6-hydroxymethyl group present in glucose. No clear evidence for further ligand units were observed. We show the positions of the xylose and HEPES residues in supplemental Fig. S1.
Structure of HirCel7A and Comparison with Related Enzymes
As expected from the high amino acid sequence identity (Fig. 3), the overall fold of HirCel7A (Fig. 4) is similar to the catalytic modules of PchCel7D (69% identity) and HjeCel7A (56% identity), with root mean square differences of 0.6 and 0.8 Å for 421 and 407 matching Cα atoms, respectively. Superposition of the HirCel7A structure, PchCel7D (1Z3V (50)), and HjeCel7A with a model of a cellulose chain bound (8CEL (17)), demonstrates that the cellulose-binding sites are highly conserved, including the catalytic triad, Glu-214 (nucleophile), Asp-216, and Glu-219 (acid/base) (positions 212, 214, and 217 in HjeCel7A and positions 207, 209, and 212 in PchCel7D) and the tryptophan platforms at subsites −7, −4, −2, and +1 (Trp-40, Trp-38, Trp-375, and Trp-384). Nearly all of the amino acids identified by Divne et al. (17) as important for cellulose binding are conserved at similar positions. Major differences, potentially related to the function of the enzyme, are observed at four regions along the substrate binding path: the tunnel entrance at subsites −7/−6 (Fig. 4B, loop A1) and loop contacts around subsite −4 (Fig. 4C, loop A3 and B2), near the catalytic center (Fig. 4D, loop A3 and B3), and adjacent to the product-binding subsites (Fig. 4E), which are discussed in turn below.
FIGURE 3.

Structure-based sequence alignment of HirCel7A, HjeCel7A, and PchCel7D with loops of interest indicated.
FIGURE 4.

A, overall structure of the HirCel7A enzyme (SX5 structure) with a docked ligand from the H. jecorina 8CEL structure (17). The ligand is shown in cyan, and loops and residues of interest are labeled. B, superposition of loop A1 at the tunnel entrance over subsite −7 from HirCel7A (violet), HjeCel7A (green), and PchCel7D (pink). The HirCel7A loop A1 contains a tyrosine residue (Tyr-101) not present in HjeCel7A or PchCel7D. Loop A1 in PchCel7D is significantly truncated. C, superposition of loop A3 and loop B2 over the −4 subsite. The enzymes exhibit different loop-loop contacts and sequence diversity. D, superposition of loops A3 and B3 near the catalytic center subsite −1 and the product sites +1/+2. HjeCel7A exhibits a longer loop B3, which forms stable contacts to loop A3 across the catalytic center. The HirCel7A loop B3 is two residues shorter and interacts via water with Glu-379 on loop A3. PchCel7D exhibits the most exposed catalytic center because of a six-residue deletion in loop B3. E, superposition of loop B4 shows the aspartate residue that may interact with the reducing end of the product at subsite +2, which is present in HirCel7A (Asp-347, D347) and PchCel7D (Asp-336, D336) but is deleted in HjeCel7A.
To further examine the structural and dynamical differences in these enzymes, we also conducted MD simulations of HirCel7A, HjeCel7A, and PchCel7D with the cellononaose ligand from the H. jecorina 8CEL structure. These simulations enable quantitative comparisons of enzyme-ligand interactions, ligand solvation, loop flexibility, and dynamical correlations within the proteins, the latter three of which have been shown to correlate with processivity (51). For each simulation, the root mean square deviation of the protein was calculated, and these results are provided in supplemental Fig. S2A. For each simulation, the protein root mean square deviation is stable. Root mean square fluctuations (RMSF) have also been determined on a per-residue basis for the HirCel7A enzyme with and without a ligand, which is shown in supplemental Fig. S2B.
Fig. 5 (A–C) shows a cluster view of ∼50 structures uniformly spaced over the MD simulations, color-coded from low (blue) to high (red) RMSF. Fig. 5D shows the protein RMSF values as a function of residue number. Generally, the more flexible regions correspond to the loop sections where sequence and structural differences are present (Figs. 3–5). In particular, there are three distinct regions that exhibit differences in fluctuations. Namely, loops B2 and A4 in the HirCel7A and loop A2 in PchCel7D exhibit the highest fluctuations during the MD simulations. We do not discuss the PchCel7D loop A2 in this study, because it does not directly contact the ligand. Furthermore, the ligand fluctuations as a function of binding site from −7 to +2 (Fig. 6A) show that the −7 and −6 subsites exhibit significant differences, whereas subsites −4 to +2 exhibit similar RMSF values for HirCel7A and PchCel7D, with HjeCel7A displaying generally lower ligand fluctuations overall. Fig. 6B illustrates that ligand solvation also differs slightly at the tunnel entrance, but the solvation values are within error.
FIGURE 5.

A–C, cluster representations of HirCel7A (A), HjeCel7A (B), and PchCel7D (C) shown over a 250-ns trajectory at 5-ns intervals. The enzymes are colored by RMSF, where red represents the highest fluctuations, and blue represents the lowest fluctuations. All RMSF values are scaled to the same scale between the three enzymes. D, RMSF as a function of residue number. Gaps in the residue numbers are inserted based on the structural alignment shown in Fig. 3.
FIGURE 6.

A, ligand RMSF as a function of binding site. The RMSF values were calculated based on the heavy atoms for each glucose. The error bars are computed with block averaging. B, ligand solvation as a function of binding site. Solvation was calculated as average number of water molecule within 3.5 Å of the heavy atoms of the d-glucose unit at each subsite. The error bars represent one standard deviation from the average value.
Comparison of the −7 Subsite
At the tunnel entrance, the cellulose chain is covered by loop A1, which differs in both length and sequence between the three enzymes (Fig. 4B). HirCel7A has the longest loop (residues 98–103) and a tyrosine (Tyr-101) at the tip that is potentially well positioned for stacking with the d-glucose residue at subsite −7, opposite to the Trp-40 platform. Similar loop length with a tyrosine at the tip is also present in the structure of M. albomyces Cel7B, and the tyrosine was proposed to direct the cellulose chain into the tunnel (19). The corresponding loop lacks a tyrosine and is shorter by one residue in HjeCel7A and shorter by four residues in PchCel7D, making the tunnel entrance much more open in the lattermost enzyme. In the HirCel7A crystal structures, we observe two distinct conformations of loop A1, with a difference between the Tyr-101 conformations of 4.9 Å for Cα and 7.0 Å for the hydroxyl group, suggesting significant flexibility in loop A1. In HjeCel7A, Lys-102 at the base of the loop binds to Glu-408, which may restrict loop movement. The other two enzymes lack this interaction because the lysine is replaced by Thr-103 in HirCel7A and Ser-99 in PchCel7D.
In the MD simulation of HirCel7A with the cellononaose ligand, we observe an almost immediate conformational change in loop A1 wherein the Tyr-101 aromatic ring binds to the glucose residue at the −7 subsite. During the vast majority of the 0.25-μs simulation, Tyr-101 remains bound to the ligand, with some fluctuations of the side chain pointing out into solution, as shown in a cluster view in Fig. 7. Trp-40 is significantly more stable than Tyr-101. We note that despite the additional interaction by Tyr-101 with the −7 glucose residue, the ligand fluctuations are higher in HirCel7A than in HjeCel7A, and the higher fluctuations of the −7 glucose residue are correlated with fluctuations in Tyr-101 (Fig. 7 and supplemental Fig. S3). PchCel7D has a deletion in loop A1 and exhibits both higher ligand RMSF values and a higher degree of solvation (Fig. 6). Conversely in HjeCel7A and PchCel7D, loop A1 is quite stable (supplemental Fig. S4A).
FIGURE 7.

A, Bottom, and B, side views of a cluster representation of Tyr-101 (yellow), Trp-40 (blue), and the cellodextrin chain (cyan and red) from the HirCel7A MD simulation. The cluster frequency is 5 ns over a 250-ns simulation.
Comparison of the Loops near the −4 Subsite
The next section of interest is around loop B2, which encloses the cellulose chain around subsite −4 (Fig. 4C). Loop B2 typically constitutes a 13–15-residue insertion in CBHs relative to GH7 EGs, which folds over the β-sandwich structure to define the “roof” of the tunnel. In HjeCel7A, two Asn residues, Asn-197 and Asn-198, at the tip of loop B2 interact with Tyr-370 on the opposing loop A3 on the other side of the β-sandwich, thus effectively closing the tunnel (17). In HirCel7A, loop B2 has the same length as HjeCel7A, but with Ser-199 and Asp-200 at its tip and His-378 on the opposing loop A3. In the SX5 structure of HirCel7A, the loop B2 conformation is similar to that of HjeCel7A, and the tunnel is closed by H-bonding of Ser-199 (2.7 Å) and van der Waals contact of Asp-200 (∼3.5 Å) with His-378. In chain A of the apo structure, loop B2 is more open; Asp-200 is still within contact distance, but Ser-199 hydrogen bonds to His-378 indirectly via a water molecule. In chain B of the apo structure, the tip of loop B2 is disordered, and residues 197–203 are not visible in the electron density, suggesting that the tunnel is open. Interestingly, this loop was also disordered in the first structure of T. emersonii Cel7A (20). Loop B2 in PchCel7D is two residues shorter, and there are no direct contacts with loop 3A. The closest distance is ∼5.5 Å from the tip of loop B2 to His-367 on loop A3.
The MD simulations suggest that loop B2 in both HirCel7A and PchCel7D exhibit significantly higher mobility than in HjeCel7A (Fig. 5 and supplemental Fig. S4B). Histograms of the minimum distances between opposing loops in the three enzymes (supplemental Fig. S5) show that the contact between loops B2 and A3 over the tunnel in HjeCel7A is quite stable over 0.25 μs of MD simulation, primarily because of a stable hydrogen bond between Tyr-370 and Asn-197. Tunnel opening occurs more frequently in both HirCel7A and PchCel7D.
Loop B2 is the region in HirCel7A that exhibits the highest flexibility and also the largest shift in RMSF between MD simulations with and without a ligand. However, rather surprisingly, the fluctuations of loop B2 were actually larger with the ligand than without (supplemental Fig. S2B). Moreover, there is a positive correlation between fluctuations in loop B2 and the adjacent loop B1 (supplemental Fig. S6). Overall, the access to the tunnel entrance appears more dynamic in PchCel7D and HirCel7A than in HjeCel7A.
Comparison of the Catalytic Center Loops
Loop B3, residues 246–253 in HirCel7A, is also referred to as the exo-loop (32). At the tip of loop B3 in HjeCel7A, Thr-246 binds to the substrate at subsite +1, and Tyr-247 interacts with both the substrate in the −2 subsite and via van der Waals contacts with Tyr-371 on loop A3 across the binding tunnel. At the end of loop B3, a conserved arginine interacts with the substrate at both subsites +1 and +2 in all three enzymes (Arg-251 in HirCel7A; Fig. 4D). Compared with HjeCel7A, loop B3 is shortened by two residues in HirCel7A and by six residues in PchCel7D, and it has no direct contact with loop A3. This is most clearly illustrated from MD simulations by examination of the minimum distance between loops A3 and B3 (supplemental Fig. S5D), where HjeCel7A maintains a minimum distance of ∼3.5 Å over the course of the simulation, whereas both HirCel7A and PchCel7D loops open as much as 12 Å. Also from the MD simulations, the fluctuations of the B3 loop are only slightly larger than for the rest of the protein in all three enzymes (Fig. 5). Thus, the catalytic center remains most closed in HjeCel7A and most exposed in PchCel7D, with HirCel7A in between.
For loop A3, the Tyr-371 in HjeCel7A is reduced to an alanine in PchCel7D. In HirCel7A, the corresponding residue Glu-379 stretches over the active site toward loop B3 but is not in direct contact (Fig. 4D). It is hydrogen-bonded via water to Asp-247 (5.4 Å), and the closest distance is to Arg-251 (4.8 Å; SX5 structure model), thus restricting the access to the active site in this region. However, the side chain of Glu-379 appears to be flexible because it displays different rotamers in the three HirCel7A structure models. There is also space for Glu-379 to bend down toward the cellulose chain. In the MD simulations, it is occasionally within contact distance to OH6 of the d-glucose unit at subsite +1 (data not shown), pointing at a putative role in substrate binding and/or mediation of cellulose sliding over the catalytic center. Glutamate at this position is found only in a few basidiomycete homologs (e.g., Puccinia graminis and Pleurotus ostreatus), and glutamine is found only in a few sequences from distantly related protists (e.g., D. discoideum and P. grassii).
Product Binding Site Differences
Loop A4 (amino acids 392–403) adjacent to the product binding sites is the second most flexible region in HirCel7A and is more flexible in HirCel7A than in the other enzymes (Fig. 5). This correlates with an up to ∼2.5 Å backbone shift in loop A4 among the HirCel7A crystal structures. The flexibility may affect product binding and expulsion, but the loop does not interact directly with substrate, and ligand fluctuations at the +1/+2 subsites are only slightly higher in HirCel7A and PchCel7D than in HjeCel7A. However, there is a conserved tyrosine residue (Tyr-389 in HirCel7A, Tyr-381 in HjeCel7A, and Tyr-378 in PchCel7D) immediately preceding loop A4 that is in contact with the +2 glucose residue. In all HirCel7A structures reported here and HjeCel7A and PchCel7D enzymes (e.g., 7CEL and 1GPI), this tyrosine residue is structurally similar.
Beyond the reducing end of the cellulose chain, the end of the active site cleft is formed by loop B4, which is anchored to the rest of the protein at both ends by conserved phenylalanine residues (Phe-345–Phe-351 in HirCel7A). HjeCel7A has a one-residue deletion in loop B4 that is unique to CBHs of Hypocrea/Trichoderma species and Thermoascus aurantiacus and Aspergillus glaucus. Most GH7 CBHs have an additional aspartate residue (at position 347 in HirCel7A and at position 336 in PchCel7D) that points toward the reducing end of a bound cellulose chain, which has been proposed to aid in product expulsion by encouraging cellobiose to tilt away from the catalytic center (50).
DISCUSSION
GH7 CBHs, like most processive cellulases and chitinases, employ a multistep mechanism to deconstruct cellulose. The steps employed include an initial complexation event via either endo- or exo-mode initiation, formation of the catalytically active complex, hydrolysis, product expulsion, and processive translation along the chain (26, 33, 52). This process continues until the enzyme reaches the end of a cellulose chain or a surface obstruction, at which point it will dissociate (27–29, 52, 53). Elucidation of the rates of each of these elementary steps and relation of structure to the complexation mode (either endo-initiation or exo-initiation) (28, 54, 55), processive action (27–29), the dissociation rate from cellulose (28, 52), and product binding and inhibition (38, 39, 56) will enable deeper understanding of CBHs. To date, structures of the GH7 CBHs reveal significant differences in the substrate accessibility to the active site, in particular with PchCel7D exhibiting the most open active site tunnel and HjeCel7A the most enclosed active site tunnel, such that these two enzymes provide a foundation to examine GH7 CBH structure-function relationships. Recently, several groups have begun to examine the rate-limiting steps involved in CBH action (28, 52, 55), which have revealed a trade-off between the ability of GH7 CBHs to be processive (to maximize hydrolytic rate) and the ability of GH7s to either complex (55) or dissociate from the substrate (to not become inactivated when blocked) (28, 29, 52, 53). One study in particular examined the differences in processivity and off rate between HjeCel7A and PchCel7D and demonstrated that PchCel7D exhibits a significantly higher dissociation rate and lower extent of processivity than HjeCel7A (28). With the development of these types of new, sophisticated analytical methods to probe time scales for GH7 CBH action, there is now a path to establish more detailed structure-activity relationships. To that end, here we have combined structural and computational studies to characterize a GH7 CBH, HirCel7A and compared this new structure to the well characterized HjeCel7A and PchCel7D enzymes.
HjeCel7A is more processive than PchCel7D on crystalline cellulose (28), and our computational results suggest that the PchCel7D ligand is both more flexible and more solvated than HjeCel7A, and HirCel7A exhibits both intermediate ligand fluctuations and solvation. It has been shown for GH18 chitinases that processivity (57, 58) is inversely correlated to ligand fluctuations and ligand solvation (51). Based on the computational results obtained here shown in Figs. 5 and 6, it is likely that HirCel7A, which exhibits intermediate ligand fluctuations and solvation between PchCel7D and HjeCel7A, may exhibit some intermediate level of processivity between these two enzymes (51). On a related note, it has long been known that GH7 CBHs can bind to insoluble cellulose in an endo-initiation mode to create new chain ends for processive action (28, 54). PchCel7D was recently shown to conduct endo-initiation more than HjeCel7A, which is likely a function of the shorter loops and more open cellulose-binding tunnel (28). The MD simulations presented here align with the experimental observations related to the accessibility of the PchCel7D active site tunnel relative to HjeCel7A, and HirCel7A exhibits a degree of tunnel opening and flexibility between HjeCel7A and PchCel7D.
Additionally, Tyr-101 in HirCel7A is observed to bind directly to the glucose in the −7 site in the MD simulation, as shown in Fig. 7. Aromatic-carbohydrate interactions are ubiquitous in tunnels and clefts of GH enzymes and have been implicated in binding, processivity, and ligand stabilization necessary for catalysis (17, 57, 59–61). In HirCel7A, Trp-40 forms the canonical GH7 binding platform at the −7 subsite, but Tyr-101 seems to form a relatively stable aromatic-carbohydrate interaction with the ligand as well. Interestingly, this binding is associated with higher ligand fluctuations in HirCel7A than in HjeCel7A. The extra binding platform provided by Tyr-101 may help to capture and guide a cellulose chain end into the active site tunnel and also affect the processive action, which we will investigate in more detail in a future study.
H. irregulare is an incredibly effective pathogen in nature because of its ability both to digest dead plant material via a saprotrophic mechanism and to infect living trees via a necrotrophic mechanism. In the genome sequencing study, many genes coding for potentially biomass-degrading enzymes were up-regulated during growth on wood, including for example the GH family 6 CBH as measured by transcript profiling (2). However, the Cel7A gene was not recognized among the up-regulated genes, although here we show that Cel7A is the main component of the H. irregulare extracellular enzyme mixture when the fungus grows on wood as a substrate, which suggests that Cel7A plays a major role in wood degradation. This in turn suggests that it would be desirable to complement transcript profiling with proteomics in future studies of the mechanisms of H. irregulare wood degradation to establish at which level the actual enzymes are present.
Additionally, HirCel7A is the only GH7 gene in the H. irregulare genome, yet it lacks a CBM despite the fact that many GHs and other carbohydrate-active enzymes in the genome exhibit family 1 CBMs (2). The aforementioned elementary steps of cellulolytic action may all be influenced to some extent by the presence or absence of a CBM attached to the GH7 module, and it is known that CBMs on GH7 CBHs can enhance degradation of solid cellulose in low solids environments (62). One possible reason that HirCel7A does not possess a CBM may be related to the environment in which it evolved. For example, despite significant differences in H. jecorina and P. chrysosporium, including their repertoire of biomass-degrading enzymes, which lignocellulose components they can utilize, and the strategies used to compete with other organisms, they both live saprotrophically on plant material. H. irregulare on the other hand, employs both necrotrophic and saprotrophic strategies toward wood utilization. Via a necrotrophic mechanism, H. irregulare is able to counterfeit and evade host defense mechanisms and thereby infect living trees, successively colonize the wood, and eventually kill the tree. As a primary colonizer, it is thereby able to exploit an ecological niche where it is practically isolated such that it does not substantially compete for food during wood degradation. Thus, the evolutionary selection pressure may have primarily been strongest on its necrotrophic ability and not on its ability to degrade wood. The other potential reason that a family 1 CBM might not be needed relates to the amount of accessible surface area of cellulose available during wood degradation (62). If there are sufficient accessible cellulose surfaces near where HirCel7A is secreted, then its ability to engage the substrate may not be a factor in the overall rate of sugar release, which would suggest no evolutionary pressure to utilize a CBM. More generally, it is noteworthy that the majority of GH7 enzymes in the Carbohydrate-Active Enzymes database (3) do not contain family 1 CBMs (14), but to our knowledge, a systematic study has not yet been conducted to understand if this is due to the environmental niches in which GH7 enzymes were evolved for particular organisms or for other potential reasons.
Lastly, we note that the observations from structural and computational work conducted here are made on GH7 enzymes not bound to a solid cellulose substrate. Initiation, processivity, and the other steps in GH7 CBH action will most usually occur at the solid-liquid interface, and enzyme binding to the cellulose surface has been previously shown to affect the dynamics of loops and binding to the ligand in HjeCel7B (63). A further extension of this study will require investigation of these three enzymes and other GH7 cellulases on the surface of cellulose to determine whether surface binding and complexation affect the dynamics of these enzymes in a manner similar to that observed for ligand binding in solution.
CONCLUSIONS
Here we have determined the structure of the cellobiohydrolase Cel7A purified from the culture filtrate of the root rot fungus H. irregulare. HirCel7A is the major secreted protein when H. irregulare was grown in static cultures with spruce as a carbon source. The enzyme activity of HirCel7A exhibits a temperature optimum at 40–45 °C and pH optimum at pH 3.6–4.5 when measured on p-nitrophenyl-β-d-lactopyranoside. The overall structure is similar to the well characterized Cel7A from ascomycete fungus H. jecorina and to the Cel7D from the basidiomycete fungus P. chrysosporium. The major differences compared with these two are within multiple loops that form the CBH tunnel structure and at the entrance of the substrate-binding tunnel, where a nonconserved tyrosine could form an additional binding platform in the −7 subsite. Building on previous observations on GH18 chitinases (51), MD simulations suggest that HirCel7A will exhibit an intermediate degree of processivity and endo-initiation between PchCel7D and HjeCel7A. Overall, understanding the structural and dynamical differences between GH7 CBHs obtained from x-ray crystallography and MD simulation in relation to experimental characterizations of these enzymes is crucial for development of structure-activity relationships and for designing enhanced biomass-degrading enzyme systems.
Supplementary Material
Acknowledgments
We thank Åke Olson (Department of Forest Mycology and Pathology, Swedish University of Agricultural Sciences, Uppsala, Sweden) for providing the H. irregulare strain TC-32-1. We thank Farid Ibatullin (Bioorganic and Medicinal Chemistry Laboratory, Molecular and Radiation Biophysics Department, B. P. Konstantinov Petersburg Nuclear Physics Institute of the Russian Academy of Sciences) for providing the xylopentaose ligand.
This work was supported in part by the Faculty for Natural Resources and Agriculture at the Swedish University of Agricultural Sciences through the research program “MicroDrivE.” This work was also supported by funds from the Department of Energy Office of the Biomass Program (to C. M. P. and G. T. B.). Computer time for this research was provided by the National Institute of Computational Science Kraken cluster under National Science Foundation Extreme Science and Engineering Discovery Environment (XSEDE) Grant MCB090159 and the National Renewable Energy Laboratory Computational Sciences Center supported by the Department of Energy Efficiency and Renewable Energy under Contract DE-AC36-08GO28308.

This article contains supplemental text and Figs. S1–S6.
The atomic coordinates and structure factors (codes 2YG1 and 2XSP) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- GH
- glycoside hydrolase
- GH7
- family 7 GH
- CBH
- cellobiohydrolase
- CBM
- carbohydrate-binding module
- EG
- endoglucanase
- SX5
- thio-β-d-xylopentaose
- RMSF
- root mean square fluctuation
- HirCel7A
- Heterobasidion irregulare Cel7A
- HjeCel7A
- Hypocrea jecorina Cel7A
- PchCel7D
- Phanerochaete chrysosporium Cel7D
- MD
- molecular dynamics.
REFERENCES
- 1. Woodward S. (1998) Heterobasidion annosum: Biology, Ecology, Impact and Control (S. Woodward J. S., Karjalainen R., Hüttermann A., eds) pp. 589, CAB International, Wallingford, UK [Google Scholar]
- 2. Olson A., Aerts A., Asiegbu F., Belbahri L., Bouzid O., Broberg A., Canbäck B., Coutinho P. M., Cullen D., Dalman K., Deflorio G., van Diepen L. T., Dunand C., Duplessis S., Durling M., Gonthier P., Grimwood J., Fossdal C. G., Hansson D., Henrissat B., Hietala A., Himmelstrand K., Hoffmeister D., Högberg N., James T. Y., Karlsson M., Kohler A., Kües U., Lee Y. H., Lin Y. C., Lind M., Lindquist E., Lombard V., Lucas S., Lundén K., Morin E., Murat C., Park J., Raffaello T., Rouzé P., Salamov A., Schmutz J., Solheim H., Ståhlberg J., Vélëz H., de Vries R. P., Wiebenga A., Woodward S., Yakovlev I., Garbelotto M., Martin F. (2012) Insight into trade-off between wood decay and parasitism from the genome of a fungal forest pathogen. New Phytol. 194, 1001–1013 [DOI] [PubMed] [Google Scholar]
- 3. Cantarel B. L., Coutinho P. M., Rancurel C., Bernard T., Lombard V., Henrissat B. (2009) The Carbohydrate-Active EnZymes database (CAZy). An expert resource for glycogenomics. Nucleic Acids Res. 37, D233–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lynd L. R., Weimer P. J., van Zyl W. H., Pretorius I. S. (2002) Microbial cellulose utilization. Fundamentals and biotechnology. Microbiol. Mol. Biol. Rev. 66, 506–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Medie F. M., Davies G. J., Drancourt M., Henrissat B. (2012) Genome analyses highlight the different biological roles of cellulases. Nat. Rev. Microbiol. 10, 227–234 [DOI] [PubMed] [Google Scholar]
- 6. Horn S. J., Vaaje-Kolstad G., Westereng B., Eijsink V. G. (2012) Novel enzymes for the degradation of cellulose. Biotechnol. Biofuels 5, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yakovlev I., Vaaje-Kolstad G., Hietala A. M., Stefanczyk E., Solheim H., Fossdal C. G. (2012) Substrate-specific transcription of the enigmatic GH61 family of the pathogenic white-rot fungus Heterobasidion irregulare during growth on lignocellulose. Appl. Microbiol. Biotechnol. 95, 979–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ilmén M., Saloheimo A., Onnela M. L., Penttilä M. E. (1997) Regulation of cellulase gene expression in the filamentous fungus Trichoderma reesei. Appl. Environ. Microbiol. 63, 1298–1306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Uzcategui E., Ruiz A., Montesino R., Johansson G., Pettersson G. (1991) The 1,4-β-d-glucan cellobiohydrolases from Phanerochaete chrysosporium. I. A system of synergistically acting enzymes homologous to Trichoderma reesei. J. Biotechnol. 19, 271–285 [DOI] [PubMed] [Google Scholar]
- 10. King A. J., Cragg S. M., Li Y., Dymond J., Guille M. J., Bowles D. J., Bruce N. C., Graham I. A., McQueen-Mason S. J. (2010) Molecular insight into lignocellulose digestion by a marine isopod in the absence of gut microbes. Proc. Natl. Acad. Sci. U.S.A. 107, 5345–5350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Himmel M. E., Ding S. Y., Johnson D. K., Adney W. S., Nimlos M. R., Brady J. W., Foust T. D. (2007) Biomass recalcitrance. Engineering plants and enzymes for biofuels production. Science 315, 804–807 [DOI] [PubMed] [Google Scholar]
- 12. Stals I., Sandra K., Geysens S., Contreras R., Van Beeumen J., Claeyssens M. (2004) Factors influencing glycosylation of Trichoderma reesei cellulases. I. Postsecretorial changes of the O- and N-glycosylation pattern of Cel7A. Glycobiology 14, 713–724 [DOI] [PubMed] [Google Scholar]
- 13. Harrison M. J., Nouwens A. S., Jardine D. R., Zachara N. E., Gooley A. A., Nevalainen H., Packer N. H. (1998) Modified glycosylation of cellobiohydrolase I from a high cellulase-producing mutant strain of Trichoderma reesei. Eur. J. Biochem. 256, 119–127 [DOI] [PubMed] [Google Scholar]
- 14. Sammond D. W., Payne C. M., Brunecky R., Himmel M. E., Crowley M. F., Beckham G. T. (2012) Cellulase linkers are optimized based on domain type and function. Insights from sequence analysis, biophysical measurements, and molecular simulation. PLoS One 7, e48615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Beckham G. T., Bomble Y. J., Matthews J. F., Taylor C. B., Resch M. G., Yarbrough J. M., Decker S. R., Bu L., Zhao X., McCabe C., Wohlert J., Bergenstråhle M., Brady J. W., Adney W. S., Himmel M. E., Crowley M. F. (2010) The O-glycosylated linker from the Trichoderma reesei family 7 cellulase is a flexible, disordered protein. Biophys. J. 99, 3773–3781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Divne C., Ståhlberg J., Reinikainen T., Ruohonen L., Pettersson G., Knowles J. K., Teeri T. T., Jones T. A. (1994) The three-dimensional crystal structure of the catalytic core of cellobiohydrolase I from Trichoderma reesei. Science 265, 524–528 [DOI] [PubMed] [Google Scholar]
- 17. Divne C., Ståhlberg J., Teeri T. T., Jones T. A. (1998) High-resolution crystal structures reveal how a cellulose chain is bound in the 50 angstrom long tunnel of cellobiohydrolase I from Trichoderma reesei. J. Mol. Biol. 275, 309–325 [DOI] [PubMed] [Google Scholar]
- 18. Muñoz I. G., Ubhayasekera W., Henriksson H., Szabó I., Pettersson G., Johansson G., Mowbray S. L., Ståhlberg J. (2001) Family 7 cellobiohydrolases from Phanerochaete chrysosporium. Crystal structure of the catalytic module of Cel7D (CBH58) at 1.32 Å resolution and homology models of the isozymes. J. Mol. Biol. 314, 1097–1111 [DOI] [PubMed] [Google Scholar]
- 19. Parkkinen T., Koivula A., Vehmaanperä J., Rouvinen J. (2008) Crystal structures of Melanocarpus albomyces cellobiohydrolase Cel7B in complex with cello-oligomers show high flexibility in the substrate binding. Protein Sci. 17, 1383–1394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Grassick A., Murray P. G., Thompson R., Collins C. M., Byrnes L., Birrane G., Higgins T. M., Tuohy M. G. (2004) Three-dimensional structure of a thermostable native cellobiohydrolase, CBH IB, and molecular characterization of the cel7 gene from the filamentous fungus, Talaromyces emersonii. Eur. J. Biochem. 271, 4495–4506 [DOI] [PubMed] [Google Scholar]
- 21. Textor L. C., Colussi F., Silveira R. L., Serpa V., de Mello B. L., Muniz J. R., Squina F. M., Pereira N., Jr., Skaf M. S., Polikarpov I. (2013) Joint x-ray crystallographic and molecular dynamics study of cellobiohydrolase I from Trichoderma harzianum. Deciphering the structural features of cellobiohydrolase catalytic activity. FEBS J. 280, 56–69 [DOI] [PubMed] [Google Scholar]
- 22. Kleywegt G. J., Zou J. Y., Divne C., Davies G. J., Sinning I., Ståhlberg J., Reinikainen T., Srisodsuk M., Teeri T. T., Jones T. A. (1997) The crystal structure of the catalytic core domain of endoglucanase I from Trichoderma reesei at 3.6 Å resolution, and a comparison with related enzymes. J. Mol. Biol. 272, 383–397 [DOI] [PubMed] [Google Scholar]
- 23. MacKenzie L. F., Sulzenbacher G., Divne C., Jones T. A., Wöldike H. F., Schülein M., Withers S. G., Davies G. J. (1998) Crystal structure of the family 7 endoglucanase I (Cel7B) from Humicola insolens at 2.2 angstrom resolution and identification of the catalytic nucleophile by trapping of the covalent glycosyl-enzyme intermediate. Biochem. J. 335, 409–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sulzenbacher G., Driguez H., Henrissat B., Schülein M., Davies G. J. (1996) Structure of the Fusarium oxysporum endoglucanase I with a nonhydrolyzable substrate analogue. Substrate distortion gives rise to the preferred axial orientation for the leaving group. Biochemistry 35, 15280–15287 [DOI] [PubMed] [Google Scholar]
- 25. Beckham G. T., Matthews J. F., Peters B., Bomble Y. J., Himmel M. E., Crowley M. F. (2011) Molecular-level origins of biomass recalcitrance. Decrystallization free energies for four common cellulose polymorphs. J. Phys. Chem. B 115, 4118–4127 [DOI] [PubMed] [Google Scholar]
- 26. Chundawat S. P., Beckham G. T., Himmel M. E., Dale B. E. (2011) Deconstruction of lignocellulosic biomass to fuels and chemicals. Annu. Rev. Chem. Biomol. Eng. 2, 121–145 [DOI] [PubMed] [Google Scholar]
- 27. Igarashi K., Koivula A., Wada M., Kimura S., Penttilä M., Samejima M. (2009) High speed atomic force microscopy visualizes processive movement of Trichoderma reesei cellobiohydrolase I on crystalline cellulose. J. Biol. Chem. 284, 36186–36190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kurasin M., Väljamäe P. (2011) Processivity of cellobiohydrolases is limited by the substrate. J. Biol. Chem. 286, 169–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Igarashi K., Uchihashi T., Koivula A., Wada M., Kimura S., Okamoto T., Penttilä M., Ando T., Samejima M. (2011) Traffic jams reduce hydrolytic efficiency of cellulase on cellulose surface. Science 333, 1279–1282 [DOI] [PubMed] [Google Scholar]
- 30. Boisset C., Fraschini C., Schülein M., Henrissat B., Chanzy H. (2000) Imaging the enzymatic digestion of bacterial cellulose ribbons reveals the endo character of the cellobiohydrolase Cel6A from Humicola insolens and its mode of synergy with cellobiohydrolase Cel7A. Appl. Environ. Microbiol. 66, 1444–1452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ståhlberg J., Divne C., Koivula A., Piens K., Claeyssens M., Teeri T. T., Jones T. A. (1996) Activity studies and crystal structures of catalytically deficient mutants of cellobiohydrolase I from Trichoderma reesei. J. Mol. Biol. 264, 337–349 [DOI] [PubMed] [Google Scholar]
- 32. von Ossowski I., Ståhlberg J., Koivula A., Piens K., Becker D., Boer H., Harle R., Harris M., Divne C., Mahdi S., Zhao Y., Driguez H., Claeyssens M., Sinnott M. L., Teeri T. T. (2003) Engineering the exo-loop of Trichoderma reesei cellobiohydrolase, Cel7A. A comparison with Phanerochaete chrysosporium Cel7D. J. Mol. Biol. 333, 817–829 [DOI] [PubMed] [Google Scholar]
- 33. Beckham G. T., Bomble Y. J., Bayer E. A., Himmel M. E., Crowley M. F. (2011) Applications of computational science for understanding enzymatic deconstruction of cellulose. Curr. Opin. Biotechnol. 22, 231–238 [DOI] [PubMed] [Google Scholar]
- 34. Beckham G. T., Matthews J. F., Bomble Y. J., Bu L., Adney W. S., Himmel M. E., Nimlos M. R., Crowley M. F. (2010) Identification of amino acids responsible for processivity in a family 1 carbohydrate-binding module from a fungal cellulase. J. Phys. Chem. B 114, 1447–1453 [DOI] [PubMed] [Google Scholar]
- 35. Bu L., Beckham G. T., Crowley M. F., Chang C. H., Matthews J. F., Bomble Y. J., Adney W. S., Himmel M. E., Nimlos M. R. (2009) The energy landscape for the interaction of the family 1 carbohydrate-binding module and the cellulose surface is altered by hydrolyzed glycosidic bonds. J. Phys. Chem. B 113, 10994–11002 [DOI] [PubMed] [Google Scholar]
- 36. Taylor C. B., Talib M. F., McCabe C., Bu L., Adney W. S., Himmel M. E., Crowley M. F., Beckham G. T. (2012) Computational investigation of glycosylation effects on a family 1 carbohydrate-binding module. J. Biol. Chem. 287, 3147–3155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nimlos M. R., Beckham G. T., Matthews J. F., Bu L., Himmel M. E., Crowley M. F. (2012) Binding preferences, surface attachment, diffusivity, and orientation of a family 1 carbohydrate-binding module on cellulose. J. Biol. Chem. 287, 20603–20612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bu L., Nimlos M. R., Shirts M. R., Ståhlberg J., Himmel M. E., Crowley M. F., Beckham G. T. (2012) Product binding varies dramatically between processive and nonprocessive cellulase enzymes. J. Biol. Chem. 287, 24807–24813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bu L., Beckham G. T., Shirts M. R., Nimlos M. R., Adney W. S., Himmel M. E., Crowley M. F. (2011) Probing carbohydrate product expulsion from a processive cellulase with multiple absolute binding free energy methods. J. Biol. Chem. 286, 18161–18169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Stenlid J. (1985) Population-structure of Heterobasidion annosum as determined by somatic incompatibility, sexual incompatibility, and isoenzyme patterns. Can. J. Bot. 63, 2268–2273 [Google Scholar]
- 41. Kremer S. M., Wood P. M. (1992) Evidence that cellobiose oxidase from Phanerochaete chrysosporium is primarily an Fe(III) reductase. Kinetic comparison with neutrophil NADPH oxidase and yeast flavocytochrome b2. Eur. J. Biochem. 205, 133–138 [DOI] [PubMed] [Google Scholar]
- 42. Shevchenko A., Wilm M., Vorm O., Mann M. (1996) Mass spectrometric sequencing of proteins from silver stained polyacrylamide gels. Anal. Chem. 68, 850–858 [DOI] [PubMed] [Google Scholar]
- 43. Grigoriev I. V., Nordberg H., Shabalov I., Aerts A., Cantor M., Goodstein D., Kuo A., Minovitsky S., Nikitin R., Ohm R. A., Otillar R., Poliakov A., Ratnere I., Riley R., Smirnova T., Rokhsar D., Dubchak I. (2011) The genome portal of the Department Of Energy Joint Genome Institute. Nucleic Acids Res., 1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. McPherson A. (1982) Preparation and Analysis of Protein Crystals, John Wiley & Sons, New York [Google Scholar]
- 45. Bailey S. (1994) The CCP4 suite. Programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 46. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Guex N., Peitsch M. C. (1997) SWISS-MODEL and the Swiss-PDBViewer. An environment for comparative protein modeling. Electrophoresis 18, 2714–2723 [DOI] [PubMed] [Google Scholar]
- 49. Emsley P., Cowtan K. (2004) COOT. Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 50. Ubhayasekera W., Muñoz I. G., Vasella A., Ståhlberg J., Mowbray S. L. (2005) Structures of Phanerochaete chrysosporium Cel7D in complex with product and inhibitors. FEBS J. 272, 1952–1964 [DOI] [PubMed] [Google Scholar]
- 51. Payne C. M., Baban J., Horn S. J., Backe P. H., Arvai A. S., Dalhus B., Bjørås M., Eijsink V. G., Sørlie M., Beckham G. T., Vaaje-Kolstad G. (2012) Hallmarks of processivity in glycoside hydrolases from crystallographic and computational studies of the Serratia marcescens chitinases. J. Biol. Chem. 287, 36322–36330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Cruys-Bagger N., Elmerdahl J., Praestgaard E., Tatsumi H., Spodsberg N., Borch K., Westh P. (2012) Pre-steady-state kinetics for hydrolysis of insoluble cellulose by cellobiohydrolase Cel7A. J. Biol. Chem. 287, 18451–18458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jalak J., Väljamäe P. (2010) Mechanism of initial rapid rate retardation in cellobiohydrolase catalyzed cellulose hydrolysis. Biotechnol. Bioeng. 106, 871–883 [DOI] [PubMed] [Google Scholar]
- 54. Ståhlberg J., Johansson G., Pettersson G. (1993) Trichoderma reesei has no true exo-cellulase. All intact and truncated cellulases produce new reducing end groups on cellulose. Biochim. Biophys. Acta 1157, 107–113 [DOI] [PubMed] [Google Scholar]
- 55. Fox J. M., Levine S. E., Clark D. S., Blanch H. W. (2012) Initial- and processive-cut products reveal cellobiohydrolase rate limitations and the role of companion enzymes. Biochemistry 51, 442–452 [DOI] [PubMed] [Google Scholar]
- 56. Gruno M., Väljamäe P., Pettersson G., Johansson G. (2004) Inhibition of the Trichoderma reesei cellulases by cellobiose is strongly dependent on the nature of the substrate. Biotechnol. Bioeng. 86, 503–511 [DOI] [PubMed] [Google Scholar]
- 57. Horn S. J., Sikorski P., Cederkvist J. B., Vaaje-Kolstad G., Sørlie M., Synstad B., Vriend G., Vårum K. M., Eijsink V. G. (2006) Costs and benefits of processivity in enzymatic degradation of recalcitrant polysaccharides. Proc. Natl. Acad. Sci. U.S.A. 103, 18089–18094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Horn S. J., Sørbotten A., Synstad B., Sikorski P., Sørlie M., Vårum K. M., Eijsink V. G. (2006) Endo/exo mechanism and processivity of family 18 chitinases produced by Serratia marcescens. FEBS J. 273, 491–503 [DOI] [PubMed] [Google Scholar]
- 59. Payne C. M., Bomble Y. J., Taylor C. B., McCabe C., Himmel M. E., Crowley M. F., Beckham G. T. (2011) Multiple functions of aromatic-carbohydrate interactions in a processive cellulase examined with molecular simulation. J. Biol. Chem. 286, 41028–41035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. van Aalten D. M., Synstad B., Brurberg M. B., Hough E., Riise B. W., Eijsink V. G., Wierenga R. K. (2000) Structure of a two-domain chitotriosidase from Serratia marcescens at 1.9 Å resolution. Proc. Natl. Acad. Sci. U.S.A. 97, 5842–5847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Koivula A., Kinnari T., Harjunpaa V., Ruohonen L., Teleman A., Drakenberg T., Rouvinen J., Jones T. A., Teeri T. T. (1998) Tryptophan 272. An essential determinant of crystalline cellulose degradation by Trichoderma reesei cellobiohydrolase Cel6A. FEBS Lett. 429, 341–346 [DOI] [PubMed] [Google Scholar]
- 62. Jalak J., Kurasin M., Teugjas H., Väljamäe P. (2012) Endo-exo synergism in cellulose hydrolysis revisited. J. Biol. Chem. 287, 28802–28815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lin Y., Silvestre-Ryan J., Himmel M. E., Crowley M. F., Beckham G. T., Chu J.-W. (2011) Protein allostery at the solid-liquid interface. Endoglucanase attachment to cellulose affects glucan clenching in the binding cleft. J. Am. Chem. Soc. 133, 16617–16624 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.