

Background: The Ebola virus matrix protein (VP40) regulates the plasma membrane assembly and egress of the Ebola virus.
Results: The plasma membrane induces membrane penetration of the VP40 C-terminal domain.
Conclusion: Membrane penetration by VP40 is important for VP40 cellular localization, oligomerization, and viral budding.
Significance: A better understanding of VP40-membrane interactions will help us to understand Ebola virus assembly and budding.
Keywords: Lipid-binding Protein, Membrane Bilayer, Membrane Biophysics, Virus, Virus Assembly, Ebola, Lipid Binding, Membrane Penetration, Oligomerization, Plasma Membrane
Abstract
Ebola, a fatal virus in humans and non-human primates, has no Food and Drug Administration-approved vaccines or therapeutics. The virus from the Filoviridae family causes hemorrhagic fever, which rapidly progresses and in some cases has a fatality rate near 90%. The Ebola genome encodes seven genes, the most abundantly expressed of which is viral protein 40 (VP40), the major Ebola matrix protein that regulates assembly and egress of the virus. It is well established that VP40 assembles on the inner leaflet of the plasma membrane; however, the mechanistic details of plasma membrane association by VP40 are not well understood. In this study, we used an array of biophysical experiments and cellular assays along with mutagenesis of VP40 to investigate the role of membrane penetration in VP40 assembly and egress. Here we demonstrate that VP40 is able to penetrate specifically into the plasma membrane through an interface enriched in hydrophobic residues in its C-terminal domain. Mutagenesis of this hydrophobic region consisting of Leu213, Ile293, Leu295, and Val298 demonstrated that membrane penetration is critical to plasma membrane localization, VP40 oligomerization, and viral particle egress. Taken together, VP40 membrane penetration is an important step in the plasma membrane localization of the matrix protein where oligomerization and budding are defective in the absence of key hydrophobic interactions with the membrane.
Introduction
Viral hemorrhagic fevers such as that caused by the Ebola virus present a serious health threat in central and eastern Africa with fatality rates as high as 90% (1, 2). The Ebola virus from the Filoviridae family uses a negative sense RNA genome encoding seven proteins to replicate in the host cell (3, 4). The nucleoprotein, VP24,4 VP30, VP35, and L protein constitute the nucleocapsid, which is critical for transcription and viral replication (4). The glycoprotein is rooted in the lipid envelope of the virus and is responsible for entry of virions (5) in an NPC1-dependent manner (6). VP40 and the enigmatic VP24 are the viral matrix proteins and are important for budding as well as virus structure and stability (7–10). VP40 is the major matrix protein, is the most abundantly expressed protein of the virus, and plays a central role in the budding of Ebola from the plasma membrane (PM). For example, expression of VP40 alone in mammalian cells is sufficient to form virus-like particles (VLPs) that have similar characteristics to the actual Ebola virus (11, 12). Additionally, studies have shown that in the absence of VP40 the nucleocapsid is not effectively transported to the PM, the site of assembly and budding, where it is incorporated into the virions (13). Therefore, understanding how VP40 regulates assembly of VLPs both in vitro and in live cells is crucial for understanding the viral life cycle and could have a significant impact in identifying potential therapeutic targets. The assembly of VLPs by Ebola VP40 also represents an attractive model for studying the assembly of the virus in a biosafety level 2 setting because the VLPs are noninfectious.
VP40 associates with the PM (14) where it initiates assembly, oligomerization (15, 16), and recruitment of the nucleoprotein. In addition to membrane association, VP40 has been shown to interact or associate with host cell factors such as the endosomal sorting complex required for transport (ESCRT) machinery (10, 17), COPII proteins (19), and actin (20, 21), which have been implicated in the budding, transport, and movement of VP40, respectively. In addition, host cell protein kinases may play an important role in Ebola infectivity as c-Abl1 has been shown to phosphorylate Tyr13 in VP40 (22). In light of the aforementioned studies, how the virus assembles on the PM prior to virion release remains poorly understood. PM localization of VP40 is thought to be an important step in this process as studies have shown that hydrophobic residues in the C-terminal domain such as Leu213 are critical to localization and budding (23). Moreover, VP40 oligomers have been detected in VLPs and UV-inactivated virions (11, 14) and reside predominately in filamentous structures emanating from the PM (24). Thus, VP40 oligomerization is thought to occur on the PM where oligomers have been selectively shown to reside (24). VP40 has primarily been shown to oligomerize into hexamers and octamers (11, 15, 16, 25), which share a similar intradimeric (monomer-monomer interface) antiparallel interface, but larger oligomeric structures have been detected in live cells and may also play a critical role in viral assembly and egress (24).
VP40 oligomers are essential for the formation of VLPs and have been found to be associated with detergent-resistant membranes (14), suggesting that the PM may play an active role in the oligomerization of VP40. Oligomerization of the matrix protein on the plasma membrane may serve as a scaffold to recruit host proteins as well as provide the necessary force to bring about membrane deformation and virus particle formation. Thus, understanding the molecular basis of VP40-PM association is critical to unraveling how the protein buds form at the PM.
In this study, we investigated the role of the VP40 C-terminal domain in membrane association and membrane penetration. Monolayer penetration analysis was used to investigate the molecular basis of VP40 membrane penetration in vitro. A combination of cellular imaging, VLP egress, and total internal reflection (TIRF) microscopy coupled with number and brightness (N&B) analysis, and site-directed mutagenesis was used to study VP40 assembly and egress in cells. N&B analysis allows for measurement of the average number of molecules as well as brightness in each pixel of a fluorescence microscopy image, allowing detection of the oligomeric state of fluorescently labeled proteins. Together, these studies revealed that a hydrophobic interface in the VP40 C-terminal domain penetrates into the PM, and this serves as an important step in VP40 oligomerization. Moreover, hydrophobic mutants that knock out PM penetration also greatly reduced VLP egress. This study demonstrates the value of the in vitro and cellular biophysical approaches to study the mechanism of VP40 membrane association and VLP formation.
EXPERIMENTAL PROCEDURES
Materials
1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE), and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylserine (POPS) were purchased from Avanti Polar Lipids, Inc. (Alabaster, AL) and used without further purification. Restriction endonucleases and enzymes for molecular biology were obtained from New England Biolabs (Beverly, MA). The QuikChange site-directed mutagenesis kit was from Agilent Technologies (Santa Clara, CA). The Nunc Lab-Tek I chambered cover glasses (8-well) and bicinchoninic acid (BCA) protein assay kit were from Thermo Fisher Scientific (Waltham, MA). Lipofectamine 2000 and Lipofectamine LTX were from Invitrogen. The lactadherin mCherry-C2 domain PS sensor was a kind gift from Dr. Sergio Grinstein (University of Toronto).
DNA Mutagenesis
VP40 mutants were generated in pcDNA3.1 using a QuikChange site-directed mutagenesis kit and then subcloned into the pGEX-4T-1 vector using the EcoRI and XhoI restriction sites. The introduced mutations were confirmed by automated DNA sequencing using the primer 5′-CCG GAA TTC GCC ATG AGG CGG GTT ATA-3′.
Protein Expression and Purification
WT VP40 and mutants were expressed and purified from Escherichia coli BL21(DE3) cells. An overnight culture (25 ml) of E. coli BL21(DE3) cells harboring the GST-VP40 plasmid was grown for 16 h at 37 °C and then added to 1 liter of LB containing 100 μg/ml ampicillin. The cells were grown at 37 °C with shaking at 250 rpm. The optical density of the solution was monitored at 600 nm, and when the absorbance reached 0.8, VP40 expression was induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside. At this time, the flask was transferred to a shaker at 25 °C with shaking at 250 rpm for 5 h. Cells were then harvested for 10 min at 6,000 × g. The resulting pellet was washed once with 20 ml of PBS and then lysed with PBS, pH 8.0 containing 50 μm phenylmethylsulfonyl fluoride (PMSF), 1 tablet of protease inhibitor mixture, 10 mg of lysozyme, and 1% Triton X-100. The suspension was then incubated on ice for 20 min. After 20 min, the sample was sonicated for 10 min using a 20-s sonication pulse followed by 40-s cooling on ice at 4 °C. This was followed by centrifugation at 50,000 × g for 30 min. The supernatant was collected and transferred to a sterile 50-ml tube, and 1 ml of GST-TagTM resin (Novagen, Madison, WI) was added. The solution was incubated at 4 °C for 2 h with stirring at 50 rpm. After this time, the mixture was poured onto a column, which was washed twice with 20 ml of PBS, pH 8.0. After the wash, the column was capped, and 10 units of thrombin were added in 1 ml of thrombin cleavage buffer to cleave the GST tag. The column was then incubated for 20 h at 4 °C for the reaction to proceed. After this time, the column was uncapped, and the flow-through was collected. 5 ml of PBS, pH 8.0 was then added to collect any cleaved VP40 that was not obtained in the flow-through. The two samples were then pooled and passed through a para-aminobenzamidine (1-ml) column to remove any residual thrombin. VP40 was collected in the flow-through and in a 10-ml wash step with PBS, pH 8.0. Samples were then pooled and concentrated using Millipore centrifugal filter units (30,000 Da) with spinning at 3,500 × g in 15-min cycles. The sample was washed three times with PBS (6 ml each) to concentrate the VP40 protein to a final volume of 1 ml. Protein concentrations were then determined with the BCA method, and protein purity was assessed using a 12% polyacrylamide gel. Protein aliquots of 2 mg/ml were made using storage buffer (PBS buffer, pH 7.4, 140 mm NaCl, 2.7 mm KCl, 10 mm Na2HPO4, 1.8 mm KH2PO4).
Monolayer Penetration Analysis
The penetration of the wild type and mutant VP40 proteins into the phospholipid monolayer was investigated by measuring the change in surface pressure (π) of invariable surface area during addition of the proteins. The experiments were performed using a 1-ml circular Teflon trough and wire probe connected to a Kibron MicroTrough X (Kibron, Inc., Helsinki, Finland). A lipid monolayer (prepared in hexane/ethanol (9:1, v/v)) containing various combinations of phospholipids was spread onto the subphase composed of 10 mm HEPES, 0.16 m KCl, pH 7.4 until the desired initial surface pressure, π0, was reached. After stabilization of the signal (∼5 min), 10 μg of protein was injected into the subphase through a hole in the wall of the trough. The surface pressure change (Δπ) was monitored for 45 min. The Δπ value reached a maximum after 20 min in all experiments.
Cell Culture and Imaging
Human embryonic kidney (HEK293) cells were cultured and maintained at 37 °C in a 5% CO2 humidified incubator supplemented with DMEM (low glucose) containing 10% FBS and 1% penicillin/streptomycin. After trypsinization, cells were transferred from a T-25 tissue culture flask to an 8-well plate used for imaging. Cells were then grown to 50–80% confluence and transfected with 1 μg of DNA/dish using Lipofectamine 2000 according to the manufacturer's protocol. Chinese hamster ovary-K1 (CHO-K1) cells were cultured and maintained at 37 °C in a 5% CO2 humidified incubator supplemented with DMEM/F-12 (low glucose) containing 10% FBS and 1% penicillin/streptomycin. After trypsinization, cells were transferred from a T-25 tissue culture flask to an 8-well plate used for imaging. Cells were then grown to 50–80% confluence and transfected with 1 μg of DNA/dish using Lipofectamine LTX according to the manufacturer's protocol. Cells were imaged using a Zeiss LSM 710 confocal microscope using a Plan Apochromat 63× 1.4 numerical aperture oil objective. The 488-nm line of the argon ion laser was used for excitation of EGFP. The laser power was maintained at 1% throughout the experiment with the emission collected through a 493–556-nm filter.
Cells were imaged to investigate the PM localization of WT VP40 and mutations in HEK293 cells to quantify the differences between WT and mutations. PM localization was quantified for each construct by imaging three independent experiments and counting at least 100 cells for each experiment (see Fig. 3H). VP40 is well established to localize to the inner leaflet of the PM of mammalian cells. PM localization was counted as positive in WT- or mutant-expressing cells when EGFP localization was observed on the PM or as puncta associated with or structures emanating from the PM. The extent or percentage of PM localization refers to the percentage of cells in which PM localization of the EGFP tag could actually be detected with confocal microscopy. PM localization was detected for VP40 by scanning and imaging cells to assess EGFP intensity at the PM compared with the cytoplasm and total cell intensity. To further demonstrate the PM localization of VP40, EGFP-VP40 was coexpressed with a robust PS sensor, the lactadherin C2 domain harboring an mCherry tag. The overlap in VP40 and lactadherin C2 signal demonstrates the predominant PM localization of VP40 (see Fig. 3B) as reported previously (18).
FIGURE 3.

VP40 cellular localization in HEK293 cells. HEK293 cells were grown in an 8-well plate and transfected with WT VP40, L213A, I293A, L295A, V298A, or L303A DNA containing an EGFP tag. Cells were imaged after 24 h using a Zeiss LSM 710 confocal microscope with a 63× 1.4 numerical aperture oil objective. A, WT VP40 exhibits extensive PM localization and strong visual evidence of VP40-enriched particles emanating from the PM. Cells were imaged to investigate the PM localization of WT VP40 and mutations in HEK293 cells to quantify the differences between WT and mutations. B, VP40 is well established to localize and associate with the inner leaflet of the PM of mammalian cells. To demonstrate the PM localization of VP40, EGFP-VP40 was coexpressed with a robust PS sensor, the lactadherin (Lact) C2 domain (18) harboring an mCherry tag. The overlap in VP40 and lactadherin C2 signal demonstrates the predominant PM localization of VP40 as reported previously (18). PM localization was counted in WT- or mutant-expressing cells when EGFP localization was observed on the PM or as puncta associated with or structures emanating from the PM. The extent or percentage of PM localization refers to the percentage of cells in which PM localization of the EGFP tag could actually be detected with confocal microscopy. C, cellular localization of L213A was nearly undetectable in the majority of cells. D, I293A had markedly reduced PM localization in HEK293 cells. E, L295A behaved similarly to I293A with markedly reduced PM localization. F, V298A exhibited some PM localization in ∼20% of HEK293 cells, but when localization was observed it was minor (only a few diffuse spots as shown) compared with WT and the L303A control. G, L303A, a residue that is important for interactions with Sec24C, displays a similar extent of PM localization. H, a histogram was plotted to demonstrate the percentage of plasma membrane localization observed for WT and mutations in HEK293 cells. Experiments were repeated in triplicate using at least 100 cells in each experiment to determine the S.D. (error bars) as shown. *, p < 0.0002; **, p < 0.0001; NS, not significant. White scale bars, 10 μm; red scale bars, 5 μm.
TIRF Imaging and Analysis
TIRF imaging was performed using a home-built TIRF imaging system (model number IX81 microscope (Olympus, Melville, NY) as described previously (26). Briefly, images were collected using a Cascade 512B electron-multiplying charge-coupled device camera. Samples were illuminated with the 488-nm line from an argon ion laser (Melles Griot, Albuquerque, NM) through a 60× 1.45 numerical aperture oil objective (Olympus). To ensure cell integrity, cells were maintained at 37 °C using a thermostated stage (Tokai Hit, Shizuoka, Japan). Images were collected at 256 × 256 pixels with a 50-ms exposure time per frame (4,000 total frames were collected). Images were saved as 16-bit unsigned and imported into the SimFCS software (Laboratory for Fluorescence Dynamics, Irvine, CA). TIRF image series were analyzed using SimFCS (Laboratory for Fluorescence Dynamics). For N&B analysis, 512 frames were analyzed per image series. HEK293 cells expressing monomeric EGFP were used as a brightness standard and imaged under the same conditions as EGFP-VP40 and respective mutations. The brightness of the EGFP was used as the brightness of the monomer as described previously (21).
Recently, determination of fluorescently labeled protein clustering has become much more robust with N&B analysis. N&B analysis is based upon moment analysis and allows for measurement of the average number of molecules as well as brightness in each pixel of a fluorescence microscopy image (27). Here the average brightness of a particle is determined from the ratio at each pixel of the variance to intensity. The number of fluctuating particles is determined by dividing the average intensity by the brightness at each pixel. N&B analysis accounts for limitations such as autofluorescence, scattering, bright immobile particles, and fast moving particles that are dim by calculating the total variance, which also incorporates detector noise. Variance is proportional to the particle brightness for particles fluctuating in the focal volume; however, the variance of the immobile particles, scattering, autofluorescence, and detector noise is proportional to the intensity of these components. Thus, only fluorescence fluctuations that are dependent upon the mobile particles (square of the brightness) have a ratio of the variance to intensity >1. Brightness maps then allow for pixel resolution of the clustering of fluorescently labeled proteins. The selection window for analysis in the brightness versus intensity plot was based on the average brightness of a monomer (0.104), which allowed for selection of oligomeric size based upon addition of each monomer to yield dimer, trimer, etc. for WT VP40 (n = 7), L213A (n = 5), and L295A (n = 5). Thus, the selection window for each species is based upon the average brightness.
For the electron-multiplying charge-coupled device camera, Equations 1 and 2 were used to compute the number and brightness,
where N and B are the apparent number and the brightness of the molecule, I is the average intensity, σ2 is the variance, and offset and σ02 are the intensity and noise variance due to the camera, respectively. With these parameters properly calibrated, we obtained the distribution of the brightness of each individual pixel in the image of the cell under investigation.
VLP Assays
To assess VLP formation, EGFP-VP40, EGFP-VP40-L213A, EGFP-VP40-I293A, EGFP-VP40-L295A, EGFP-VP40-V298A, EGFP-VP40-L303A, or EGFP was transfected into CHO-K1 cells using a standard transfection protocol. VLPs were isolated from cellular media as described previously (11) 48 h post-transfection. Cells were collected and lysed to measure the cellular concentration of each construct using the BCA protein assay method as a control for total protein content. Cell lysates and VLP samples were mixed with SDS loading buffer and boiled before loading on an 8% SDS-polyacrylamide gel. The gel was then transferred to a nitrocellulose membrane (Bio-Rad) that was later incubated with primary rabbit polyclonal anti-EGFP antibody (Thermo Scientific) (1:1,000 dilutions) or rabbit polyclonal anti-GAPDH (Santa Cruz Biotechnology) followed by goat anti-rabbit HRP (Bio-Rad) conjugate as secondary antibody according to the manufacturer's protocol (Thermo Scientific). Blots were exposed using ImageQuant LAS 4000 (GE Healthcare). This method allows a comparison of VP40 in VLPs versus cell lysate using GAPDH as a loading control. Experiments were performed in triplicate on different days.
RESULTS
Ebola VP40 Penetrates Membranes That Recapitulate the Plasma Membrane
It is well established that VP40 associates with the plasma membrane (3, 4), and this has been proposed and shown to be mediated by its C-terminal domain (28). VP40 is composed of two domains that have unique functions in the viral life cycle (see Fig. 1). The N-terminal domain has been shown to mediate oligomerization of VP40 in vitro and in cells (16) through an intradimeric interface consisting of Trp95, Arg149, Arg151, Glu160, and Gln184. The C-terminal domain of VP40 has been shown to mediate the membrane association of VP40 (28), but the molecular basis of this interaction is still poorly understood. A number of lipid-binding proteins including modules such as FYVE, PX, and ENTH (epsin N-terminal homology) domains that associate strongly with cellular membranes have been shown to penetrate membranes following the recognition of specific lipids (29–31). This lipid coordination partially reduces positive potential surrounding the binding pocket, facilitating insertion of the adjacent hydrophobic residues into the bilayer. Other proteins harboring C2 lipid-binding domains are inserted in response to Ca2+ coordination (32), whereas other proteins harboring C1 domains may be promiscuous with their insertion but still have some selectivity in this process as they more robustly penetrate membranes harboring anionic lipids (33). Initially, the ability of VP40 to penetrate membranes was examined by monitoring changes in surface tension of lipid monolayers. Phospholipid monolayers at the air-water interface serve as a highly sensitive tool for measuring the membrane penetration by peripheral proteins (34). A PM POPC/POPE/POPS/POPI/cholesterol (12:35:22:9:22) or nuclear membrane (NM) POPC/POPE/POPS/POPI/cholesterol (61:21:4:7:7) monolayer mimetic of initial surface pressure π0 was spread at constant area, and the change in surface pressure (Δπ) after the injection of the protein was monitored (Fig. 2A). Δπ is inversely proportional to π0, and an extrapolation of Δπ versus π0 yields the critical surface pressure (πc), which specifies an upper limit of π0 into which a protein can penetrate (34). As shown in Fig. 2A, VP40 has low intrinsic membrane-penetrating ability into the NM. The πc value of an NM mimetic monolayer was found to be ∼25 mN/m. In contrast, with the PM mimetic, VP40 substantially penetrated into the monolayer with a πc value of ∼34 dynes/cm (Fig. 2A). This is significant as biological membranes have been shown to have surface pressure values in the 30–35 mN/m range (35, 36). Thus, at low levels of abundant PM lipids such as PS, phosphatidylethanolamine, and/or cholesterol, VP40 does not significantly insert into membranes.
FIGURE 1.

VP40 x-ray structure depicting the N- and C-terminal domains and hydrophobic residues mutated in this study. A, the VP40 x-ray structure (Protein Data Bank code 1ES6) (40) is shown with the N-terminal domain in dark gray and the C-terminal domain in light gray. The N-terminal domain has been shown to be involved in oligomerization, and the C-terminal domain has been deemed important for membrane binding. A hydrophobic interface composed of two loops harboring Leu213, Ile293, Leu295, and Val298 was the focus of this study. All four residues were mutated to Ala. As a negative control, Leu303, adjacent but more buried than this hydrophobic interface, was also mutated to Ala. Leu303 has been shown to be part of a region in VP40 that interacts with Sec24C and was not expected to be involved in membrane penetration. B, the VP40 x-ray structure is presented looking down onto the hydrophobic interface in the C-terminal domain with key residues mutated in this study. Leu213, Ile293, Leu295, and Val298 were mutated to Ala to test their role in membrane penetration. A control residue, Leu303, is shown in cyan and was also mutated to Ala. Leu303 is just below and to the right of the hydrophobic patch we propose is involved in membrane penetration. In addition, Leu303 was a viable negative control as it has been shown to be part of a region that interacts with Sec24C. Images were generated in Mac PyMOL.
FIGURE 2.
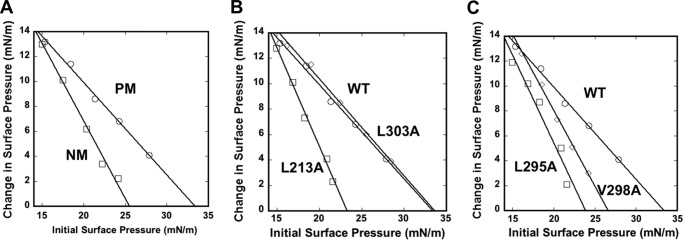
Monolayer penetration of VP40 and hydrophobic mutations. A, insertion of WT VP40 into a PM mimetic (POPC/POPE/POPS/POPI/cholesterol (12:35:22:9:22)) monolayer (circles) or an NM mimetic (POPC/POPE/POPS/POPI/cholesterol (61:21:4:7:7)) monolayer (squares) monitored as a function of π0. B, WT VP40 (circles), L213A (squares), and L303A (diamonds) were monitored for insertion into a PM mimetic monolayer as a function of π0. C, WT VP40 (circles), L295A (squares), and V298A (diamonds) were monitored for insertion into a PM mimetic monolayer as a function of π0. The subphase was 10 mm HEPES buffer, pH 7.4 with 0.16 m KCl for all measurements.
To investigate the molecular basis of VP40 PM penetration, we prepared mutants of hydrophobic residues present in a patch in the C-terminal domain (See Fig. 1). Previously, Leu213 has been shown to abrogate VP40 PM localization and egress when mutated, although the molecular basis of its role has not been delineated (23). Leu213 (shown in Fig. 1, A and B) resides on the same interface as Ile293, Leu295, and Val298, which are in a second loop region exposed on the same surface of VP40. To investigate whether these residues are involved in PM penetration by VP40, we prepared mutants of each amino acid to Ala. In addition, we prepared a control mutation at the Leu303 position. Leu303 is adjacent to the hydrophobic interface we propose to penetrate the membrane, is slightly more buried, and has been shown to interact with the protein Sec24C (19). Thus, L303A was not expected to affect membrane penetration but serve as a control for hydrophobic mutagenesis within the VP40 C-terminal domain.
Monolayer penetration studies performed at saturating concentrations of mutants at the monolayer interface revealed that L213A significantly reduced membrane penetration with a πc value of 24 mN/m for the PM mimetic (Fig. 2B), reminiscent of the low intrinsic penetrating ability of VP40 into the NM mimetic. In addition, L295A and V298A greatly reduced the πc for the PM to 24 and 26 mN/m, respectively. However, the L303A mutant behaved similarly to WT VP40 with a πc value of ∼34 mN/m. This strongly suggests that the VP40 PM penetration is mediated by a hydrophobic interface in the C-terminal domain.
VP40 Cellular Localization
VP40 is well established to localize and associate with the inner leaflet of the PM of mammalian cells (14, 24) where Ebola assembly and egress occur. In addition, VP40 is often found enriched in puncta at the PM and membrane tubules or sites of viral egress that extend from the PM of mammalian cells. In addition, VP40 oligomers, which are critical to egress, are found highly enriched on the PM specifically at these membrane protrusion sites (24). To analyze the effect of the hydrophobic mutants on the PM localization of VP40 in human cells, we transfected HEK293 cells with plasmids encoding WT EGFP-VP40, L213A, I293A, L295A, V298A, and L303A. The cells were imaged 18 h post-transfection using confocal microscopy. As shown in Fig. 3A, WT EGFP-VP40 was predominantly localized to the PM with strong visual evidence of VLP extensions stemming from the PM (Fig. 3A). The PM localization of VP40 was further demonstrated when EGFP-VP40 was coexpressed in HEK293 cells with an mCherry-lactadherin C2 domain, which binds and marks PS on the inner leaflet of the PM as well as the cytoplasmic face of internal organelles (Fig. 3B). Note that both VP40 and lactadherin C2 were found enriched at the PM, but only lactadherin C2 was significantly detected at internal membranes.
In contrast to WT, L213A displayed a diffuse cytosolic localization and a drastic reduction in PM localization (Fig. 3C). I293A, L295A, and V298A also significantly reduced PM localization and evidence of VLP egress (Fig. 3, D–F). To quantify the effect of these mutations on PM localization of VP40, experiments were performed in triplicate with at least 100 cells imaged for each respective construct in each experiment. PM localization was counted as positive in WT- or mutant-expressing cells when EGFP localization was observed on the PM or as puncta associated with or structures emanating from the PM. The extent or percentage of PM localization refers to the percentage of cells in which PM localization of the EGFP tag could actually be detected with confocal microscopy. For I293A, L295A, and V298A, only 16–21% of cells exhibited detectable PM localization, and for those cells where PM localization was detectable, the number of PM spots was minimal as represented in Fig. 3F for V298A. These three mutations also exhibited a loss of evident VLP egress, consistent with their role in PM penetration. L303A (shown in Fig. 3G) displayed similar PM localization and VLP egress in HEK293 cells and when analyzed for more than 100 cells repeated in triplicate did not display a statistically significant reduction in PM localization (Fig. 3H). Cellular studies support the in vitro findings as PM targeting efficiency of the mutations correlates with their ability to penetrate the PM.
VP40 Oligomerization
VP40 oligomers are critical to Ebola assembly and egress because when oligomers are abolished via mutagenesis viral infectivity and egress are drastically reduced (16). VP40 oligomerizes on the inner leaflet of the PM into hexamers, octamers, and larger oligomers that are enriched in PM protrusion sites (24). VP40 oligomerization was previously resolved with a combination of confocal and TIRF microscopy to elucidate the localization and origin of VP40 oligomers. TIRF is well suited for this approach as it selectively excites fluorescent molecules on or near the PM. However, the evanescent wave critical to fluorophore detection in TIRF can still protrude past the PM of cells and excite fluorophores in a premembrane zone. Thus, a small portion of EGFP intensity in these experiments comes from VP40 that is not bound to the PM; however, when confocal microscopy was used to further analyze VP40 oligomers and rule out oligomers in this premembrane zone, it was shown that VP40 oligomers reside exclusively on the PM. Specifically, VP40 oligomers were found to be enriched in PM protrusion sites consistent with viral egress (24) because when mutants that abrogate oligomerization were expressed the PM localization, membrane protrusion sites, and VP40 oligomerization were no longer detected in live human cells (24).
To understand how oligomerization of VP40 is related to membrane penetration in HEK293 cells, we used TIRF microscopy and N&B analysis to study the oligomerization state of VP40 and hydrophobic mutants on the PM. N&B analysis is based upon moment analysis and allows for measurement of the average number of molecules as well as brightness in each pixel of a fluorescence microscopy image (27). Here the average brightness of a particle is determined from the ratio at each pixel of the variance to intensity. The average number of fluctuating particles is determined by dividing the average intensity by the brightness at each pixel. The cellular system is calibrated with monomeric EGFP to determine the brightness versus intensity plot for a monomer, which then allows for selection of oligomers in a brightness versus intensity plot (see Fig. 4D) based upon the brightness of the monomer. This allows visualization of monomers, dimers, trimers, etc. and their specific cellular localization as shown in Fig. 4. Thus, this method is well suited to resolve the spatial distribution of VP40 and VP40 mutant oligomers in live cells (24).
FIGURE 4.

Brightness analysis of VP40 in HEK293 cells. A, membrane protrusion sites emanating from the PM were inspected with TIRF microscopy. B, TIRF average intensity image of an HEK293 cell transfected with plasmid expressing EGFP-VP40 demonstrates sites of signal enrichment and a number of sites of membrane protrusions and viral egress. C, brightness image (variance/intensity) of the same cell demonstrates the enriched sites of VLP egress where significant EGFP signal (red) is detected. D, brightness versus intensity plot displaying monomers (brightness of 1.104) (red box), dimers (blue box), and hexamers (green box). E, brightness distribution of VP40 with selected pixels from D displaying localization of monomers (red), dimers (blue), and hexamers (green). F, frequency versus apparent brightness plot demonstrates the extensive oligomerization of VP40 at or near the PM of HEK293 cells. The apparent brightness of a monomer is 1.104, indicating the significant frequency of a monomer but extensive enrichment of oligomers up to an apparent brightness of 12. Scale bars, 18 μm.
Shown in Fig. 4B is an average intensity plot of EGFP-VP40 from a TIRF image of an HEK293 cell (Fig. 4A) that demonstrates a number of extensions protruding from the PM. When the same cell is shown in a variance/intensity plot (Fig. 4C), a significant enrichment of EGFP intensity is observed at discrete sites, and when the brightness versus intensity (Fig. 4D) of this signal is plotted, it is evident that there is significant oligomerization of VP40 in these cells as the brightness of the calibrated EGFP monomer under the same conditions is 1.104 (y coordinate). Further analysis of this plot demonstrates as reported previously (24) that the predominant forms of VP40 in the TIRF images were monomers and dimers (see Fig. 4, D and E), although hexamers, octamers, and higher order oligomers (>8) were also highly enriched but localized almost exclusively to membrane regions extending from the PM (Fig. 5). A frequency versus apparent brightness (Fig. 4F) plot reiterates the significant oligomerization of VP40 from the TIRF images as the monomeric EGFP-VP40 is significant (apparent brightness of 1.104), but an extensive frequency signal is observed from VP40 from an apparent brightness of 1.104 up to nearly 12. In contrast, hydrophobic mutations described below have frequency versus apparent brightness plots that cluster around the monomeric apparent brightness signal of 1.104. This along with previous studies implies that the monomers and dimers build higher order oligomers that are then involved in VLP egress. Previously, the sites of protrusion were lost when VP40 mutants that abolished oligomerization were used (24), strongly suggesting that the membrane protrusion sites require VP40 membrane association and oligomerization to form.
FIGURE 5.

Brightness versus intensity analysis of EGFP-VP40 in HEK293 cells displaying oligomers. A, brightness versus intensity plot of the HEK293 cell shown in Fig. 4 highlighting hexamers (red box), octamers (blue box), and oligomers larger than octamers (green box). B, brightness distribution of VP40 with selected pixels from A displaying hexamer (red), octamers (blue), and oligomers larger than octamers (green). Oligomeric VP40 structures are enriched on the PM and filaments protruding from the cell PM. C, brightness versus intensity plot of the HEK293 cell shown in Fig. 4 highlighting oligomers larger than octamers (green box). D, brightness distribution of VP40 with selected pixels from C displaying oligomers larger than octamers (green). Scale bars, 18 μm.
To test the role of C-terminal hydrophobic residues in VP40 oligomerization, we used TIRF and N&B analysis to assess the oligomerization of L213A (Fig. 6) and L295A (Fig. 7). Both mutations displayed a very low fluorescence intensity of EGFP on the PM in a similar fashion to cellular localization experiments shown in Fig. 3. L213A and L295A also exhibited a drastic loss off brightness or oligomers (variance/intensity) at the PM (see Figs. 6B and 7B) or in the premembrane zone as well as a lack of evidence of membrane protrusion (VLP egress) sites. This suggests a lack of EGFP clustering and oligomerization for these mutations. Indeed, brightness versus intensity plots reveal little to no oligomerization for these mutants (Figs. 6, C and D, and 7C) with the predominant species in these cells being monomers and dimers. A comparison of frequency versus apparent brightness plots for WT and mutations also clearly demonstrates the significant reduction in VP40 oligomerization when membrane penetration is ablated (Figs. 4E, 6E, and 7D). It should be noted that the oligomerization of these hydrophobic mutants may be limited by the amount of EGFP signal that actually associates with the PM. Thus, it cannot be completely ruled out that lack of oligomerization in cells is solely due to lack of membrane penetration. That being said, the majority of VP40 in the cytoplasm or premembrane zone in the current and previous study (24) is monomeric or dimeric as were the signals detected for L213A and L295A, strongly suggesting that the hydrophobic amino acids are critical to PM localization, membrane penetration, and oligomerization of VP40. Thus, membrane penetration by the VP40 C-terminal domain is a prerequisite for oligomerization on the PM and formation of sites of VLP egress. These results suggest that even loss of one key hydrophobic amino acid can result in a loss of PM association and oligomerization for VP40.
FIGURE 6.
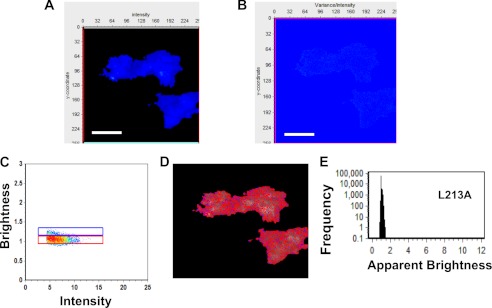
Brightness analysis of L213A in HEK293 cells. A, TIRF average intensity image of an HEK293 cell transfected with plasmid expressing L213A-EGFP demonstrates little localization on the PM, similar to that shown in Fig. 3. B, brightness image of the same cell demonstrates a lack of EGFP clustering or PM extension in a variance/intensity plot, indicating little oligomerization of L213A on the PM or from the premembrane zone. C, brightness versus intensity plot displaying monomers (red box) and dimers (blue box). D, brightness distribution of L213A with selected pixels from C displaying monomers (red) or dimers (blue). E, frequency versus apparent brightness plot demonstrates a lack of oligomerization of L213A at or near the PM of HEK293 cells. Scale bars, 18 μm.
FIGURE 7.

Brightness analysis of L295A in HEK293 cells. A, TIRF average intensity image of an HEK293 cell transfected with plasmid expressing L295A-EGFP demonstrates little L295A localization on the PM. B, brightness image of the same cell demonstrates a lack of EGFP clustering or PM extensions for L295A. C, brightness distribution of VP40 with selected pixels displaying monomers (red) or dimers (blue). D, frequency versus apparent brightness plot demonstrates a lack of oligomerization of L295A at or near the PM of HEK293 cells with the majority of the L295A apparent brightness signal clustered around the monomeric value of 1.104. Scale bars, 18 μm.
Viral Egress Studies
To investigate the role of membrane penetration in VLP egress, we collected VLPs for each construct 48 h post-transfection as described under “Experimental Procedures.” The isolated VLPs and cell lysates were subjected to Western blot (Fig. 8) for the EGFP tag using blotting for GAPDH as a loading control. Western blot results confirmed the previously obtained membrane penetration, cellular imaging, and oligomerization studies. Release of VLPs was not detectable for L213A or L295A, whereas I293A and V298A had significantly reduced VLP release. L303A, which did not influence membrane penetration of VP40, exhibited a similar level of VLP release. Taken together, C-terminal domain membrane penetration is important for VP40 plasma membrane localization and critical to viral egress.
FIGURE 8.
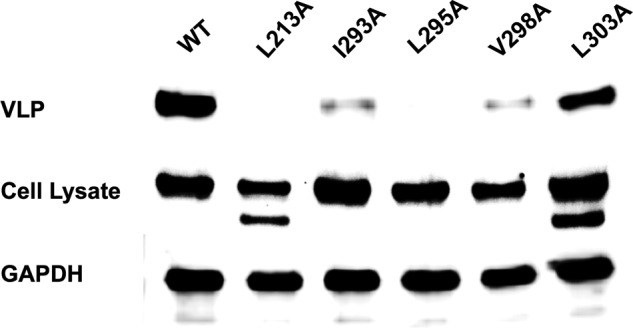
VLP egress studies of WT VP40 and hydrophobic mutations. CHO-K1 cells were transfected with WT VP40, L213A, I293A, L295A, V298A, or L303A DNA. The cell lysate and VLPs were collected after 48 h as described earlier and subjected to Western blot with anti-EGFP. The ratio of cell lysate to VLPs was maintained for each sample with GAPDH used as a loading control for total cell density. L213A and L295A VLP intensity was not detectable in this assay, whereas I293A and V298A exhibited markedly reduced VLPs.
DISCUSSION
VP40 is the major matrix protein and is the most abundantly expressed protein of the Ebola virus. To date, membrane association of VP40 is appreciated as interactions with anionic lipids have been observed (28); however, there is a paucity of quantitative biophysical information on the interactions of VP40 with biological membranes in vitro and in cells. In addition, VP40 lipid specificity or specific amino acids that interact with the membrane bilayer have yet to be elucidated. A number of studies have identified point mutations that are able to abrogate PM localization of VP40 and VLP egress (3, 16, 23), but evidence of these amino acids being involved in membrane association is also lacking. A key previous study identified the hydrophobic amino acid Leu213 to be important to VP40 PM localization, oligomerization, and VLP egress (23). This finding was intriguing to us as Leu213 resides on an interface where a cluster of hydrophobic residues are located including Ile293, Leu295, and Val298 (Fig. 1). Insertion of loops enriched in hydrophobic amino acids is often an important mechanism for peripheral proteins to achieve high affinity interactions with cellular membranes that may aid in eliciting their biological function (37). This led us to hypothesize that hydrophobic interactions between the C-terminal domain and the membrane may be important for PM association and subsequent VLP egress.
To this end, studies with phospholipid monolayers that recapitulate the PM and NM of mammalian cellular membranes demonstrated that VP40 could selectively robustly penetrate into monolayers that were enriched with lipids more abundant in the PM (notably anionic lipids like PS). Biological membranes and lipid vesicles have been shown to have surface pressures in the 30–35 mN/m range (35, 36), and VP40 is able to penetrate up to a πc of 34 mN/m. In contrast, penetration into the NM mimetic was not physiologically significant for VP40 with a πc value of 25 mN/m. It is often the case that a peripheral protein weakly associates with monolayers under conditions where they do not have high affinity for the membrane or a key cognate lipid ligand is absent. Subsequently, monolayer studies with hydrophobic mutations demonstrated their importance in membrane penetration as L213A, L295A, and V298A abrogated the selective PM-induced penetration, whereas the L303A control mutation did not (Fig. 2). These mutations also significantly reduced PM localization and VLP egress of VP40, indicating an important role in VP40-mediated membrane association.
Oligomerization of VP40 represents a critical step in the assembly of the Ebola virus (11, 16). Inhibition of this step of the viral life cycle has been found to abolish the formation and release of new virions and VLPs (3, 11, 13, 14–16). N&B analysis, a recent application, demonstrated that the predominant forms of VP40 in the cell were monomers and dimers (24). Interestingly, it was also shown that the majority of VP40 proteins on the membrane were monomers and dimers, suggesting that monomeric VP40 is recruited from the cytosol or trafficked on transport vesicles to serve as building blocks of multimerization for VP40 oligomers found predominantly in filamentous PM protrusion sites. Here we again demonstrate that VP40 oligomers are found highly enriched in PM protrusion sites and are a prerequisite for egress because when oligomerization is precluded in hydrophobic mutations so is VLP egress. L213A and L295A did not have detectable oligomers larger than dimers and in addition to loss of oligomerization did not have filamentous protrusion sites emanating from the PM. Although this could be attributed to lack of signal for the mutations penetrating into the PM, even signal detected in the premembrane zone for these mutations was primarily monomeric, analogous to WT VP40. The main determinant of VP40-mediated penetration is still unknown, and it is tempting to propose that high affinity of VP40 for an enriched PM component such as PS is sufficient to induce penetration and oligomerization. Indeed, VP40 harbors three residues adjacent to Leu213 that could interact with PS or other anionic lipids including His210, Lys212, and Arg214, all of which have been shown to lower VLP egress when mutated (23). Although the molecular basis of lipid recognition or even lipid specificity of VP40 is unknown, the investigation of specific VP40-lipid and VP40-protein interactions at the PM is of great interest as these interactions may serve as pharmacological targets to inhibit Ebola budding and egress.
In conclusion, VP40 associates with nanomolar affinity to PM-like vesicles5 where it penetrates membranes that recapitulate the PM but not the NM with physiological significance. VP40 membrane penetration is attributed to the C-terminal domain where a hydrophobic interface composed of residues including Leu213, Ile293, Leu295, and Val298 (see Fig. 1) is inserted into the membrane. We have demonstrated that these residues are important for PM localization of VP40 and critical to VP40 oligomerization and VLP egress. We also show that an adjacent hydrophobic residue at position Leu303, which is near the hydrophobic interface but more buried in the structure (See Fig. 1) and has been shown to interact with Sec24C (19), did not play an important role in membrane penetration. Although this study and previous work do not rule out lipid binding properties for the N-terminal domain or the unstructured region of ∼40 amino acids at the N terminus of VP40, they strongly suggests that the C-terminal domain hydrophobic insertion is a key determinant of localization, membrane association, and viral egress (Fig. 9).
FIGURE 9.
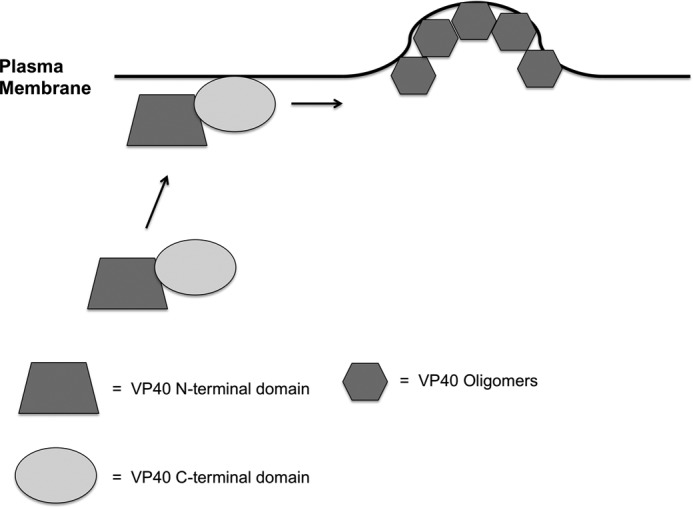
VP40 model of PM assembly, oligomerization, and egress. A model is depicted in which the C-terminal domain of VP40 is responsible for membrane penetration into the plasma membrane of the host cell. Membrane penetration of the VP40 C-terminal domain is important for VP40 oligomerization and VLP budding from the PM of host cells. In the absence of membrane penetration by VP40, VP40 oligomers and sites of VLP egress are not detectable.
Recently, the N-terminal unstructured region and the C-terminal tail have been shown to have latchlike properties that keep VP40 in a monomeric closed conformation by stabilizing the monomeric form (38). These authors also have shown that RNA binding by VP40 can regulate VP40 oligomerization by releasing the latch properties of the two termini. Similarly, membrane insertion by the C-terminal domain may be sufficient to induce dissociation of this latch and prompt VP40 oligomerization. Indeed, Scianimanico et al. (28) demonstrated that membrane association of VP40 was able to induce a conformational change of VP40 from monomeric to hexameric forms. Our studies support this finding and demonstrate that membrane insertion is a prerequisite for oligomerization and that, in addition to hexamers, VP40 also forms larger oligomeric structures. In closing, VP40 behaves similarly to a large number of peripheral proteins (37, 39) where hydrophobic interactions with the membrane bilayer regulate not only membrane penetration but also cellular localization and biological function (37, 39). Moreover, in the case of VP40, membrane penetration may induce conformational changes that can induce oligomerization through loss of N- and C-terminal latch properties.
This work was supported, in whole or in part, by National Institutes of Health Grants AI081077 (to R. V. S.) and P41-RRO3155 and P50-GM076516 (to E. G. and M. A. D.). This work was also supported by the Notre Dame Center for Rare and Neglected Diseases, the Indiana University School of Medicine-South Bend Imaging and Flow Cytometry Core Facility, and the Notre Dame Integrated Imaging Facility (to R. V. S.).
E. Adu-Gyamfi, S. P. Soni, and R. V. Stahelin, unpublished data.
- VP
- viral protein
- BCA
- bicinchoninic acid
- NM
- nuclear membrane
- PC
- phosphatidylcholine
- POPC
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- POPE
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine
- POPS
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylserine
- PM
- plasma membrane
- PS
- phosphatidylserine
- VLP
- virus-like particle
- TIRF
- total internal reflection
- N&B
- number and brightness
- EGFP
- enhanced GFP
- mN
- millinewtons.
REFERENCES
- 1. Johnson K. M., Lange J. V., Webb P. A., Murphy F. A. (1977) Isolation and partial characterisation of a new virus causing acute haemorrhagic fever in Zaire. Lancet 1, 569–571 [DOI] [PubMed] [Google Scholar]
- 2. Elliott L. H., Kiley M. P., McCormick J. B. (1985) Descriptive analysis of Ebola virus proteins. Virology 147, 169–176 [DOI] [PubMed] [Google Scholar]
- 3. Harty R. N. (2009) No exit: targeting the budding process to inhibit filovirus replication. Antiviral Res. 81, 189–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Olejnik J., Ryabchikova E., Corley R.B., Mühlberger E. (2011) Intracellular events and cell fate in filovirus infection. Viruses 3, 1501–1531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Feldmann H., Volchkov V. E., Volchkova V. A., Klenk H. D. (1999) The glycoproteins of Marburg and Ebola virus and their potential roles in pathogenesis. Arch. Virol. Suppl. 15, 159–169 [DOI] [PubMed] [Google Scholar]
- 6. Côté M., Misasi J., Ren T., Bruchez A., Lee K., Filone C. M., Hensley L., Li Q., Ory D., Chandran K., Cunningham J. (2011) Small molecule inhibitors reveal Niemann-Pick C1 is essential for Ebola virus infection. Nature 477, 344–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Reynard O., Nemirov K., Page A., Mateo M., Raoul H., Weissenhorn W., Volchkov V. E. (2011) Conserved proline-rich region of Ebola virus matrix protein VP40 is essential for plasma membrane targeting and virus-like particle release. J. Infect. Dis. 204, Suppl. 3, S884–S891 [DOI] [PubMed] [Google Scholar]
- 8. Makino A., Yamayoshi S., Shinya K., Noda T., Kawaoka Y. (2011) Identification of amino acids in Marburg virus VP40 that are important for virus-like particle budding. J. Infect. Dis. 204, Suppl. 3, S871–S877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Harty R. N., Brown M. E., Wang G., Huibregtse J., Hayes F. P. (2000) A PPxY motif within the VP40 protein of Ebola virus interacts physically and functionally with a ubiquitin ligase: implications for filovirus budding. Proc. Natl. Acad. Sci. U.S.A. 97, 13871–13876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Licata J. M., Simpson-Holley M., Wright N. T., Han Z., Paragas J., Harty R. N. (2003) Overlapping motifs (PTAP and PPEY) within the Ebola virus VP40 protein function independently as late budding domains: involvement of host proteins TSG101 and VPS-4. J. Virol. 77, 1812–1819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hoenen T., Volchkov V., Kolesnikova L., Mittler E., Timmins J., Ottmann M., Reynard O., Becker S., Weissenhorn W. (2005) VP40 octamers are essential for Ebola virus replication. J. Virol. 79, 1898–1905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu Y., Cocka L., Okumura A., Zhang Y. A., Sunyer J. O., Harty R. N. (2010) Conserved motifs within Ebola and Marburg virus VP40 proteins are important for stability, localization, and subsequent budding of virus-like particles. J. Virol. 84, 2294–2303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Noda T., Ebihara H., Muramoto Y., Fujii K., Takada A., Sagara H., Kim J. H., Kida H., Feldmann H., Kawaoka Y. (2006) Assembly and budding of Ebolavirus. PLoS Pathog. 2, e99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Panchal R. G., Ruthel G., Kenny T. A., Kallstrom G. H., Lane D., Badie S. S., Li L., Bavari S., Aman M. J. (2003) In vivo oligomerization and raft localization of Ebola virus protein VP40 during vesicular budding. Proc. Natl. Acad. Sci. U.S.A. 100, 15936–15941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Timmins J., Schoehn G., Kohlhaas C., Klenk H.-D., Ruigrok R. W., Weissenhorn W. (2003) Oligomerization and polymerization of the filovirus matrix protein VP40. Virology 312, 359–368 [DOI] [PubMed] [Google Scholar]
- 16. Hoenen T., Biedenkopf N., Zielecki F., Jung S., Groseth A., Feldmann H., Becker S. (2010) Oligomerization of Ebola virus VP40 Is essential for particle morphogenesis and regulation of viral transcription. J. Virol. 84, 7053–7063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Neumann G., Ebihara H., Takada A., Noda T., Kobasa D., Jasenosky L. D., Watanabe S., Kim J. H., Feldmann H., Kawaoka Y. (2005) Ebola virus VP40 late domains are not essential for viral replication in cell culture. J. Virol. 79, 10300–10307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fairn G. D., Schieber N. L., Ariotti N., Murphy S., Kuerschner L., Webb R. I., Grinstein S., Parton R. G. (2011) High-resolution mapping reveals topologically distinct cellular pools of phosphatidylserine. J. Cell Biol. 194, 257–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yamayoshi S., Noda T., Ebihara H., Goto H., Morikawa Y., Lukashevich I. S., Neumann G., Feldmann H., Kawaoka Y. (2008) Ebola virus matrix protein VP40 uses the COPII transport system for its intracellular transport. Cell Host Microbe 3, 168–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Han Z., Harty R. N. (2005) Packaging of actin into Ebola virus VLPs. Virol. J. 2, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Adu-Gyamfi E., Digman M. A., Gratton E., Stahelin R. V. (2012) Single-particle tracking demonstrates that actin coordinates the movement of the Ebola virus matrix protein. Biophys. J. 103, L41–L43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. García M., Cooper A., Shi W., Bornmann W., Carrion R., Kalman D., Nabel G. J. (2012) Productive replication of Ebola virus is regulated by the c-Abl1 tyrosine kinase. Sci. Transl. Med. 4, 123ra24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McCarthy S. E., Johnson R. F., Zhang Y. A., Sunyer J. O., Harty R. N. (2007) Role for amino acids 212KLR214 of Ebola virus VP40 in assembly and budding. J. Virol. 81, 11452–11460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Adu-Gyamfi E., Digman M. A., Gratton E., Stahelin R. V. (2012) Investigation of Ebola VP40 assembly and oligomerization in live cells using number and brightness analysis. Biophys. J. 102, 2517–2525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gomis-Rüth F. X., Dessen A., Timmins J., Bracher A., Kolesnikowa L., Becker S., Klenk H. D., Weissenhorn W. (2003) The matrix protein VP40 from Ebola virus octamerizes into pore-like structures with specific RNA binding properties. Structure 11, 423–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ross J. A., Digman M. A., Wang L., Gratton E., Albanesi J. P., Jameson D. M. (2011) Oligomerization state of dynamin 2 in cell membranes using TIRF and number and brightness analysis. Biophys. J. 100, L15–L17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Digman M. A., Dalal R., Horwitz A. F., Gratton E. (2008) Mapping the number of molecules and brightness in the laser scanning microscope. Biophys. J. 94, 2320–2332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Scianimanico S., Schoehn G., Timmins J., Ruigrok R. H., Klenk H. D., Weissenhorn W. (2000) Membrane association induces a conformational change in the Ebola virus matrix protein. EMBO J. 19, 6732–6741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Stahelin R. V., Long F., Diraviyam K., Bruzik K. S., Murray D., Cho W. (2002) Phosphatidylinositol 3-phosphate induces the membrane penetration of the FYVE domains of Vps27p and Hrs. J. Biol. Chem. 277, 26379–26388 [DOI] [PubMed] [Google Scholar]
- 30. Stahelin R. V., Burian A., Bruzik K. S., Murray D., Cho W. (2003) Membrane binding mechanisms of the PX domains of NADPH oxidase p40phox and p47phox. J. Biol. Chem. 278, 14469–14479 [DOI] [PubMed] [Google Scholar]
- 31. Stahelin R. V., Long F., Peter B. J., Murray D., De Camilli P., McMahon H. T., Cho W. (2003) Contrasting membrane interaction mechanisms of AP180 N-terminal homology (ANTH) and epsin N-terminal homology (ENTH) domains. J. Biol. Chem. 278, 28993–28999 [DOI] [PubMed] [Google Scholar]
- 32. Bittova L., Sumandea M., Cho W. (1999) A structure-function study of the C2 domain of cytosolic phospholipase A2. Identification of essential calcium ligands and hydrophobic membrane binding residues. J. Biol. Chem. 274, 9665–9672 [DOI] [PubMed] [Google Scholar]
- 33. Medkova M., Cho W. (1999) Interplay of C1 and C2 domains of protein kinase C-α in its membrane binding and activation. J. Biol. Chem. 274, 19852–19861 [DOI] [PubMed] [Google Scholar]
- 34. Cho W., Bittova L., Stahelin R. V. (2001) Membrane binding assays for peripheral proteins. Anal. Biochem. 296, 153–161 [DOI] [PubMed] [Google Scholar]
- 35. Marsh D. (1996) Lateral pressure in membranes. Biochim. Biophys. Acta 1286, 183–223 [DOI] [PubMed] [Google Scholar]
- 36. Demel R. A., Geurts van Kessel W. S., Zwaal R. F., Roelofsen B., van Deenen L. L. (1975) Relation between various phospholipase actions on human red cell membranes and the interfacial phospholipid pressure in monolayers. Biochim. Biophys. Acta 406, 97–107 [DOI] [PubMed] [Google Scholar]
- 37. Cho W., Stahelin R. V. (2005) Membrane-protein interactions in cell signaling and membrane trafficking. Annu. Rev. Biophys. Biomol. Struct. 34, 119–151 [DOI] [PubMed] [Google Scholar]
- 38. Silva L. P., Vanzile M., Bavari S., Aman J. M., Schriemer D. C. (2012) Assembly of Ebola virus matrix protein VP40 is regulated by latch-like properties of N and C terminal tails. PLoS One 7, e39978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Scott J. L., Musselman C. A., Adu-Gyamfi E., Kutateladze T. G., Stahelin R. V. (2012) Emerging methodologies to investigate lipid-protein interactions. Integr. Biol. 4, 247–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dessen A., Volchkov V., Dolnik O., Klenk H. D., Weissenhorn W. (2000) Crystal structure of the matrix protein VP40 from Ebola virus. EMBO J. 19, 4228–4236 [DOI] [PMC free article] [PubMed] [Google Scholar]